

Abstract
Norcoclaurine synthase (NCS) is a Pictet-Spenglerase that catalyzes the first key step in plant benzylisoquinoline alkaloid metabolism, a compound family that includes bioactive natural products such as morphine. The enzyme has also shown great potential as a biocatalyst for the formation of chiral isoquinolines. Here we present new high-resolution X-ray crystallography data describing Thalictrum flavum NCS bound to a mechanism-inspired ligand. The structure supports two key features of the NCS “dopamine-first” mechanism: the binding of dopamine catechol to Lys-122 and the position of the carbonyl substrate binding site at the active site entrance. The catalytically vital residue Glu-110 occupies a previously unobserved ligand-bound conformation that may be catalytically significant. The potential roles of inhibitory binding and alternative amino acid conformations in the mechanism have also been revealed. This work significantly advances our understanding of the NCS mechanism and will aid future efforts to engineer the substrate scope and catalytic properties of this useful biocatalyst.
Norcoclaurine synthase (NCS) catalyzes the formation of (1S)-substituted tetrahydroisoquinolines via a Pictet-Spengler reaction between a β-arylethylamine and a carbonyl-containing compound (Figure 1a).1 The natural reaction catalyzed by NCS involves the condensation of dopamine 1 and 4-hydroxyphenylacetaldehyde (4-HPAA) 2, forming (S)-norcoclaurine 4 (Figure 1b).1−3 This is the first committed step into the plant benzylisoquinoline alkaloids (BIA), a diverse family of bioactive natural products that includes morphine and berberine.4
Figure 1.

NCS-catalyzed reaction. (a) General reaction catalyzed by NCS. (b) NCS reactions with 4-hydroxyphenylacetaldehyde 2 (4-HPAA) and 4-methoxyphenylacetaldehyde (4-MOPAA) 3, and the intermediate mimic 6. (c) Outline of the proposed dopamine-first mechanism prior to these results.
NCS has also played a key role in in vivo heterologous reconstitutions of BIA biosynthesis,5−8 and because of its considerable carbonyl substrate promiscuity, it has been used for the in vitro production of alkaloids.9−18 Therefore, a thorough understanding of the NCS mechanism will both shed light on a key aspect of plant specialized metabolism and enable rational enzyme engineering to improve the catalytic efficiency and substrate scope of a promising biocatalyst.
Two enzyme mechanisms have been proposed, which differ most simply by the order in which the substrates bind to the enzyme. In the “HPAA-first” mechanism, the carbonyl substrate binds prior to dopamine.19,20 This mechanism was inspired by the observed binding modes of dopamine and the nonproductive aldehyde p-hydroxybenzaldehyde in structural data from X-ray crystallography [Protein Data Bank (PDB) entry 2VQ5].19 However, this mechanism cannot account for the carbonyl substrate promiscuity of the enzyme,9,10 nor does it provide a residue to deprotonate the dopamine 3-OH, which had previously been shown to be a key reaction step.21
Computational docking and mutagenesis experiments led to the development of an alternative dopamine-first mechanism, in which dopamine binds deep in the active site and the carbonyl-derived substituent is positioned at the active site entrance, partially exposed to the solvent.10,12 The location of this carbonyl substrate binding site can account for the enzyme promiscuity and has been supported by amino acid substitutions that have altered the enzyme carbonyl substrate tolerance.12,17
The dopamine-first mechanism (Figure 1c) involves initial binding of dopamine 3-OH to residue K122, and binding of the nitrogen to E110 and D141. The carbonyl substrate binds subsequently, and iminium formation is catalyzed by Y108, E110, and D141. Electrophilic addition is triggered by deprotonation of 3-OH by K122, and this is followed by deprotonation of the quinone by E110, revealing the product.
