

Abstract

Photoinitiated silane–ene chemistry has the potential to pave the way toward spatially resolved organosilicon compounds, which might find application in biomedicine, microelectronics, and other advanced fields. Moreover, this approach could serve as a viable alternative to the popular photoinitiated thiol–ene chemistry, which gives access to defined and functional photopolymer networks. A difunctional bis(trimethylsilyl)silane with abstractable hydrogens (DSiH) was successfully synthesized in a simple one-pot procedure. The radical reactivity of DSiH with various homopolymerizable monomers (i.e., (meth)acrylate, vinyl ester, acrylamide) was assessed via 1H NMR spectroscopic studies. DSiH shows good reactivity with acrylates and vinyl esters. The most promising silane–acrylate system was further investigated in cross-linking formulations toward its reactivity (e.g., heat of polymerization, curing time, occurrence of gelation, double-bond conversion) and compared to state-of-the-art thiol–acrylate resins. The storage stability of prepared resin formulations is greatly improved for silane–acrylate systems vs thiol–ene resins. Double-bond conversion at the gel point (DBCgel) and overall DBC were increased, and polymerization-induced shrinkage stress has been significantly reduced with the introduction of silane–acrylate chemistry. Resulting photopolymer networks exhibit a homogeneous network architecture (indicated by a narrow glass transition) that can be tuned by varying silane concentration, and this confirms the postulated regulation of radical network formation. Similar to thiol–acrylate networks, this leads to more flexible photopolymer networks with increased elongation at break and improved impact resistance. Additionally, swelling tests indicate a high gel fraction for silane–acrylate photopolymers.
Introduction
Radical photopolymerization1 finds application in various fields from classical coatings2 and adhesives to more advanced technologies such as biomaterials3,4 or 3D structuring.5,6 The underlying photoinitiated radical polymerization mechanism enables rapid formation of polymer networks within seconds. Usually, radical photopolymerization proceeds in a chain growth manner yielding highly cross-linked polymers that exhibit inhomogeneous network architectures. The rapid formation of such photopolymer networks results in high internal shrinkage stress and incomplete conversions. As a consequence, the polymerization-induced shrinkage stress often leads to stress cracking and mechanical failure of the photopolymer, and residual unreacted double bonds within the material may cause toxic side reactions, especially when applied in biomedicine.7 Moreover, brittle and glassy materials are fabricated, and this limits the application of such materials in more advanced fields such as biomaterials, microelectronics, dental materials, and stereolithography.8
Gaining regulation over this radical network formation has been emphasized in the past through chain transfer techniques (e.g., thiol–ene/yne chemistry,9,10 regulation via addition–fragmentation chain transfer11−14). By introduction of a chain transfer agent (CTA), the radical chain growth process can be altered toward a mixed chain growth/step growth-like mechanism, thus giving access to a more regulated network formation with reduced shrinkage stress and improved conversions. The final materials exhibit easily tunable thermomechanical properties and improved toughness.
Thiol–ene/yne chemistry represents a unique approach in photopolymerization targeting major challenges such as oxygen inhibition15 and opening new possibilities for the toughening of materials,7,16 two-stage reactive systems for functional materials,17,18 and applications in biomedicine.19 However, drawbacks such as strong odor of thiols20 and limited storage stability of the thiol–ene formulations21,22 have motivated further development of these materials. Inspired by thiol–ene chemistry and its vast potential, phosphane–,23 germane–,24 and iodo–ene25 polymerizations have recently been introduced as possible alternatives.
Another promising candidate would be silane–ene chemistry, which has already been proposed in the literature.26 By exploring various silanes with Si–H bonds reactive toward radical abstraction, a suitable silane–ene system could be developed. A reactive silane–ene system could also serve as a powerful tool for photografting applications on silicon surfaces27 and in silicon polymer science.28 Such silanes have also shown potential to reduce oxygen inhibition29 due to the high reactivity of silane radicals toward molecular oxygen (kox ∼ 3 × 109 L mol–1 s–1) and might act as radical reducing agents30 or type II co-initiators.31,32 Tris(trimethylsilyl)silane (TTMSSiH) has been identified as the most promising candidate for radical silane–ene chemistry due to its comparatively low bond dissociation energy33 and the high reactivity of the respective silyl radical toward addition to enes.34 Nevertheless, multifunctional derivatives based on TTMSSiH are not easily accessible, thus limiting the use in photopolymer networks.
In a recent study we have shown the synthesis and assessed the reactivity of a monosubstituted bis(trimethylsilyl)silane with various enes.35 Efficient chain transfer activity of the tested silane could be confirmed, and the radical reactivity with acrylates has shown the most promising results. A first regulated acrylate-based photopolymer network with reduced cross-linking density and a more defined thermal polymer phase transition has been fabricated.
In this paper we present the synthesis of a difunctional bis(trimethylsilyl)silane (DSiH), which has been accomplished using a divinyl ether as precursor. The reactivity of DSiH in photoinitiated radical reactions with various homopolymerizable monomers (i.e., (meth)acrylate, vinyl ester, acrylamide) was assessed by determining the respective silane SiHC and double-bond conversions DBC via 1H NMR spectroscopy. The most promising silane–acrylate system has been studied in greater detail. Difunctional silane–acrylate formulations have been prepared, and their storage stability was assessed via rheometry and NMR spectroscopy. Then, the photopolymerization reaction of the respective formulations was studied (photo-DSC, real time (RT)-NIR-photorheology), and the thermomechanical and mechanical properties of the final photopolymer networks were investigated (DMTA, tensile testing, Dynstat impact test, swelling tests). The obtained results were compared to a thiol–acrylate reference system.
Experimental Section
Materials and General Methods
The chemicals tris(trimethylsilyl)silane (TTMSSiH, abcr), 1,4-butanediol divinyl ether (BDE, Sigma-Aldrich), benzyl acrylate (BA, abcr), benzyl methacrylate (BMA, Sigma-Aldrich), vinyl benzoate (VB, Sigma-Aldrich), N-acryloylmorpholine (NAM, Sigma-Aldrich), pyrogallol (Sigma-Aldrich), 1,10-decanediol diacrylate (D3A, TCI), triethylene glycol dithiol (DSH, Sigma-Aldrich), and the photoinitiator 2-hydroxy-2-methyl-1-phenylpropan-1-one (Darocur 1173, Ciba) were purchased from the respective companies and used without further purification. The photoinitiator bis(4-methoxybenzoyl)diethylgermane36 (BMDG) and the strongly acidic monomer (2-((2-(ethoxycarbonyl)allyl)oxy)ethyl)phosphonic acid (MA) were kindly provided by Ivoclar Vivadent AG.
