

Abstract
Three new positron emission tomography (PET) radiotracers of interest to our functional neuroimaging and translational oncology programs have been prepared through new developments in [11C]CO2 fixation chemistry. [11C]QZ (glutaminyl cyclase) was prepared via a tandem trapping of [11C]CO2/intramolecular cyclization; [11C]tideglusib (glycogen synthase kinase-3) was synthesized through a tandem trapping of [11C]CO2 followed by an intermolecular cycloaddition between a [11C]isocyanate and an isothiocyanate to form the 1,2,4-thiadiazolidine-3,5-dione core; [11C]ibrutinib (Bruton’s tyrosine kinase) was synthesized through a HATU peptide coupling of an amino precursor with [11C]acrylic acid (generated from [11C]CO2 fixation with vinylmagnesium bromide). All radiochemical syntheses are fully-automated on commercial radiochemical synthesis modules and provide radiotracers in 1 – 5% radiochemical yield (non-corrected, based upon [11C]CO2). All three radiotracers have advanced to rodent imaging studies and preliminary PET imaging results are also reported.
TOC image
INTRODUCTION
Positron emission tomography (PET) imaging is a type of functional molecular imaging using probes, known as radiotracers, composed of a bioactive molecule conjugated with a positron-emitting radionuclide.1 The functional information garnered from a PET scan, when combined with the anatomical information from the co-registered CT or MRI scan, provide unprecedented insight into biochemical pathways, mechanisms of disease pathology, and in vivo behavior of drug molecules. Reflecting this, PET imaging is having far reaching impact on personalized medicine2 and drug discovery.3
Commonly used PET radionuclides include carbon-11 (t1/2 = 20 min), fluorine-18 (t1/2 = 110 min), and gallium-68 (t1/2 = 68 min). The choice of radionuclide depends on a number of factors ranging from synthetic considerations about how it will be incorporated into the bioactive molecule of choice, to practical aspects associated with intended application (e.g. the short half-life of 11C allows patients to receive multiple PET scans in a single hospital visit, while the half-life of 18F facilitates radiotracer distribution from centralized manufacturing facilities).
Carbon-11 is an attractive choice of PET radionuclide because multiple scans can be conducted in series during a single patient visit (e.g. scans with 2 different radiotracers, baseline and intervention studies with 1 tracer). Moreover, it can be frequently incorporated into bioactive or endogenous molecules without any structural modification to the original (non-radioactive) molecule, which may or may not be the case with other radionuclides (e.g. use of radioactive metal ions such as 68Ga require decoration of the bioactive molecule with a suitable metal-chelating group prior to radiolabeling). Carbon-11 is produced by a cyclotron, reacting with oxygen added to the cyclotron target gas to generate [11C]CO2, which is delivered to the radiochemistry laboratory and used to synthesize radiotracers. The short half-life of carbon-11 is advantageous for the reasons outlined above, but it presents challenges. Most notably, the short half-life necessitates that all reactions used to synthesize 11C-radiotracers are reasonably high yielding over a very short time course (e.g. 2–10 min) so that they provide usable amounts of radiotracer, thereby limiting the number of reactions that are practical. Typically, [11C]CO2 is converted into a secondary synthon such as [11C]CH3I, [11C]CH3OTf or [11C]KCN, which is then reacted with a suitable precursor to yield the 11C-labeled compound. Such radiochemical reactions have been used to great effect to synthesize 11C-radiotracers (for recent reviews of carbon-11 radiochemistry, see:4,5,6). However, there are limitations in the types of radiotracers that can be accessed from such synthons. For example, there must be a place to introduce a methyl group if [11C]CH3I or [11C]CH3OTf are to be used for labeling. Given the prevalence of carbonyl groups in bioactive molecules (e.g. many of the best-selling drugs contain one or more C=O bonds7), there is significant interest in developing methods that enable incorporation of a 11C-carbonyl unit into bioactive molecules to increase the number and diversity of available PET radiotracers. One such approach involves synthesis of PET radiotracers directly from [11C]CO2.