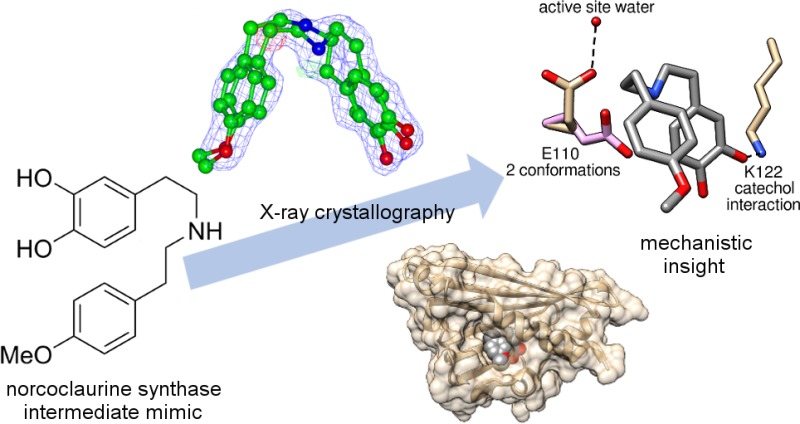
Here we provide the first experimental structural evidence at high resolution for the dopamine-first mechanism. We have crystallized Thalictrum flavum NCS (TfNCS) and used a nonproductive mechanism-inspired ligand to generate structures with an intermediate mimic bound, which validate key predictions of the dopamine-first mechanism: the binding of dopamine 3-OH to K122 and the position of the carbonyl substituent at the active site entrance. The structures also reveal two distinct conformations of the mechanistically vital E110 side chain.
To minimize flexible residues to promote crystallization, a truncated TfNCS1 construct was designed, lacking 33 residues from the N-terminus and residues 196–210 from the C-terminus (ΔN33C196TfNCS) (Figure S1). The truncations did not affect the enzyme activity compared to that of Δ29TfNCS (Figure S2). The structure of ΔN33C196TfNCS was determined in apo form at 2.00 Å resolution, with three copies in the asymmetric unit (PDB entry 5N8Q, Table S1).
The apo monomer structures were similar to the previously published structures of Δ19TfNCS; the Cα root-mean-square deviation (RMSD) calculated among all possible monomer pairs of the apo structure 5N8Q and 2VQ5 was just 0.5–0.8 Å. The highest RMSD was found for residues G102 and E103 that reside in a flexible loop region with high B factors. Active site residue F112 was present in all new structures as a single rotamer, as predicted by molecular dynamics simulations.12
Attempts to observe dopamine 1 or (S)-norcoclaurine 4 bound in the active site by soaking or co-crystallization were not successful. Instead, the binding of a reaction intermediate mimic was investigated. The reaction mechanism is expected to proceed via an iminium intermediate (Figure 1b,c).21 It was hypothesized that a secondary amine would mimic the behavior of the iminium species in the active site (Figure 1b). The mimic was based on the iminium intermediate of the reaction between dopamine 1 and 4-methoxyphenylacetaldehyde 3 (4-MOPAA), producing (S)-argemexirine 5. 4-MOPAA 3 differs from natural substrate 4-HPAA 2 by only a single methyl group and is less sensitive to oxidation. This reaction was previously demonstrated to be catalyzed efficiently by both TfNCS9 and Coptis japonica NCS2,10 equaling or exceeding the conversion of 4-HPAA 2.
Mimic 6 was synthesized in three steps from the reported amine 3,4-bis(benzoyloxy)dopamine via amide coupling, reduction, and deprotection (overall three-step purified yield of 25%). Co-crystallization of the protein and mimic 6 generated a mimic-bound structure at 1.85 Å resolution (PDB entry 5NON, Table S1). The structures showed 6 residing in the active site, with the catechol group bound to K122 (distance of 2.6 Å) (Figure 2a,b). The presence of this protein–ligand interaction is unequivocal and verifies a key interaction predicted by the dopamine-first mechanism.
Figure 2.