A Bruker Avance DRX-400 was used for NMR spectroscopy at 400 MHz for 1H (100 MHz for 13C, 79.5 MHz for 29Si), and chemical shifts were reported in ppm. They were referenced to the solvent residual peak for 1H and 13C nuclei (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm). Chemical shifts of 29Si nuclei are reported to SiMe4 as external standard and further an INEPT pulse sequence was used for enhancement of the 29Si signals. Multiplicities are referred to as s (singlet), d (doublet), and m (multiplet) and coupling constants (J values) are given in hertz. Silica gel chromatography was performed with a Büchi MPLC-system equipped with the control unit C-620, fraction collector C-660, and RI-detector Refractom. Commercial grade reagents (potassium tert-butoxide KOtBu, Sigma-Aldrich; HCl, VWR) and solvents (THF, Acros; petrol ether and ethyl acetate, Donau Chemie) were used without further purification. An Ocean Optics USB 2000+ spectrometer was used to measure the total irradiation intensities at the position of the samples.
Synthesis of a Difunctional Silane (DSiH)
A simple one-pot synthesis of the difunctional silane (DSiH) was performed in two steps. The first step was conducted in an Ar-flushed flask, which was charged with the photoinitiator Daraocur 1173 (115.2 mg, 0.7 mmol, 0.02 equiv), TTMSSiH (17.42 g, 70 mmol, 2 equiv), and BDE (4.98 g, 35 mmol, 1 equiv). The reaction solution was irradiated for 2 h with an Omnicure EXFO 2000 light source (Hg broadband lamp, 320–500 nm, ∼10 mW cm–2 on the surface of the reaction solution) using a quick-fit with integrated quartz glass window. After irradiation, KOtBu (8.27 g, 73.5 mmol) was added to the reaction together with 100 mL of absolute THF. The solution was stirred for another 6 h at ambient temperature and then quenched by pouring it onto 300 mL of ice-cold 2 N HCl. The aqueous phase was extracted with petrol ether (5 × 200 mL), and the combined organic phases were dried over Na2SO4. The solvent was evaporated, and the crude product was purified via silica column chromatography (7% ethyl acetate in petrol ether).
2,2,15,15-Tetramethyl-3,14-bis(trimethylsilyl)-6,11-dioxa-2,3,14,15-tetrasilahexadecane (DSiH): colorless liquid; 54% yield (9.33 g). 1H NMR (400 MHz, CDCl3, δ, ppm): 3.49–3.39 (m, 8H; −CH2–O–CH2−), 2.95 (t, 2J = 4.7 Hz, 2H; Si–H), 1.66–1.61 (m, 4H; −CH2–CH2−), 1.18–1.12 (m, 4H; −CH2–Si), 0.16 (s, 36H; −Si(CH3)3). 13C NMR (100 MHz, CDCl3, δ, ppm): 70.5 (C2), 70.2 (C2), 26.7 (C2), 9.0 (C2), 0.2 (C1). 29Si INEPT NMR (79.5 MHz, CDCl3, δ, ppm): −12.6, −15.1. The respective NMR spectra are displayed in the Supporting Information (Figures S1–S3).
Preparation of Resin Formulations and Photopolymer Specimens
For first photoreactivity tests formulations of various homopolymerizable monomers (i.e., acrylate BA, methacrylate BMA, vinyl ester VB, and acrylamide NAM) with TTMSSiH or DSiH (ratio of double bond/SiH = 1/1) were prepared, and 0.5 wt % Darocur 1173 was added as photoinitiator. Reference formulations of monomer and photoinitiator (0.5 wt %) were prepared as well.
For photo-DSC and RT-NIR-photorheology experiments the difunctional acrylate D3A was used as reference mixed with 1 wt % photoinitiator (BMDG). Formulations with 5, 20, and 50 mol % chain transfer agent (i.e., silane DSiH or thiol DSH) were prepared and homogenized in an ultrasonic bath for 30 min. All thiol-based formulations were additionally stabilized with pyrogallol (9 mM) and the acidic monomer MA (90 mM).22
For the preparation of photopolymer specimens for mechanical testing formulations based on D3A and with the respective amounts of CTA (i.e., DSH or DSiH) were prepared analogously to the photo-DSC and RT-NIR-photorheology experiments. Here, 0.2 wt % of BMDG was added to the formulations and photopolymer specimens of all D3A-based formulations were prepared by casting the respective resins in silicone molds (rectangular-shaped 5 × 2 × 40 mm3 for DMTA and 10 × 4 × 15 mm3 for Dynstat test, dumbbell-shaped with a total length of 35 mm and a parallel constriction region dimension of 2 × 2 × 12 mm3 for tensile test, disc-shaped ϕ = 4 mm, h = 2 mm for swelling tests). The formulations were photocured in a Lumamat 100 light oven provided by Ivoclar Vivadent AG. Osram Dulux L Blue lamps were used as irradiation source (18 W, 400–580 nm). A total intensity of ∼20 mW cm–2 was determined at the position of the silicone molds with an Ocean Optics USB 2000+ spectrometer. All samples were irradiated for 2 × 10 min and flipped in between irradiation periods. After curing, the test specimens were sanded to ensure uniform sample geometries.
Photoreactivity Tests via 1H NMR Spectroscopy
Photoinitiated radical reactions with acrylate BA, methacrylate BMA, vinyl ester VB, and acrylamide NAM were performed. Mixtures (∼100 mg) of monomer (1 equiv), silane (i.e., 1 equiv TTMSSiH or 0.5 equiv DSiH ), and Darocur 1173 (0.5 mol %) were prepared (1/1 molar ratio of double bond DB/silane SiH). The respective formulations were purged with Ar and then divided into two NMR tubes. One of the tubes was exposed to filtered UV light (5 min, 320–500 nm, ∼26 mW cm–2 on the surface of the NMR tube) from an Exfo OmniCure S2000 broadband Hg lamp. After the irradiation period the reactions were quenched with CDCl3 (0.5 mL, nonirradiated reference samples were diluted immediately). From the measured 1H NMR spectra the overall conversions for monomer (double-bond conversion, DBC) and silane (SiHC) were derived. Conversion data assessed with NMR spectroscopy are reliable with an error of <1%. Moreover, the accuracy of the adapted NMR spectroscopic method can be assumed with ±3%.
Storage Stability Tests
Storage stability tests of the prepared monomer formulations were conducted on a modular compact rheometer MCR 300 by Physica Anton Paar. Rheology measurements were performed at the start and after a storage period of 1 and 20 days, respectively. Samples were stored in the dark at ambient conditions. The viscosity of the formulations was measured at 20 °C with a CP-25-1 measuring system (cone–plate, diameter 25 mm, angle 1°) at a gap of 48 μm and a shear rate of 100 s–1. Thiol-based formulations were not additionally stabilized in this case.
Photo-DSC
A Netzsch DSC 204 F1 coupled with an Omnicure light source (Hg broadband, filtered UV light 400–500 nm, 1 W cm–2 at the exit of the light guide corresponding to ∼20 mW cm–2 on the surface of the sample) was used for photo-DSC measurements. All samples were exactly weighed into aluminum pans (10 ± 1 mg) and irradiated for 2 × 5 min at 25 °C under inert atmosphere (N2 flow rate = 20 mL min–1). The second irradiation period of 5 min was conducted to eliminate the influence of heat effects coming from light absorption of the sample or the aluminum pan. Triplicate measurements were performed for each sample formulation.