The electrophilic carbon in [11C]CO2 means it can be used as a carbonyl source, and can be trapped by an appropriate nucleophilic component. For example, this approach can be used to synthesize radiolabeled carboxylic acids, such as [11C]acetate and [11C]palmitate, by reacting [11C]CO2 with an appropriate Grignard reagent.8 New advances in the synthesis of [11C]carboxylic acids involve treating organoboron precursors with [11C]CO2 in the presence of a copper catalyst.9,10 More recently, there has also been a surge in [11C]CO2 fixation chemistry (for a review of current developments, see:11). For example, [11C]CO2 fixation chemistry has recently been employed in the synthesis of [11C]ureas (both symmetrical12 and unsymmetrical13,14,15,16,17) and [11C]carbamates.14,17,18,19,20,21 In an interesting variant of the latter, Miller also demonstrated that analogous reactions with [11C]CS2 can be employed to generate [11C]dithiocarbamates.22
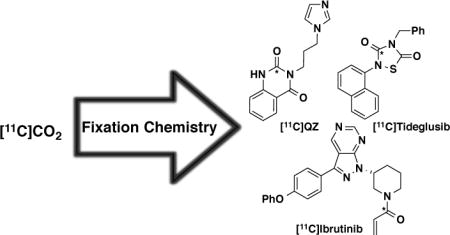
These impressive new developments in [11C]CO2 fixation chemistry were of particular interest to us because they have greatly opened up the synthetic transformations possible with carbon-11, and we believed that we could now employ [11C]CO2 fixation to synthesize three radiotracers of interest to our neuroimaging and translational oncology programs that would be extremely challenging to prepare by other means (Figure 1). From a neuroimaging perspective, we were interested in accessing [11C]3-(3-(1H-imidazol-1-yl)propyl)quinazoline-2,4(1H,3H)-dione ([11C]QZ, 1) and [11C]tideglusib (2) as potential radiotracers for glutaminyl cyclase (QC) and glycogen synthase kinase-3 (GSK-3), respectively.23,24 In our growing translational oncology program, we were also interested in a method for synthesizing [11C]ibrutinib (3), a radiolabeled version of the anticancer drug targeting B-cell malignancies.25 We reasoned that all three radiotracers could be synthesized from labeled building blocks accessible using [11C]CO2 fixation. Herein we report three new variants of [11C]CO2 fixation chemistry to generate intermediates that can be used in intramolecular cyclization-, intermolecular cycloaddition- and peptide coupling-reactions to generate [11C]QZ, [11C]tideglusib and [11C]ibrutinib, respectively.
Figure 1.
New radiotracers labeled by [11C]CO2 fixation (* = 11C)
RESULTS AND DISCUSSION
Automation and Specific Activity Considerations
We initially wished to have a means of automating [11C]CO2 fixation chemistry using our General Electric (GE) TRACERLab FXC-Pro systems and made modifications to the synthesis modules accordingly (see ESI for detailed description). Briefly, because of the harsh, often sparingly soluble, reagents used in [11C]CO2 fixation chemistry, we initially replaced the reagent reservoirs of the synthesis module with disposable v-vials to enable handling of air sensitive reagents and to prevent cross contamination between syntheses. Moreover, valves were modified or installed to allow delivery of [11C]CO2 directly to the TRACERLab reaction vessel. [11C]CO2 was produced with a GE PETTrace cyclotron via the 14N(p,α)11C reaction. Carbon-11 produced in this manner has very high specific activity (for example, previously reported specific activities for 11C-labeled tracers produced in our laboratory range from 1 – 40 Ci/μmol26). While high specific activities are required for potent radiotracers such as [11C]carfentanil (low specific activity doses could elicit unwanted pharmacological responses), in our hands very high specific activity 11C has proven problematic for [11C]CO2 fixation chemistry. For example, we have previously reported the need to include carrier [12C]CO2 when producing [11C]acetate to avoid contaminating doses with [11C]acetone and [11C]tert-butanol resulting from over alkylation of [11C]CO2 with the Grignard reagent.8 No-carrier-added (NCA) reactions proved to be an issue in the present work too (vide infra). For example, while [11C]ibrutinib could be prepared carrier-free, [11C]tideglusib could not. The synthesis of [11C]ibrutinib was also notably cleaner and higher yielding when carrier [12C]CO2 was added. Our standard protocol is to introduce 0.5 – 1.0 mL (~22.5 – 45 μmoles) of [12C]CO2 to the molecular sieves prior to delivery of [11C]CO2 from the cyclotron.