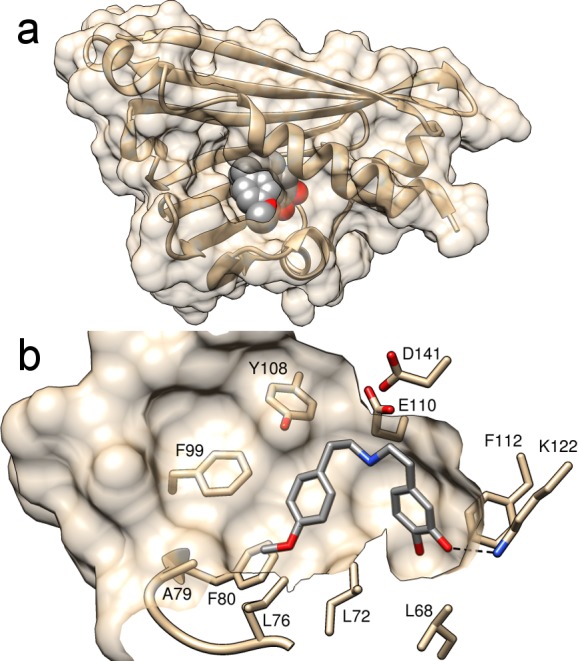
Mimic bound in the active site of structure 5NON. (a) Overall structure, solvent-excluded surface, and active site entrance. The mimic is depicted as gray spheres. (b) The major conformation of the mimic is shown as gray sticks. Key active site side chains and loops are depicted. The distance between dopamine 3-OH and K122 (black dashed line) is 2.6 Å. The protein solvent-excluded surface has been clipped to show the active site cavity.
Interestingly, 6 appeared to adopt two possible orientations within the active site, one productive orientation in which 3-OH was closest to K122 and an inhibitory orientation in which 4-OH was closest (Figure 3a,b). The proposed mechanism requires the 3-OH to be deprotonated by K122 (Figure 1c), which is supported by the loss of detectable product formation when the 3-OH substitution of β-arylethylamine substrates is absent or modified.9,10 The contribution of the orientations was estimated by refining the occupancy constrained to a total of 1.0 of the two ligand conformations in each site (Figure 3a,b). This method showed the productive and inhibitory orientations accounted for approximately 70 and 30% of the density, respectively (see Figure S4 for different interpretations of the ligand electron density). It is unclear whether the inhibitory orientation of the ligand is biologically relevant; the more rigid iminium reaction intermediate may not be able to occupy such a conformation. However, computational docking studies with 5NON suggested that dopamine alone could bind in equivalent productive and inhibitory orientations (Figure 3c,d). The productive binding mode is predicted to be only slightly more favorable than the inhibitory mode (Table S2). The presence of these inhibitory binding modes may reduce the catalytic efficiency of the enzyme.22
Figure 3.
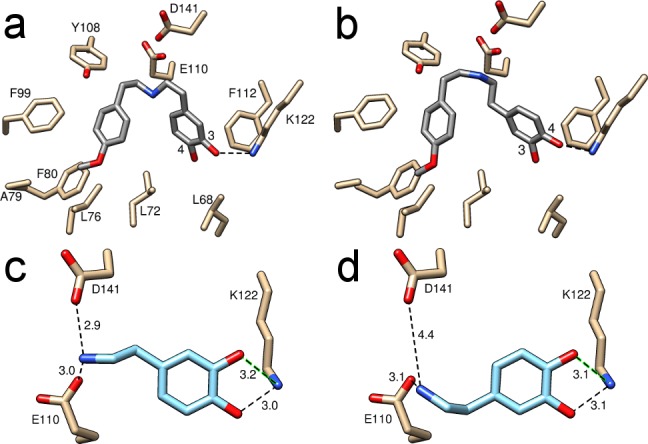
Interactions between catechol and K122. (a) Productive and (b) inhibitory mimic binding orientations, accounting for approximately 70 and 30% occupancy, respectively. The arrangement is productive when 3-OH is bound to K122. (c) Productive and (d) inhibitory dopamine binding orientation generated by computational docking. Numbers show the lengths in angstroms of the dotted lines.
The binding of ligand 6 to the protein caused changes to amino acid side chain orientations, most significantly to the catalytically vital residue E110. Two distinct conformations were apparent in the structures. The apo structure featured the same E110 conformation as observed in previously published structures (Figure 4a),19 while in the mimic-bound structure, a novel ligand-bound conformation was present (Figure 4b). The ligand-bound conformation of E110 is characterized by a H-bond to water 201, which in turn was bound to the main chains of V124 and M126 and the side chain of D141, all conserved residues throughout NCSs (Figure 4b and Figure S1).23 This mimic-bound structure conformation brings the carboxyl group of E110 close to D141, forming a negatively charged region in which a positively charged ligand nitrogen can bind. The productive binding mode of dopamine generated by computational docking to the mimic-bound structure highlights the fact that both E110 and D141 are within H-bonding distance of the substrate nitrogen (Figure 3c).