RT-NIR-Photorheology
Experiments were performed on a hyphenated RT-NIR-photorheology setup with a Bruker Vertex 80 FTIR spectrometer and an Anton Paar MCR302 WESP rheometer equipped with a PP-25 steel measurement plate and a P-PTD 200/GL Peltier element.37 The temperature at the optical rheometer plate was set to 20 °C, and a sample volume of 140 μL was placed at the center of the plate. Then the measurement gap was set to 200 μm. Before UV irradiation, the respective samples were measured via NIR spectroscopy and analyzed by rheology. The formulations were oscillated with a strain of 1% and a frequency of 1 Hz. To start the reaction, UV-light was projected onto the sample from the underside of the optical plate using an Exfo OmniCureTM 2000 with a broadband Hg lamp (300 s, 400–500 nm, ∼10 mW cm–2 on the surface of the sample). The curing reaction is monitored by recording the storage modulus G′ and loss modulus G″ of the sample as well as time-resolved NIR spectra. The gel point of each reaction was derived from the intersection of storage and loss modulus (G′/G″ = 1). The chemical conversion (double-bond conversion DBC of the acrylate functionality) was determined by recording a set of single spectra with a time interval of ∼0.2 s using the software OPUS 7.0. The relevant peak area for the reactive acrylate double bonds (i.e., overtone at 6080–6250 cm–1) was then evaluated. The ratio of the peak areas from the start to the end of the measurement gave the DBC plot. The conversion at the gel point (DBCgel) and final double-bond conversion (DBCfinal) were derived from this plot. All measurements were performed in duplicate giving satisfactory reproducibility.
Dynamic Mechanical Thermal Analysis (DMTA)
For DMTA measurements an Anton Paar MCR 301 with a CTD 450 oven and an SRF 12 measuring system was used. The experiments were performed in torsion mode with a frequency of 1 Hz, strain of 0.1%, constant normal force of −1 N, and a set temperature program (−100 to 200 °C, 2 °C min–1). The software Rheoplus/32 V3.40 from Anton Paar was used to record the storage modulus and the loss factor of the polymer samples.
Mechanical Testing
Tensile Test
A Zwick Z050 testing machine, equipped with a 1 kN load cell, was used for tensile tests (ISO 527). For each sample, five dumbbell specimens were tested, and the respective specimens were strained with a crosshead speed of 5 mm min–1. A stress–strain plot was recorded simultaneously.
Dynstat Impact Test
Dynstat impact testing was performed according to DIN 51230. The prepared polymer specimens were broken with a 1 J hammer. For each sample four specimens were tested, and the acquired impact resistance value was normalized to the width and thickness of the tested specimens. The impact resistance is determined by the ratio of work required to break the respective specimen to the cross section of the sample at the fracture site.
Swelling Tests
The disc-shaped polymer specimens were submerged in ethanol and stored at ambient conditions for 7 days. The inhibitor hydroquinone monomethyl ether (200 ppm) was added to the ethanol to prevent additional free-radical reactions in the dark. The ethanol was replaced twice after 1 and 3 days. The polymer discs were dried using a paper towel and then weighed. Afterward, the discs were placed in a 60 °C vacuum oven and dried until a constant weight was reached.
Results and Discussion
Synthesis of the Difunctional Silane DSiH
Based on the previously reported synthetic procedure,35 a difunctional silane (DSiH) was synthesized in one pot starting from butanediol divinyl ether BDE and tris(trimethylsilyl)silane TTMSSiH (Scheme 1). The photoinitiator Darocur 1173 was added to the reaction, and the mixture was irradiated with broadband UV light. The photoinitiator is cleaved homolytically, and the formed radicals can abstract the hydrogen from TTMSSiH. The silyl radical then attacks the double bond of BDE, and in the following a hydrogen atom of the next TTMSSiH molecule is abstracted. As BDE does not undergo homopolymerization the synthesis follows a clean radical addition and hydrogen abstraction process leading to the intermediate product DSi. Treating this intermediate with KOtBu in absolute THF yields the desired product in satisfactory yield of >50% after silica column chromatography.
Scheme 1. Synthetic Route to a Difunctional Bis(trimethylsilyl)silane with Abstractable Hydrogens (DSiH).

Reactivity of DSiH with Different Enes in Photoinitiated Radical Reactions
The synthesized difunctional silane DSiH was tested toward its coreactivity with various homopolymerizable enes (0.5 equiv of DSiH in acrylate BA, methacrylate BMA, vinyl ester VB, and acrylamide NAM) in a radical photopolymerization using Darocur 1173 as photoinitiator. The pure monomers and equimolar mixtures of monomer and silane TTMSSiH were studied previously35 and used as references. After UV irradiation (5 min, 320–500 nm, ∼26 mW cm–2) the final polymers were analyzed via 1H NMR spectroscopy to assess final double-bond DBC and silane conversion SiHC. DSiH–acrylate (97% DBC, 61% SiHC) and DSiH–vinyl ester formulations (77% DBC, 62% SiHC) show the highest coreactivity with silane conversions >60% and a ratio of DBC/SiHC < 2. The BMA- (25% DBC, 10% SiHC) and NAM-based fomulations (87% DBC, 30% SiHC) showed low silane conversion and high ratios of DBC/SiHC > 2 and thus have not been studied in further detail. With acrylates as the most relevant photopolymerizable functionality the potential for silane–acrylate photopolymerization is most promising and has been the focus within this publication. Nevertheless, all studied monomers showed radical reactivity with DSiH (Figure S4), and investigating such systems under a wider scope could be interesting especially when thinking about polymerizations in solution and thermal initiation.
Radical Mechanism of Silane–Ene Network Formation
After preliminary studies of the synthesized silane DSiH in monofunctional monomers, the focus was shifted toward formation of photopolymer networks via the promising silane–acrylate chemistry. The underlying radical reaction mechanism has been previously reported by the research group of Fouassier,26 and a schematic illustration of the desired silane–ene network formation is displayed in Figure 1a. Photopolymerization is initiated by the activation of a photoinitiator via light impulse and subsequent homolytical bond cleavage. The formed radicals start the polymerization reaction, and in the case of homopolymerizable acrylates a rapid and unregulated radical chain growth reaction is started and continued via propagation with additional monomers. Similarly to the well-known thiol–ene concept, the silane DSiH is expected to introduce a step growth-like radical polymerization with the termination of growing radical fragments via hydrogen abstraction and a reinitiation step of the formed silyl radical with acrylates. Consequently, a mixed radical chain growth/step growth-like process is generated, and the ratio of propagation to chain transfer determines the resulting polymer network architecture. In thiol–acrylate systems the radical chain growth propagation step of acrylates is favored, thus limiting the ability of the thiol to regulate the network formation.38,39 From the above-mentioned preliminary results a more homogeneous silane–acrylate polymerization can be expected and will be tested for its ability to increase the final acrylate conversion, reduce polymerization induced shrinkage stress, and regulate the final network architecture.
Figure 1.