[11C]QZ (1)
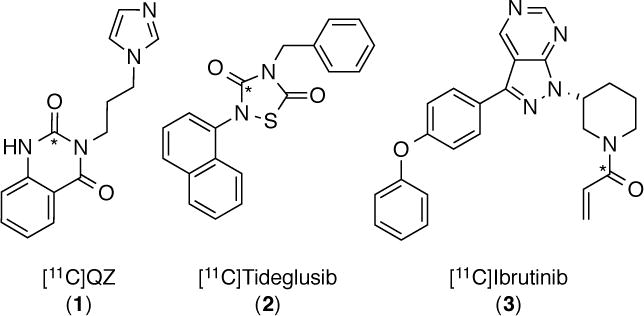
Increased levels of glutaminyl cyclase (QC) in AD are thought to result in the formation of pyroglutamate (pGlu)-modified Aβ peptides. These derivatives of Aβ are suspected to be more neurotoxic and resistant to clearance than regular amyloid-β, and they are prion-like, functioning as a template for misfolding and as a nucleus for aggregation with unmodified amyloid-β. In a post-mortem study of human neocortical brain samples it was found AD patients had upregulated levels of QC mRNA compared to patients without dementia, while immunohistochemistry suggests 20–30% higher expression of QC in all cortical layers of AD patients compared to controls.27,28 We postulate that upregulated QC could therefore be a prodromal marker for AD and were extremely interested to determine if this upregulation could be detected using PET. Our initial efforts to develop a PET radiotracer for QC concentrated on [11C]PBD150.29 However, despite strong literature precedent for the compound (PBD150 is the only QC inhibitor to date that has been shown to reduce the formation of Aβ aggregates in vitro and in vivo27,30), we were disappointed to discover that [11C]PBD150 does not cross the blood-brain barrier in rodents. Given the importance of QC as both a therapeutic and diagnostic target in AD, our efforts to develop a radiotracer for QC continue. It is unclear why [11C]PBD150 did not enter the CNS. P-glycoprotein transporter involvement was ruled out,29 but one potential explanation is the compound is too hydrophilic to cross the BBB (CLogP = 0.9). Reflecting this, we were interested in next evaluating QC radiotracers that were more lipophilic. To this end, we selected [11C]QZ (1) as a 2nd generation QC radiotracer for further evaluation (CLogP = 1.5). QZ is a closely related desmethyl analog of a recently reported QC inhibitor (IC50 = 123 nM), Figure 2.23
Figure 2.
QC Inhibitors
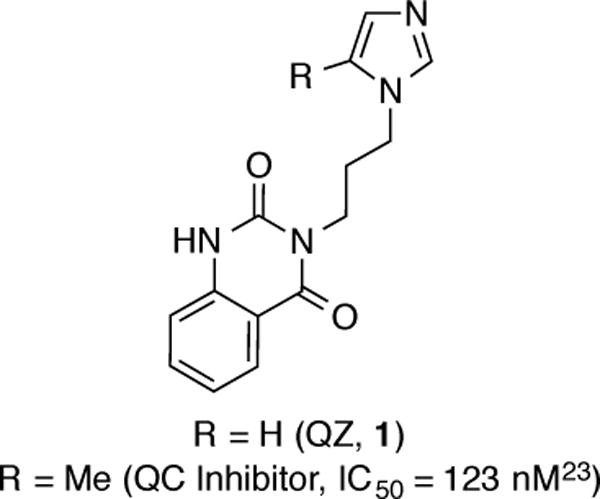
Our strategy for accessing [11C]QZ (1) was through an intramolecular [11C]CO2 fixation reaction with precursor 5 (Scheme 1). Precursor 5 was synthesized in 39% yield from isatoic anhydride (4) by coupling with 1-(3-aminopropyl)imidazole. Treatment with [11C]CO2/[12C]CO2 and BEMP, followed by POCl3, generated carrier-added [11C]QZ (CA-[11C]QZ, 1). Purification by semi-preparative HPLC provided 2 mCi of the isolated and formulated radiotracer (non-decay corrected radiochemical yield (RCY) based upon [11C]CO2 = 1%, radiochemical purity (RCP) >99% and specific activity = 3 Ci/mmol).