Figure 4.
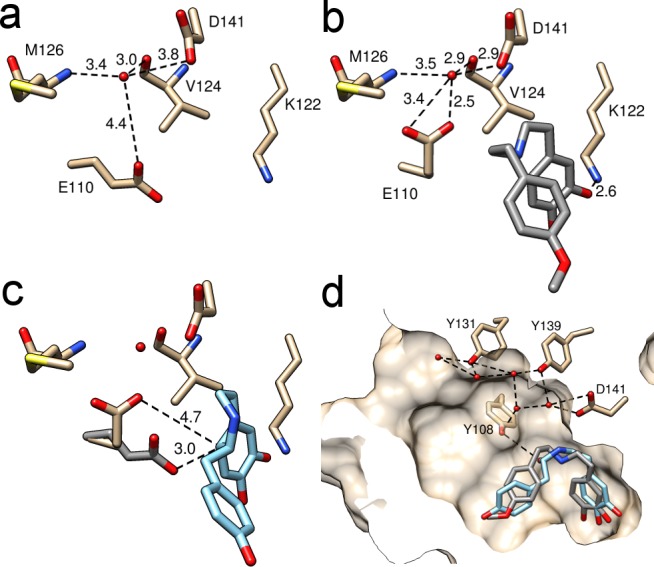
Water in the active site. Water 201 interactions in (a) apo and (b) mimic-bound structures. (c) Quinone intermediate (light blue) docked into the 5NON active site (residues colored beige). Apo residues are colored gray to show proximity of E110 to C8a. Numbers are distances in angstroms. (d) Water channel mediated by hydrophilic active site residues. Gray sticks show the bound mimic; light blue sticks show (S)-hemiacetal docked into the active site, highlighting the position of the water leaving group. The protein solvent-excluded surface has been clipped to show the active site cavity.
A proposed mechanistic role of E110 is the deprotonation of the quinone intermediate at C8a, which results in the formation of the aromatic THIQ product (Figure 1c). Binding modes of the quinone intermediate generated by computational docking suggest the non-water-bound apo E110 conformation is required for this reaction step (Figure 4c and Table S2). This suggests that both conformations of E110 may be mechanistically important but required for different catalytic steps. Such a role of E110 is supported by previous experiments in which the E110D variant had no measurable activity;12 the exact side chain length is necessary for E110 to perform its precise role.
Adjacent to D141 are the fully conserved residues Y108 and Y139, and the partially conserved Y131 (Figure S1). These hydrophilic residues may aid in the removal of water from the active site at the initial condensation step (Figure 4d).
The carbonyl substrate-derived portion of the ligand is oriented toward the active site entrance (Figure 2a). This supports a key prediction of the dopamine-first mechanism: the location of the carbonyl substrate binding site near the bulk solvent, which accounts for the substrate promiscuity of the enzyme. The ligand interaction is slightly more constrained than predicted by computational docking experiments; the ligand methoxy group is bound between L72 and L76. It is unclear how such a conformation could be adopted when bulkier carbonyl substituents are present (e.g., citronellal12).
Ligand binding triggered subtle changes to amino acid conformations, as judged by RMSD comparisons with the apo structure. In subunits A and B, there appear to be changes in the active site entrance loop (residues 76–80) both in the side chains and in the Cα positions (Figure S5). This region is known to influence carbonyl substrate specificity, with amino acid substitutions shown to increase the levels of conversion with some unnatural substrates.12,17,18 Amino acids in this loop, with the exception of A79, are fully conserved among known NCSs.23 There were also changes in the conformation and increased flexibility in loop 99–103 upon ligand binding (Figure S5). Within this loop, F99 shows particular proximity to the aldehyde substituent and, along with G102, is conserved among NCSs.23 In all three subunits, the positions of residues 177 and 180 also appear to shift upon ligand binding. These residue motions upon ligand binding are corroborated by previous nuclear magnetic resonance experiments that detected ligand-dependent chemical shift perturbations of residues 99, 100, 177, and 180.24
The cyclization step in the reaction mechanism requires C6 and the iminium carbon to come into the proximity before C–C bond formation can occur (Figure 1c). It is possible that the subtle amino acid movements of loops 76–80 and 99–103 are coupled to this change in the intermediate conformation. Such a hypothesis could account for the observation that increased bulk in loop 76–80 (mutants A79I and A79F) improved conversions17 and enzyme activities (Figure S3) with certain substrates (for the proposed mechanism, see Figure S6).