(a) Mechanism for radical silane–ene network formation. (b) Reference monomer D3A, thiol reference DSH, difunctional silane DSiH, and photoinitiator BMDG.
Storage Stability Assessment of a Silane–Acrylate System
Premixed photopolymerizable formulations are required for convenient and user-friendly application, thus making storage stability a crucial aspect when designing new resins. Thiol–ene formulations are known for their poor stability as they tend to form charge-transfer complexes and undergo spontaneous polymerization in the dark. Research has taken efforts to control or even prevent certain thiol–ene formulations from pregelation.21,22 While systems such as thiol–yne show fairly good stability,40 the thiol–acrylate system tends to gel after only a few hours or sometimes even within minutes. This has limited the commercial exploitation of the otherwise quite promising thiol–ene concept.
We have prepared silane–acrylate formulations with the commercially available difunctional acrylate D3A and the synthesized difunctional silane DSiH (20 mol %). Pure D3A was employed as acrylate reference, and a mixture of D3A and dithiol DSH (20 mol %), with a comparable structure to DSiH, was used as CTA-based reference formulation (Figure 1b). All three formulations (D3A, D3A/DSiH 20, and D3A/DSH 20) were premixed with BMDG as photoinitiator, and the viscosity at ambient conditions was assessed with a rheometer. Then, the formulations were stored in the dark at ambient conditions, and the viscosity was reevaluated after storage periods of 1 and 20 days, respectively (Figure 2). As expected, the pure acrylate resin exhibits good storage stability, while the thiol–acrylate system exhibits a constant increase in viscosity and shows gelation after 20 days. However, the respective silane–acrylate formulation shows great storage stability during the entire experiment (no significant viscosity change). Additionally, NMR spectroscopic measurements of the stored silane–acrylate formulation after 64 days confirm a good storage stability of >2 months (Figure S5).
Figure 2.

Viscosity for reference D3A (green), D3A/DSiH 20 (blue), and D3A/DSH 20 (gray) at start (solid), after 1 day (dense pattern), and after 20 days (sparse pattern).
Reactivity of Silane–Acrylate Formulations in Photoinitiated Radical Reactions
Photo-DSC
In order to assess the reactivity of the prepared acrylate-based formulations, photo-DSC experiments were performed at 25 °C under a nitrogen atmosphere. Formulations with 5, 20, and 50 mol % CTA (i.e., DSiH or DSH) were studied. The samples were irradiated with visible light (400–500 nm, ∼20 mW cm–2, 5 min), and the evolved heat of polymerization upon irradiation was measured. The pure D3A reference exhibits the highest heat of polymerization, which was to be expected due to the rapid gelation of the cross-linking photopolymerization. The values for tmax (time to maximum polymerization rate, 2.6 s) and t95% (time to 95% of heat evolution, 20 s) are measures for the reaction rate and further confirm the rapid gelation and final reaction time for the D3A formulation (Table 1). The value for tmax can be viewed as an indication for the gelation of the respective photopolymerizations. As expected, the addition of chain transfer agents to the photopolymerizable resins results in reduced polymerization heat and a delay of gelation. This effect is more pronounced with increasing CTA content (e.g., for DSiH in Figure 3a), but most importantly the final reaction time is only slightly increased by the addition of CTA (t95% values: 20 s for D3A, 23 s for D3A/DSH 20 mol %, and 33 s for D3A/DSiH 20 mol %, Table 1).
Table 1. Results from Photo-DSCa and RT-NIR-Photorheologyb.
| Photo-DSC | RT-NIR-Photorheology | |||||||
|---|---|---|---|---|---|---|---|---|
| formulation | ΔH (J g–1) | tmaxc (s) | t95%b (s) | G′final (MPa) | tgelc (s) | DBCgel (%) | DBCfinal (%) | FN (%) |
| D3A | 472 | 2.6 | 20 | 0.63 | 1.0 | 22 | 93 | 100 |
| D3A/DSH 5 | 447 | 3.1 | 23 | 0.61 | 2.2 | 39 | 95 | 90 |
| D3A/DSH 20 | 442 | 3.9 | 23 | 0.65 | 2.9 | 51 | >99 | 76 |
| D3A/DSH 50 | 310 | 3.9 | 20 | 0.44 | 4.2 | 82 | >99 | 41 |
| D3A/DSiH 5 | 445 | 5.2 | 22 | 0.72 | 3.1 | 26 | 95 | 84 |
| D3A/DSiH 20 | 398 | 9.1 | 33 | 0.65 | 8.1 | 40 | 97 | 65 |
| D3A/DSiH 50 | 231 | 11.3 | 28 | 0.28 | 16.8 | 75 | 97 | 26 |
ΔH is the measured heat of polymerization; tmax is the time to maximum of polymerization rate; t95% is the time to 95% of heat evolution.
G′final is the final storage modulus; tgel is the time to gel point; DBCgel is the double-bond conversion at gel point; DBCfinal is the final double-bond conversion; FN is the measured normal force. Standard deviation for final values can be seen in Tables S1 and S2.
The initial measurement period of 5 s before light irradiation has been subtracted.
Figure 3.
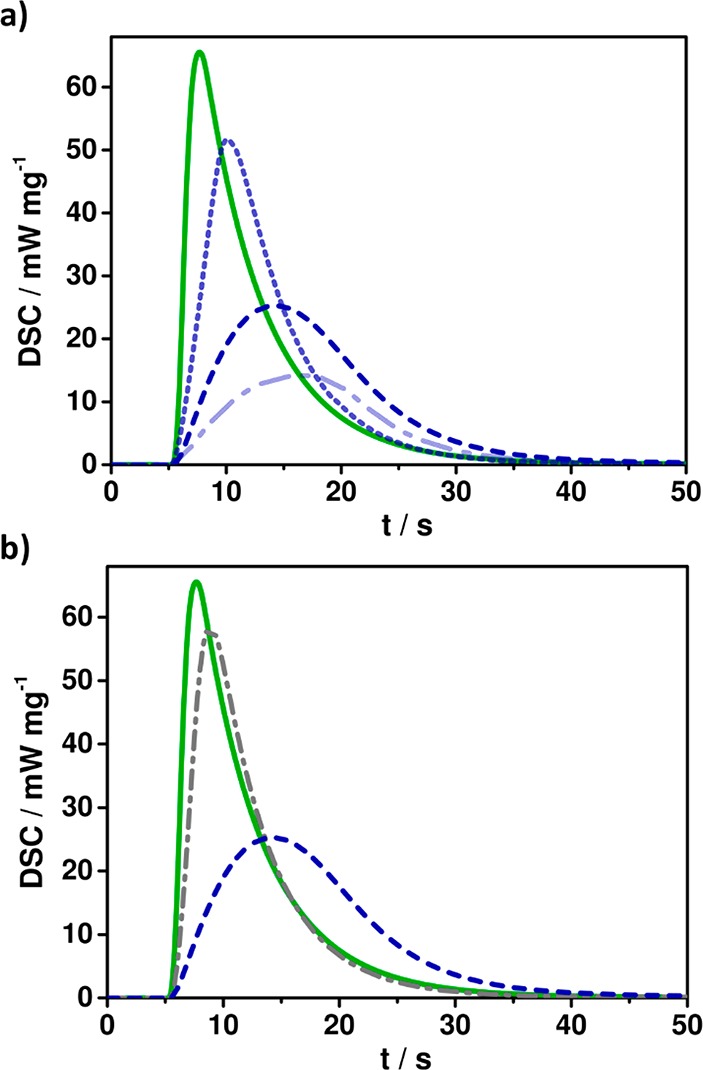
Photo-DSC plots (a) for reference D3A (solid), D3A/DSiH 5 (short dash), 20 (dash), and 50 (dash-dot) and (b) for reference D3A (solid), D3A/DSH 20 (dash-dot), and D3A/DSiH 20 (dash); light starts at 5 s.