Scheme 1.
Synthesis of [11C]QZ (1)
A preliminary rodent microPET imaging study was conducted with CA-[11C]QZ (Figure 3). CA-[11C]QZ was administered to a female Sprague Dawley rat and dynamic PET scans were acquired for 60 min post-injection of the radiotracer (n = 1). Unfortunately, like [11C]PBD150 previously,29 analysis of summed frame images showed no brain uptake and early frames showed no first pass uptake in the brain. Given these findings, at this time we do not recommend the imidazolyl propyl urea or thiourea scaffolds for further development of a radiotracer for quantifying brain QC activity without further work to address brain permeability of the compounds.
Figure 3.
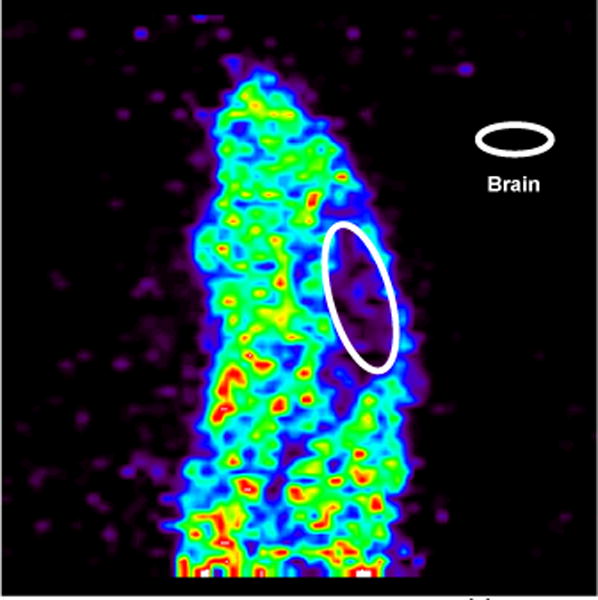
Summed sagittal rat PET image with [11C]QZ (1) (0 – 60 min after tail-vein injection of the radiotracer)
[11C]Tideglusib (2)
Glycogen synthase kinase-3β (GSK-3β) dysregulation is hypothesized to play an important role in a number of disease states, and inhibitors of GSK-3 are being explored as potential therapeutics.31 Notably, increased levels of the active form of the GSK-3β isoform are linked to hyperphosphorylation and subsequent toxic aggregation of tau protein in Alzheimer’s disease and related tauopathies.32,33 In connection with our program developing radiotracers for PET imaging of GSK-3β,34,35 we were interested in radiolabeling tideglusib, an irreversible inhibitor of GSK-3β that was shown to reduce tau hyperphosphorylation in murine models and has recently been used in phase 2 clinical trials for treatment of progressive supranuclear palsy (PSP)36 and AD.37 Radiolabeled tideglusib could also be used as a companion diagnostic during development of the therapeutic allowing, for example, study of brain uptake, target engagement, receptor occupancy and biodistribution, as well as potentially serving as a high affinity GSK-3β probe for future research studies.