The investigation of the TfNCS structure with a mechanistically inspired ligand has provided structural data in support of the dopamine-first mechanism. The two key observations supporting this model are the interactions between the K122 and the dopamine-derived catechol moiety, and the orientation of the aldehyde substituent toward the active site entrance. The study has also revealed novel features of the NCS mechanism, including the variable conformations of E110 and the possibility of inhibitory substrate binding modes. Overall, this improved understanding of the enzyme structure and mechanism will aid rational engineering approaches to increasing the kinetic parameters and widening the substrate scope of an important biocatalyst.
Acknowledgments
We gratefully acknowledge Jianxiong Zhao for help in confirming the chemical analysis and beamline scientists at Soleil Proxima 1 and Diamond I0-2 beamlines. We also acknowledge K. Karu (University College London Mass Spectrometry Facility) and A. E. Aliev (University College London NMR Facility) in the Department of Chemistry for their assistance.
Glossary
Abbreviations
- 4-HPAA
4-hydroxyphenylacetaldehyde
- 4-MOPAA
4-methoxyphenylacetaldehyde
- BIA
benzylisoquinoline alkaloid
- NCS
norcoclaurine synthase
- RMSD
root-mean-square deviation
- Tf
T. flavum.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00769.
Experimental procedures and supplemental figures and tables (PDF)
Accession Codes
Crystal structures reported here have been deposited in the Protein Data Bank as entries 5N8Q (ΔN33C196TfNCS apo) and 5NON (mimic-bound).
Author Present Address
∥ B.R.L.: John Innes Centre, Norwich Research Park, Norwich NR4 7UH, U.K.
Author Contributions
B.R.L. and A.S. designed research and performed protein expression and purification. B.R.L. performed enzyme assays and computational docking. A.S. performed crystallization and collected X-ray diffraction data. A.S. and N.H.K. determined and refined crystal structures. T.P. synthesized ligands. H.C.H. verified the ligand structure. H.C.H., J.M.W., and N.H.K. supervised the project. All authors contributed to the writing of the manuscript. B.R.L. and A.S. contributed equally to this work.
This research was supported in part by studentship funding from the Wellcome Trust to B.R.L.
The authors declare no competing financial interest.
Supplementary Material
References
- Samanani N.; Liscombe D. K.; Facchini P. J. (2004) Plant J. 40, 302–313. 10.1111/j.1365-313X.2004.02210.x. [DOI] [PubMed] [Google Scholar]
- Stadler R.; Kutchan T. M.; Zenk H. (1989) Phytochemistry 28, 1083–1086. 10.1016/0031-9422(89)80187-6. [DOI] [Google Scholar]
- Stadler R.; Kutchan T. M.; Loeffler S.; Nagakura N.; Cassels B.; Zenk M. H. (1987) Tetrahedron Lett. 28, 1251–1254. 10.1016/S0040-4039(00)95338-3. [DOI] [Google Scholar]
- Hagel J. M.; Facchini P. J. (2013) Plant Cell Physiol. 54, 647–672. 10.1093/pcp/pct020. [DOI] [PubMed] [Google Scholar]