From the previously conducted model reactions with DSiH an increased ratio of chain transfer to acrylate propagation is expected for formulations with D3A compared to the reference D3A with dithiol DSH. The photo-DSC experiments with the silane DSiH confirm these expectations by the increased shift in gelation (higher tmax) compared to thiol-based resins (Figure 3b). The reduced polymerization heat can be also interpreted as additional indication for more chain transfer reactions and the suppression of acrylate propagation in the case for D3A/DSiH samples. Nevertheless, the heat of polymerization is difficult to interpret, as the knowledge of the real ratio between propagation and chain transfer and also the reaction heat values for each individual reaction step would need to be considered for a true estimation.
RT-NIR-Photorheology
Photo-DSC measurements gave a first real indication of the good reactivity of photoinitiated radical silane–acrylate chemistry with the difunctional silane DSiH in a cross-linking matrix. RT-NIR-photorheology could further confirm these findings by recording characteristics for mechanical curing (via storage modulus G′ and shrinkage force FN) and chemical conversion (via NIR tracking) in situ during the photopolymerization reactions. This yields a more elaborate understanding for the effects of chain transfer reactions on the acrylate network formation. While photo-DSC measurements gave an indication about the time to gelation for each photopolymerization, the monitoring of storage G′ and loss modulus G″ during the reaction allows a direct evaluation of gel time, determined by the intersection of G′ and G″. Moreover, the respective conversion of acrylate groups at the gel point DBCgel can be assessed through NIR spectroscopic tracking. In principle, the slower the initial curing reaction, the later gelation is reached (higher DBCgel), and this has been found to directly correlate with the concentration of chain transfer content, further confirming this assumption from photo-DSC.
The concentration series of 5, 20, and 50 mol % of the disilane DSiH in D3A-based resins show that the addition of DSiH yields a significant increase in gel time (tgel increased by a factor of >10 for D3A/DSiH 50), and simultaneously the double-bond conversion at the gel point is vastly increased (DBCgel > 70%, Table 1). This not only leads to an increase in overall double-bond conversion (DBCfinal > 95% in DSiH-based formulations, Figure 4a) but also significantly reduces polymerization induced shrinkage stress. Once the gel point is reached, polymerization induced shrinkage stress is formed within the respective polymer networks. A shift of gelation toward higher conversion results in less chemical reactions in the gel state, which inherently leads to a reduction of shrinkage stress for the respective photopolymerizations. This effect can be monitored in situ by following the evolution of the normal force FN, which is recorded by the rheology measurement system. From the resulting FN plots the expected reduction of shrinkage stress with the introduction of DSiH in acrylate photopolymerization is shown (Figure 4b). An addition of only 5 mol % of DSiH already yields a 20% reduction of shrinkage stress, which can be further reduced to as little as ∼25% of initial shrinkage in D3A/DSiH 50 formulations. With DSiH contents >20% the final storage modulus G′ is reduced (Figure 4c). Nevertheless, formulations with up to 20 mol % DiSH exhibit a comparably high final G′ value as reference resin D3A.
Figure 4.

(a) DBC, (b) shrinkage force FN, and (c) storage modulus G′ plots for reference D3A (solid), D3A/DSiH 5 (short dash), 20 (dash), and 50 (dash-dot); light starts at 5 s.
The comparison with DSH-based resins also shows that the conversion of acrylate double bonds is slower for D3A/DSiH 20 compared to the acrylate-based reference and thiol-based resins (Figure 5a). However, this can be expected as the preliminary tests showed a much more uniform reaction of DSiH with acrylates. In the case of thiol–acrylate formulations the fast double-bond conversion goes hand in hand with a low chain transfer reactivity of thiols, which has been stated in the literature.38,39 This assumption is further confirmed by the significantly reduced shrinkage force for DSiH-based resins compared to their thiol-based analogues (Figure 5b). Moreover, while gelation is delayed for DSiH-based samples, the final G′ remains comparably high (Figure 5c).
Figure 5.

(a) DBC, (b) shrinkage force FN, and (c) storage modulus G′ plots for reference D3A (solid), D3A/DSH 20 (dash-dot), and D3A/DSiH 20 (dash); light starts at 5 s.
Thermomechanical Properties
Dynamic Mechanical Thermal Analysis (DMTA)
Following the detailed investigation of photoinitiated radical silane–acrylate network formation, the resulting photopolymer networks were investigated toward their thermomechanical properties. A crucial characteristic for homogeneous polymer networks is their thermomechanical behavior and the underlying thermal polymer phase transitions. While inhomogeneous acrylate-based photopolymer networks tend to exhibit rather broad thermal polymer phase transitions, very defined and sharp transitions can be expected for photopolymers with more homogeneous network architectures.13 All prepared D3A-based photopolymerizable resins were cast into silicone molds and characterized by DMTA in a temperature region of −100 to 200 °C. The storage modulus (G′) and loss factor (tan δ) plots of the newly synthesized silane–acrylate networks were studied in comparison to the unregulated polyD3A matrix (Figure 6a) and the respective thiol–acrylate networks (Figure 6b). Photopolymer networks with 50 mol % CTA content were not included in mechanical studies as the final cross-linking density was not high enough to form durable specimens. The chain transfer reaction results in rather soft, lightly cross-linked photopolymer networks with low rigidity in both cases, for silane– and thiol–acrylate systems.
Figure 6.

Storage modulus G′ and tan δ plots (a) for reference polyD3A (solid), polyD3A/DSiH 5 (short dash), and 20 (dash) and (b) for reference polyD3A (solid), polyD3A/DSH 20 (dash-dot), and polyD3A/DSiH 20 (dash).
As expected, with increasing content of silane DSiH the Tg of the resulting photopolymer network is reduced. At the same time a more defined thermal polymer phase transition is evident, which hints toward a more homogeneous network architecture with a smaller kinetic backbone length and further proves the assumed silane–ene radical mechanism (smaller full width at half-maximum fwhm, Table S3). The decreased storage modulus at the rubbery state G′r for photopolymers with 5 and 20 mol % DSiH further confirms a reduced cross-linking density for the silane–ene networks compared to the polyD3A reference.