We investigated the synthesis of [11C]tideglusib (2) from naphthalen-1-amine (6), Scheme 2. We hypothesized that carbon-11 fixation chemistry could be used to generate [11C]naphthylisocyanate 7, and that the 1,2,4-thiadiazolidine-3,5-dione core of [11C]tideglusib could then be accessed through an intermolecular cycloaddition between [11C]isocyanate 7 and (isothiocyanatomethyl)benzene (8), analogous to reported cold syntheses of such compounds.38 Initially treating 6 with carrier-added [11C]CO2/[12C]CO2 and BEMP followed by POCl3 generated CA-[11C]naphthylisocyanate 7. Gratifyingly, exposing 7 to (isothiocyanatomethyl)benzene (8) in the presence of additional POCl3, followed by hydrolysis with water/atmospheric oxygen, promoted cyclization to CA-[11C]tideglusib (2) as expected. However, yields using this method were poor (0.2% non-decay corrected RCY based upon [11C]CO2), likely due to inefficient chlorination of 8 with POCl3 and, consequently, a poor cyclization yield. Considering alternative chlorinating agents identified in the literature,39 we investigated whether N-chlorosuccinimide would be more effective at promoting the cyclization. This proved to be the case, and CA-[11C]tideglusib (2) was synthesized in 2% non-decay corrected RCY based upon [11C]CO2. Attempts to synthesize non-carrier-added [11C]tideglusib (NCA-[11C]tideglusib) from [11C]CO2 were unsuccessful, possibly because of competing side reactions of NCA-[11C]7, and studies to address this will be conducted in due course. We therefore focused upon automating the procedure using CA-[11C]CO2 on a GE TRACERLab FXC-Pro synthesis module (see SI for further details), which included purification by semi-preparative HPLC. However, initial attempts at automation employing a 60% acetonitrile buffered eluent for semi-preparative HPLC purification led to rapid decomposition of 2 and a RCP of only 25% at 10 min post-end-of-synthesis (EOS). Additional investigation of this issue revealed that decomposition was likely occurring due to radiolysis and not chemical instability of 2, as dilute samples (0.0001mg/mL) of non-radioactive reference standard did not show decomposition in this eluent system after 48 hours. The addition of ascorbic acid did not retard the decomposition of CA-[11C]tideglusib, so the eluent composition was switched from acetonitrile to ethanol. A 70% ethanol buffered eluent led to stabilization of CA-[11C]tideglusib, and 1 – 2 mCi were obtained following semi-preparative purification. The isolated product, formulated in 70% ethanolic HPLC mobile phase, exhibited stability over time with no decrease in RCP after ~30 min. Reformulation of a 550 μCi sample provided 150 μCi of CA-[11C]tideglusib in 10% ethanol in USP saline suitable for preclinical use (>99% RCP, specific activity: 37 Ci/mmol). No change in RCP of the reformulated sample was observed over 60 min, indicating radiolytic stability of CA-[11C]tideglusib in the injectable formulation at these levels of radioactivity.
Scheme 2.
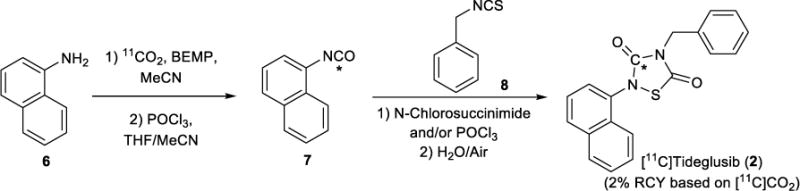
Synthesis of [11C]Tideglusib (2)
CA-[11C]Tideglusib was advanced to a microPET imaging study in a female Sprague Dawley rat. CNS permeability was observed with peak brain uptake of 200 nCi/cc, corresponding to a peak SUV of 0.7, after 3 minutes, followed by washout (Figure 4). In the whole brain time-radioactivity curve (Figure 4B), there is an apparent increase in SUV during the last two time points. We see this frequently in our data analysis, independent of radiotracer, and attribute it to increased noise resulting from the low residual counts that remain in the brain during the late frames of the PET scans. The imaging results are somewhat inconsistent with reported pharmacokinetic data, which suggests that tideglusib is an irreversible inhibitor of GSK-3β (Ki= 60nM in vitro, [35S]tideglusib/GSK-3β binding study), albeit with a time-dependent inhibition profile (IC50 = 5 nM with 60 min pre-incubation, 105 nM without pre-incubation in a double ATP and inhibitor study).40 Thus, it was expected that brain uptake should increase, or at least not decrease, over time. It is uncertain whether washout was due to A) the low specific activity of the radiotracer resulting in GSK-3β saturation, B) rapid metabolism to a non-GSK-3β active agent, or C) that tideglusib PK in vivo differ from PK in vitro for another reason. As such, studies are currently underway to improve RCY and specific activity of [11C]tideglusib, so that a better understanding of the in vivo characteristics of this clinically-relevant pharmaceutical agent can be realized.