- Minami H.; Kim J.-S.; Ikezawa N.; Takemura T.; Katayama T.; Kumagai H.; Sato F. (2008) Proc. Natl. Acad. Sci. U. S. A. 105, 7393–7398. 10.1073/pnas.0802981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A.; Minami H.; Kim J.-S.; Koyanagi T.; Katayama T.; Sato F.; Kumagai H. (2011) Nat. Commun. 2, 326. 10.1038/ncomms1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLoache W. C.; Russ Z. N.; Narcross L.; Gonzales A. M.; Martin V. J. J.; Dueber J. E. (2015) Nat. Chem. Biol. 11, 465–471. 10.1038/nchembio.1816. [DOI] [PubMed] [Google Scholar]
- Galanie S.; Thodey K.; Trenchard I. J.; Filsinger Interrante M.; Smolke C. D. (2015) Science 349, 1095–1100. 10.1126/science.aac9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff B. M.; Bräse S.; O’Connor S. E. (2012) Tetrahedron Lett. 53, 1071–1074. 10.1016/j.tetlet.2011.12.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesnot T.; Gershater M. C.; Ward J. M.; Hailes H. C. (2012) Adv. Synth. Catal. 354, 2997–3008. 10.1002/adsc.201200641. [DOI] [Google Scholar]
- Nishihachijo M.; Hirai Y.; Kawano S.; Nishiyama A.; Minami H.; Katayama T.; Yasohara Y.; Sato F.; Kumagai H. (2014) Biosci., Biotechnol., Biochem. 78, 701–707. 10.1080/09168451.2014.890039. [DOI] [PubMed] [Google Scholar]
- Lichman B. R.; Gershater M. C.; Lamming E. D.; Pesnot T.; Sula A.; Keep N. H.; Hailes H. C.; Ward J. M. (2015) FEBS J. 282, 1137–1151. 10.1111/febs.13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonamore A.; Rovardi I.; Gasparrini F.; Baiocco P.; Barba M.; Molinaro C.; Botta B.; Boffi A.; Macone A. (2010) Green Chem. 12, 1623–1627. 10.1039/c0gc00036a. [DOI] [Google Scholar]
- Maresh J. J.; Crowe S. O.; Ralko A. A.; Aparece M. D.; Murphy C. M.; Krzeszowiec M.; Mullowney M. W. (2014) Tetrahedron Lett. 55, 5047–5051. 10.1016/j.tetlet.2014.07.043. [DOI] [Google Scholar]
- Lichman B. R.; Lamming E. D.; Pesnot T.; Smith J. M.; Hailes H. C.; Ward J. M. (2015) Green Chem. 17, 852–855. 10.1039/C4GC02325K. [DOI] [Google Scholar]
- Bonamore A.; Calisti L.; Calcaterra A.; Ismail O. H.; Gargano M.; D’Acquarica I.; Botta B.; Boffi A.; Macone A. (2016) ChemistrySelect 1, 1525–1528. 10.1002/slct.201600134. [DOI] [Google Scholar]
- Lichman B. R.; Zhao J.; Hailes H. C.; Ward J. M. (2017) Nat. Commun. 8, 14883. 10.1038/ncomms14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann V.; Lichman B. R.; Zhao J.; Simon R. C.; Kroutil W.; Ward J. M.; Hailes H. C.; Rother D. (2017) Angew. Chem., Int. Ed. 10.1002/anie.201705855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilari A.; Franceschini S.; Bonamore A.; Arenghi F.; Botta B.; Macone A.; Pasquo A.; Bellucci L.; Boffi A. (2009) J. Biol. Chem. 284, 897–904. 10.1074/jbc.M803738200. [DOI] [PubMed] [Google Scholar]
- Bonamore A.; Barba M.; Botta B.; Boffi A.; Macone A. (2010) Molecules 15, 2070–2078. 10.3390/molecules15042070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk L. Y. P.; Bunn S.; Liscombe D. K.; Facchini P. J.; Tanner M. E. (2007) Biochemistry 46, 10153–10161. 10.1021/bi700752n. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Milo R.; Noor E.; Tawfik D. S. (2015) Biochemistry 54, 4969–4977. 10.1021/acs.biochem.5b00621. [DOI] [PubMed] [Google Scholar]
- Li J.; Lee E.-J.; Chang L.; Facchini P. J. (2016) Sci. Rep. 6, 39256. 10.1038/srep39256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkner H.; Schweimer K.; Matecko I.; Rösch P. (2008) Biochem. J. 413, 281–290. 10.1042/BJ20080306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.