Compared to the respective thiol–acrylate counterparts, the silane–acrylate photopolymer networks exhibit narrower fwhm values, which leads to the assumption that the prepared silane–ene networks exhibit a more homogeneous network architecture. This can be explained by a more uniform silane–acrylate reaction, which would also point toward a higher silane conversion that can be expected from previous work.35 Overall, the final Tg of silane–acrylate networks is significantly higher compared to the thiol–ene references, which tend to show soft behavior due to their flexible thioether bonds.
The silane-based photopolymer D3A/DSiH 20 broke during the DMTA measurement at the rubbery state. In order to prove sufficient thermal stability of the prepared silane-based photopolymer networks, thermogravimetric analysis (TGA) was performed. Silane-based networks exhibit slightly lower thermal stability compared to the polyD3A reference. Nevertheless, thermal stability up to 350 °C is evident, and silane-based photopolymer networks exhibit a higher thermal stability compared to their thiol-based analogues (Figure S6).
Mechanical Properties
Tensile Strength
The mechanical properties of the fabricated photopolymer specimens were also tested at ambient conditions by conducting a tensile test. The recorded stress–strain plots for the evaluated networks show that CTA-based networks tend to exhibit lower tensile strength with increasing CTA content, but this evidently results in an increased elongation at break (from ∼9% for poly D3A to ∼23% for polyD3A/DSiH 20; Figure 7 and Table S4). Silane–acrylate networks show higher tensile strength compared to the thiol–acrylate references, which was expected from their higher storage modulus at ambient conditions G′20 and Tg (Table S3).
Figure 7.

Stress–strain plot for reference polyD3A (solid), polyD3A/DSH 5 (dash-dot-dot) and 20 (dash-dot), and polyD3A/DSiH 5 (short dash) and 20 (dash).
Impact Resistance
Highly cross-linked, acrylate-based photopolymer networks are known to be rather brittle and thus exhibit low impact toughness. This can mostly be explained by the glassy nature and inhomogeneous network architecture originating from the unregulated radical curing process. Thiol–ene networks have been numerously stated in the literature as tough materials with improved impact resistance,41,42 which is achieved by a mixed chain growth/step growth-like radical polymerization yielding more homogeneous networks. Similarly, silane–ene networks are expected to enhance material toughness due to their regulated network architecture. In order to prove this assumption, Dynstat impact tests were performed on the synthesized photopolymer networks (Figure 8). While the impact resistance of polymer networks with the addition of 5 mol % thiol is increased compared to the acrylate reference, the addition of 20 mol % DSH shows a surprisingly low impact resistance. The rather low impact resistance for thiol-based D3A/DSH 20 mol % could be an indication for a high number of thiol-based dangling chains, which can be expected from the low chain transfer constant Ctr < 0.2 for thiol–acrylate systems.38 The silane-based networks show a different trend with no significant change in impact resistance for networks with 5 mol % silane and an increased impact resistance for specimens with 20 mol % silane (factor of almost 3). The tested impact resistance of D3A/DSiH 20 mol %, while significantly increased, was tested at temperatures >20 mol %, while significantly increased, was tested at temperatures >Tg, which has a significant influence on the experimental outcome. Therefore, the acquired results are solely an indication for improved impact resistance but need to be confirmed in more elaborate studies of optimized photopolymer networks.
Figure 8.

Dynstat impact resistance for reference polyD3A (green, solid), polyD3A/DSH (gray, solid), and polyD3A/DSiH (blue, striped).
Swelling Tests
In order to prove a covalently linked silane–ene network structure and eliminate the suspicion of potential migratable components, disc-shaped photopolymer networks were fabricated and subjected to swelling studies. The respective polymer discs were weighed (mstart), stored in ethanol for 7 days, and then weighed to determine the mass of the swollen network (mswollen). Afterward, the polymers were dried until constant weight (mdry), and the degree of swelling (S) and the gel fraction (GF) of the respective samples were evaluated using eqs 1 and 2.
| 1 |
| 2 |
Compared to the pure acrylate-based reference polyD3A, the evaluated silane–acrylate networks exhibit a higher degree of swelling. This can be explained by the low Tg of 14 °C and a reduced cross-linking density due to chain transfer reactions during curing (Figure 9a, also confirmed by the low G′r of 16 MPa). In addition, the increased double-bond conversion (see RT-NIR-Photorheology section) and the assumption for comparably high silane conversion can be confirmed by a higher gel fraction for silane-based polymers compared to the acrylate-based reference D3A (Figure 9b). With gel fractions >98%, it can be concluded that the difunctional silane DSiH is covalently incorporated into the respective silane–acrylate networks. All in all, the investigated silane–acrylate networks show comparable degrees of swelling and gel fractions to the respective thiol–acrylate references. This further proves the fabrication of cross-linked silane–acrylate networks with high conversion and reduced cross-linking density.
Figure 9.

(a) Degree of swelling S and (b) gel fraction GF in ethanol for reference polyD3A (green, solid), polyD3A/DSH (gray, solid), and polyD3A/DSiH (blue, striped).
Conclusions
A new difunctional silane DSiH has been synthesized via a simple one-pot procedure and was introduced as chain transfer agent in photoinitiated radical reactions with various homopolymerizable monomers. The silane–acrylate system showed the highest coreactivity and was further investigated in radical network formation. With the use of DSiH the radical curing mechanism is regulated by changing the mechanism toward a radical step growth-like process analogously to the popular thiol–ene chemistry. The comparison with the state-of-the-art thiol–acrylate concept shows the vast potential of the tested silane–acrylate resins. The respective silane-based formulations exhibit neutral odor and improved storage stability. Silane–acrylate network formation with DSiH yields rapid radical network formation with delayed gelation at higher double-bond conversions. This results in photopolymer networks with increased overall double-bond conversion (>95%) and reduced shrinkage stress (∼35% reduction of initial stress for D3A/DSiH 20). These effects can be tuned by changing the silane content. A sharp glass transition for the fabricated silane-based photopolymer networks points toward the expected more homogeneous network architecture, and final photopolymer networks exhibit improved impact resistance. The increased degree of swelling for silane-based networks is another indication for the designed homogeneous photopolymer networks with reduced cross-linking density, and the high gel fraction proves a good incorporation of the difunctional silane into the photopolymer network. In addition to the variability of silane content, further studies with silanes of different functionality could also be targeted in the future. This would provide another lever for the fabrication of precisely designed silane–acrylate photopolymer networks. The concept of silane–ene chemistry has potential in surface modification of silicon, silicon polymer science, and most importantly photopolymerization. Such silane-based photopolymer networks could soon play a major role in various fields such as lithography-based 3D printing, photopatterning for microelectronics, coatings, or adhesives.
Acknowledgments
Funding by the Christian-Doppler Research Association and the company Ivoclar Vivadent within the framework of the Christian-Doppler laboratory “Photopolymers in digital and restorative dentistry” is gratefully acknowledged. We also thank the Austrian Science fund (FWF) for funding project P27059 and Prof. Robert Liska for productive discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.7b01399.