Figure 4.
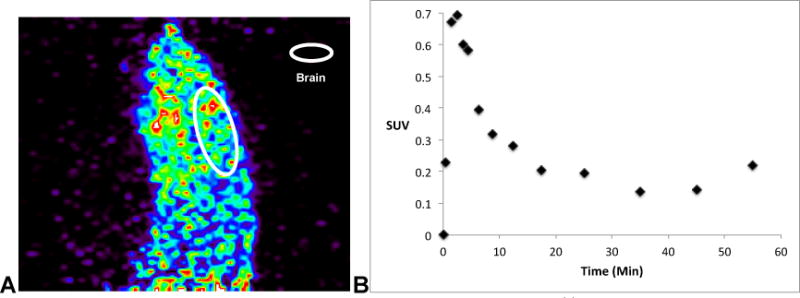
A: Summed sagittal rat PET image with CA-[11C]tideglusib (2) (0 – 60 min after tail-vein injection of the radiotracer); B: whole brain time-radioactivity curve.
[11C]Ibrutinib (3)
Bruton’s tyrosine kinase (BTK) is a mediator of the B-cell-receptor signaling pathway implicated in the pathogenesis of B-cell cancers. Early-stage clinical trials found ibrutinib, a BTK inhibitor, showed antitumor activity in several types of B cell lymphoma (for a review of the clinical use of ibrutinib, see:25), and is approved by the U.S. Food and Drug Administration (FDA) for the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. Radiolabeled ibrutinib has the potential to function as a companion diagnostic for predicting response to ibrutinib therapy (using, for example, comparable approaches to those recently employed for [11C]docetaxel41), as well as to support future drug discovery efforts around the BTK pathway.
Synthesis of [11C]ibrutinib (Scheme 3) commenced from vinylmagnesium bromide (8). The Grignard reagent was treated with [11C]CO2 directly. In our first approach, we then generated [11C]acryloyl chloride 10 by treatment with phthaloyl dichloride, per a literature procedure.42 We next attempted to distill 10 into a solution of precursor 11 and 2,6-di-tert-butylpyridine (DTBP), and were encouraged to generate [11C]ibrutinib (3), albeit in low yields (15 mCi, ~0.5% non-decay corrected RCY based upon [11C]CO2). Analysis of reaction mixtures suggested that ~90% of the [11C]acryloyl chloride polymerized during distillation. To avoid this polymerization issue at elevated temperatures, we next used oxalyl chloride to generate 10, and then transferred the entire [11C]acryloyl chloride reaction mixture into a vial containing amino precursor 11 and DTBP at room temperature. We manually produced injectable doses of 30mCi (~1% non-decay corrected RCY, based upon [11C]CO2) with >90% RCP, but had a difficult time adapting this method to the automated radiochemistry synthesis modules. Both the Grignard reagent and oxalyl chloride are very reactive and are sensitive to air, or can react with each other. In addition, any excess of either reagent would negatively impact the next step of the reaction, so quantitative quenching was required for each step. Although the synthetic method works, we anticipated that these issues would make the method practically unreliable and complicate routine automated production.
Scheme 3.