1H, 13C, and 29Si NMR spectra of DSiH; conversion analysis of photopolymerization reactions with monofunctional monomers; 1H NMR spectra of D3A/DSiH 20 before and after storage period; numerical data for photo-DSC, RT-NIR-photorheology, DMTA, and tensile test; TGA plots of studied networks (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fouassier J.-P.Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications; Hanser: Munich, 1995; p 388 ff. [Google Scholar]
- Abe Y. Current status and updates on the radiation curing technology for polymers for coatings. DIC Technol. Rev. 2005, 11, 1–20. [Google Scholar]
- Moszner N.; Hirt T. New polymer-chemical developments in clinical dental polymer materials: Enamel-dentin adhesives and restorative composites. J. Polym. Sci., Part A: Polym. Chem. 2012, 50 (21), 4369–4402. 10.1002/pola.26260. [DOI] [Google Scholar]
- Roche E. T.; Polygerinos P.; Whyte W.; Mooney D. J.; Walsh C. J.; Fabozzo A.; Lee Y.; Karp J. M.; Friehs I.; Casar B. A. M.; Bueno A.; Lang N.; Feins E.; Vasilyev N. V.; Del N. P. J.; Schuster L.; Pereira M. J. N.; Wasserman S.; O’Cearbhaill E. D. A light-reflecting balloon catheter for atraumatic tissue defect repair. Sci. Transl. Med. 2015, 7 (306), 306ra149. 10.1126/scitranslmed.aaa2406. [DOI] [PubMed] [Google Scholar]
- Tumbleston J. R.; Shirvanyants D.; Ermoshkin N.; Janusziewicz R.; Johnson A. R.; Kelly D.; Chen K.; Pinschmidt R.; Rolland J. P.; Ermoshkin A.; Samulski E. T.; DeSimone J. M. Continuous liquid interface production of 3D objects. Science 2015, 347 (6228), 1349–1352. 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]
- Zanchetta E.; Cattaldo M.; Franchin G.; Schwentenwein M.; Homa J.; Brusatin G.; Colombo P. Stereolithography of SiOC Ceramic Microcomponents. Adv. Mater. 2016, 28 (2), 370–376. 10.1002/adma.201503470. [DOI] [PubMed] [Google Scholar]
- Mautner A.; Steinbauer B.; Orman S.; Russmueller G.; MacFelda K.; Koch T.; Stampfl J.; Liska R. Tough photopolymers based on vinyl esters for biomedical applications. J. Polym. Sci., Part A: Polym. Chem. 2016, 54 (13), 1987–1997. 10.1002/pola.28065. [DOI] [Google Scholar]
- Ligon-Auer S. C.; Schwentenwein M.; Gorsche C.; Stampfl J.; Liska R. Toughening of photo-curable polymer networks: a review. Polym. Chem. 2016, 7 (2), 257–286. 10.1039/C5PY01631B. [DOI] [Google Scholar]
- Hoyle C. E.; Bowman C. N. Thiol-Ene Click Chemistry. Angew. Chem., Int. Ed. 2010, 49 (9), 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- Lowe A. B. Thiol-yne ’click’/coupling chemistry and recent applications in polymer and materials synthesis and modification. Polymer 2014, 55 (22), 5517–5549. 10.1016/j.polymer.2014.08.015. [DOI] [Google Scholar]
- Kloxin C. J.; Scott T. F.; Bowman C. N. Stress Relaxation via Addition-Fragmentation Chain Transfer in a Thiol-ene Photopolymerization. Macromolecules 2009, 42 (7), 2551–2556. 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. Y.; Kloxin C. J.; Abuelyaman A. S.; Oxman J. D.; Bowman C. N. Stress Relaxation via Addition-Fragmentation Chain Transfer in High Tg, High Conversion Methacrylate-Based Systems. Macromolecules 2012, 45 (14), 5640–5646. 10.1021/ma300228z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsche C.; Koch T.; Moszner N.; Liska R. Exploring the benefits of β-allyl sulfones for more homogeneous dimethacrylate photopolymer networks. Polym. Chem. 2015, 6 (11), 2038–2047. 10.1039/C4PY01582G. [DOI] [Google Scholar]
- Gorsche C.; Seidler K.; Knaack P.; Dorfinger P.; Koch T.; Stampfl J.; Moszner N.; Liska R. Rapid formation of regulated methacrylate networks yielding tough materials for lithography-based 3D printing. Polym. Chem. 2016, 7 (11), 2009–2014. 10.1039/C5PY02009C. [DOI] [Google Scholar]
- Ligon S. C.; Husar B.; Wutzel H.; Holman R.; Liska R. Strategies to Reduce Oxygen Inhibition in Photoinduced Polymerization. Chem. Rev. 2014, 114 (1), 557–589. 10.1021/cr3005197. [DOI] [PubMed] [Google Scholar]
- Oesterreicher A.; Gorsche C.; Ayalur-Karunakaran S.; Moser A.; Edler M.; Pinter G.; Schloegl S.; Liska R.; Griesser T. Exploring Network Formation of Tough and Biocompatible Thiol-yne Based Photopolymers. Macromol. Rapid Commun. 2016, 37 (20), 1701–1706. 10.1002/marc.201600369. [DOI] [PubMed] [Google Scholar]
- Lee T. Y.; Smith Z.; Reddy S. K.; Cramer N. B.; Bowman C. N. Thiol-Allyl Ether-Methacrylate Ternary Systems. Polymerization Mechanism. Macromolecules 2007, 40 (5), 1466–1472. 10.1021/ma062494b. [DOI] [Google Scholar]
- Peng H.; Nair D. P.; Kowalski B. A.; Xi W.; Gong T.; Wang C.; Cole M.; Cramer N. B.; Xie X.; McLeod R. R.; Bowman C. N. High Performance Graded Rainbow Holograms via Two-Stage Sequential Orthogonal Thiol-Click Chemistry. Macromolecules 2014, 47 (7), 2306–2315. 10.1021/ma500167x. [DOI] [Google Scholar]
- Nguyen K. D. Q.; Megone W. V.; Kong D.; Gautrot J. E. Ultrafast diffusion-controlled thiol-ene based crosslinking of silicone elastomers with tailored mechanical properties for biomedical applications. Polym. Chem. 2016, 7 (33), 5281–5293. 10.1039/C6PY01134A. [DOI] [Google Scholar]
- Podgorski M.; Becka E.; Chatani S.; Claudino M.; Bowman C. N. Ester-free thiol-X resins: new materials with enhanced mechanical behavior and solvent resistance. Polym. Chem. 2015, 6 (12), 2234–2240. 10.1039/C4PY01552E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belbakra Z.; Cherkaoui Z. M.; Allonas X. Photocurable polythiol based (meth)acrylate resins stabilization: New powerful stabilizers and stabilization systems. Polym. Degrad. Stab. 2014, 110, 298–307. 10.1016/j.polymdegradstab.2014.09.012. [DOI] [Google Scholar]
- Esfandiari P.; Ligon S. C.; Lagref J. J.; Frantz R.; Cherkaoui Z.; Liska R. Efficient stabilization of thiol-ene formulations in radical photopolymerization. J. Polym. Sci., Part A: Polym. Chem. 2013, 51 (20), 4261–4266. 10.1002/pola.26848. [DOI] [Google Scholar]
- Guterman R.; Rabiee Kenaree A.; Gilroy J. B.; Gillies E. R.; Ragogna P. J. Polymer Network Formation Using the Phosphane-ene Reaction: A Thiol-ene Analogue with Diverse Postpolymerization Chemistry. Chem. Mater. 2015, 27 (4), 1412–1419. 10.1021/cm504784e. [DOI] [Google Scholar]
- Guterman R.; Harrison T. D.; Gillies E. R.; Ragogna P. J. Synthesis and functionalization of polymer networks via germane-ene chemistry. Polym. Chem. 2017, 8, 3425. 10.1039/C7PY00438A. [DOI] [Google Scholar]
- Scott T. F.; Furgal J. C.; Kloxin C. J. Expanding the Alternating Propagation-Chain Transfer-Based Polymerization Toolkit: The Iodo-Ene Reaction. ACS Macro Lett. 2015, 4 (12), 1404–1409. 10.1021/acsmacrolett.5b00640. [DOI] [PubMed] [Google Scholar]
- El-Roz M.; Lalevee J.; Allonas X.; Fouassier J.-P. The silane-ene and silane-acrylate polymerization process: a new promising chemistry?. Macromol. Rapid Commun. 2008, 29 (10), 804–808. 10.1002/marc.200800052. [DOI] [Google Scholar]
- Souane R.; Lalevee J.; Allonas X.; Fouassier J.-P. H-Si Functionalized Silicon Surfaces and Powders as Photoinitiators of Polymerization. Macromol. Mater. Eng. 2010, 295 (4), 351–354. 10.1002/mame.200900352. [DOI] [Google Scholar]
- Marchi S.; Sangermano M.; Ligorio D.; Meier P.; Kornmann X. Impressive Rate Raise of the Hydrosilation Reaction Through UV-Activation: Energy and Time Saving. Macromol. Mater. Eng. 2016, 301 (5), 610–613. 10.1002/mame.201500413. [DOI] [Google Scholar]
- Lalevee J.; Tehfe M. A.; Morlet-Savary F.; Graff B.; Allonas X.; Fouassier J. P. Radical photopolymerization reactions under air upon lamp and diode laser exposure: The input of the organosilane radical chemistry. Prog. Org. Coat. 2011, 70 (2), 83–90. 10.1016/j.porgcoat.2010.10.008. [DOI] [Google Scholar]
- Chatgilialoglu C.; Lalevee J. Recent applications of the (TMS)3SiH radical-based reagent. Molecules 2012, 17, 527–555. 10.3390/molecules17010527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalevee J.; Dirani A.; El-Roz M.; Allonas X.; Fouassier J. P. Silanes as New Highly Efficient Co-initiators for Radical Polymerization in Aerated Media. Macromolecules 2008, 41 (6), 2003–2010. 10.1021/ma702301x. [DOI] [Google Scholar]
- Lalevee J.; Fouassier J. P. Recent advances in sunlight induced polymerization: role of new photoinitiating systems based on the silyl radical chemistry. Polym. Chem. 2011, 2 (5), 1107–1113. 10.1039/c1py00073j. [DOI] [Google Scholar]
- Chatgilialoglu C.; Guerrini A.; Lucarini M. The trimethylsilyl substituent effect on the reactivity of silanes. Structural correlations between silyl radicals and their parent silanes. J. Org. Chem. 1992, 57 (12), 3405–3409. 10.1021/jo00038a031. [DOI] [Google Scholar]
- Lalevee J.; Allonas X.; Fouassier J. P. Tris(trimethylsilyl)silane (TTMSS)-Derived Radical Reactivity toward Alkenes: A Combined Quantum Mechanical and Laser Flash Photolysis Study. J. Org. Chem. 2007, 72 (17), 6434–6439. 10.1021/jo0706473. [DOI] [PubMed] [Google Scholar]
- Steindl J.; Svirkova A.; Marchetti-Deschmann M.; Moszner N.; Gorsche C. Light-Triggered Radical Silane-Ene Chemistry Using a Monosubstituted Bis(trimethylsilyl)silane. Macromol. Chem. Phys. 2017, 218 (9), 1600563. 10.1002/macp.201600563. [DOI] [Google Scholar]
- Moszner N.; Zeuner F.; Lamparth I.; Fischer U. K. Benzoylgermanium Derivatives as Novel Visible-Light Photoinitiators for Dental Composites. Macromol. Mater. Eng. 2009, 294 (12), 877–886. 10.1002/mame.200900181. [DOI] [Google Scholar]
- Gorsche C.; Harikrishna R.; Baudis S.; Knaack P.; Husar B.; Laeuger J.; Hoffmann H.; Liska R. Real Time-NIR/MIR-Photorheology: A Versatile Tool for the in Situ Characterization of Photopolymerization Reactions. Anal. Chem. 2017, 89 (9), 4958–4968. 10.1021/acs.analchem.7b00272. [DOI] [PubMed] [Google Scholar]
- Cramer N. B.; Reddy S. K.; O’Brien A. K.; Bowman C. N. Thiol-Ene Photopolymerization Mechanism and Rate Limiting Step Changes for Various Vinyl Functional Group Chemistries. Macromolecules 2003, 36 (21), 7964–7969. 10.1021/ma034667s. [DOI] [Google Scholar]
- Sahin M.; Ayalur-Karunakaran S.; Manhart J.; Wolfahrt M.; Kern W.; Schloegl S. Thiol-Ene versus Binary Thiol-Acrylate Chemistry: Material Properties and Network Characteristics of Photopolymers. Adv. Eng. Mater. 2017, 19 (4), 1600620. 10.1002/adem.201600620. [DOI] [Google Scholar]
- Oesterreicher A.; Ayalur-Karunakaran S.; Moser A.; Mostegel F. H.; Edler M.; Kaschnitz P.; Pinter G.; Trimmel G.; Schloegl S.; Griesser T. Exploring thiol-yne based monomers as low cytotoxic building blocks for radical photopolymerization. J. Polym. Sci., Part A: Polym. Chem. 2016, 54 (21), 3484–3494. 10.1002/pola.28239. [DOI] [Google Scholar]
- Hoyle C. E.; Lee T. Y.; Roper T. Thiol-enes: Chemistry of the past with promise for the future. J. Polym. Sci., Part A: Polym. Chem. 2004, 42 (21), 5301–5338. 10.1002/pola.20366. [DOI] [Google Scholar]
- Mautner A.; Qin X.; Wutzel H.; Ligon S. C.; Kapeller B.; Moser D.; Russmueller G.; Stampfl J.; Liska R. Thiol-ene photopolymerization for efficient curing of vinyl esters. J. Polym. Sci., Part A: Polym. Chem. 2013, 51 (1), 203–212. 10.1002/pola.26365. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.