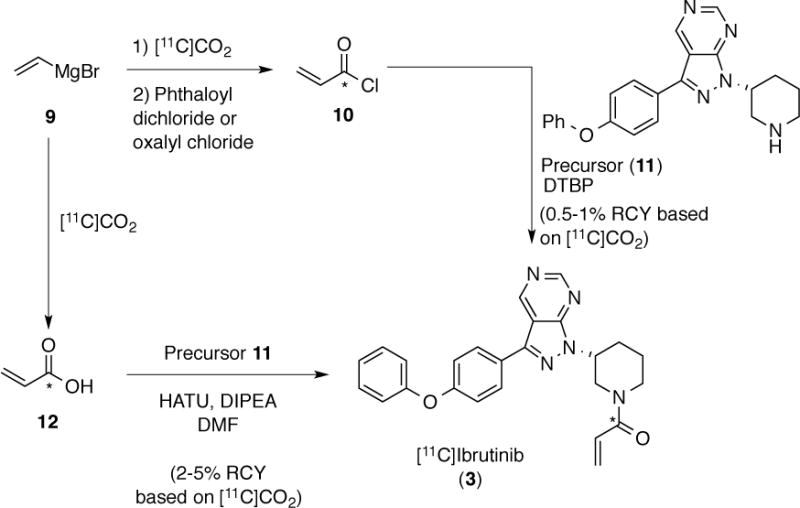
Approaches to [11C]Ibrutinib (3)
To address these issues, we next explored the possibility of generating [11C]acrylic acid 12 and then using it in a peptide coupling with precursor 11 (Scheme 3). Quenching the carboxylate, resulting from treatment of vinylmagnesium bromide with [11C]CO2 (diluted with 0.5 mL of 12CO2), using HCl efficiently generated CA-[11C]acrylic acid (12). We selected HATU as the peptide-coupling reagent, and efficient coupling of precursor 11 with CA-[11C]acrylic acid 12 generated CA-[11C]ibrutinib (3). Purification by semi-preparative HPLC was followed by reconstitution into ethanolic saline using a C18 Sep-Pak cartridge and gave 150 mCi of isolated and formulated product (5% non-decay corrected RCY based upon [11C]CO2, RCP >99% and specific activity ~55 mCi/μmol). Total synthesis time was 70 minutes from end of beam and the procedure was fully automated on a GE TRACERLab FXC-Pro synthesis module. We also tried this method no-carrier added, obtaining similar amounts (60 – 150 mCi) of isolated and formulated NCA-[11C]ibrutinib (2 – 5% non-decay corrected RCY based upon [11C]CO2) but with higher specific activities (600 – 2100 mCi/μmol). Notably however, RCP (>90%) and chemical purity were lower for the NCA product (see SI for further details of both CA and NCA syntheses).
In an initial baseline PET study in a female CD-1 mouse, NCA-[11C]ibrutinib showed significant uptake and retention in spleen, liver and intestines (Figure 5), consistent with the known distribution and excretion of ibrutinib.43 Further investigations of the biodistribution and metabolism of [11C]ibrutinib using normal and tumor bearing rodents, as well as nonhuman primates, are underway and will be reported in due course.
Figure 5.
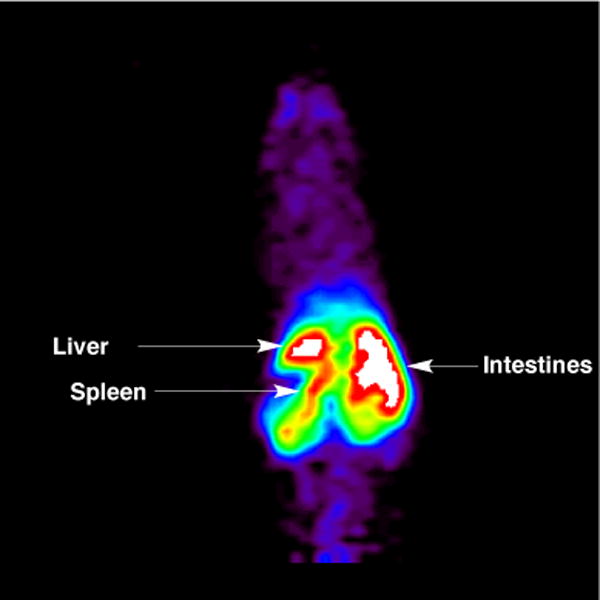
Summed coronal mouse PET image with [11C]ibrutinib (3) (0 – 60 min after tail-vein injection of the radiotracer)
In summary we have demonstrated the synthesis of complex PET radiotracers using different building blocks all accessed by [11C]CO2 fixation chemistry. This work demonstrates that the ability to rapidly trap [11C]CO2 and incorporate it into building blocks using a number of different approaches allows quite sophisticated downstream chemistry to take place in spite of the 20 min half-life of carbon-11. For example, we demonstrate that intramolecular cyclizations, intermolecular cycloaddition reactions and peptide coupling reactions are all possible. Each process is fully automated through new modifications to a standard radiochemical synthesis module, and the radiochemical syntheses provide isolated and formulated doses of radiotracers, which are appropriate for pre-clinical in vivo imaging in rodents and non-human primates. To date, radiochemical yields are somewhat low but reflect the challenging chemistry being conducted with short-lived carbon-11. Notably, yields could be improved by adding carrier [12C]CO2 to the reactions rather than using NCA-[11C]CO2, which we attribute to competing side reactions that become significant when working with NCA carbon-11. This is a current limitation of using this new chemistry with very high specific activity carbon-11, and should be taken into consideration when considering [11C]CO2 fixation approaches for radiolabeling. Exploring the scope of [11C]CO2 fixation reactions is ongoing in our laboratory, including efforts to determine the minimal amount of [12C]CO2 that can be added to minimize side reactions, and maximize specific activities of the resulting radiotracers. Nevertheless, the new reactions reported herein have enabled access to 3 new radiotracers inaccessible by other carbon-11 radiochemistry in amounts suitable for initial pre-clinical evaluation. [11C]QZ does not cross the BBB and so is unsuitable for imaging glutaminyl cyclase in the CNS. Contrastingly, initial results with [11C]tideglusib and [11C]ibrutinib are promising and suggest that additional research with both radiotracers is warranted.
EXPERIMENTAL PROCEDURES
Chemistry
General considerations
All solvents and reagents were commercially available and used without further purification unless otherwise stated. NMR spectra were recorded with a Varian 400 MHz instrument at room temperature with tetramethylsilane (TMS) as an internal standard. Mass spectra were performed on an Agilent Q-TOF HPLC-MS or VG (Micromass) 70-250-S Magnetic sector mass spectrometer employing the electrospray ionization (ESI) method or electron ionization (EI) method. High performance liquid chromatography (HPLC) was performed using a Shimadzu LC-2010A HT system equipped with a Bioscan B-FC-1000 radiation detector. All procedures including anhydrous solvents were performed with rigorously dried glassware under inert atmosphere.
Precursors
Precursors 6 and 8 were commercially available (Aldrich); precursor 11 was obtained from Bristol-Myers Squibb and used as received; precursor 5 was synthesized as described below.
N-(3-(1H-imidazol-1-yl)propyl)-2-aminobenzamide (5)
Isatoic anhydride 4 (0.300 g, 2.4 mmol) was dissolved in THF (14.4 mL, 0.17 M) and 1-(3-aminopropyl)imidazole (0.286 mL, 2.4 mmol) was added. The solution was stirred overnight at room temperature, at which point solvent was removed under vacuum. The resulting product was extracted with DCM and the organic layer was washed with 5 mL saturated sodium bicarbonate solution, 2× with water, dried (MgSO4), and concentrated in vacuo. The product was purified by silica gel chromatography (DCM/MeOH gradient). After purification, product 5 was collected as an oil (0.23 g, 39% yield); Rf 0.17 (9:1 DCM:MeOH); 1H NMR (400 MHz d4-MeOD): 7.68 (1H, s), 7.40 (1H, d, J=7.8), 7.17-7.14 (2H, m), 6.95 (1H, br), 6.72 (1H, d, J=8.1), 6.60 (1H, t, J=7.5), 4.08 (2H, t, J=7.0), 3.31 (2H, t, J=6.8), 2.05 (2H, quin, J=6.9); HRMS: calculated for C13H16N4O: 244.1397, found: 245.1394 ([M+H]+).
Reference Standards
The identity of all 11C-labeled radiotracers was confirmed by co-injection onto HPLC with the corresponding unlabeled reference standard. Details on commercial suppliers and/or synthesis of reference standards is described in the supporting information (SI).
Radiochemistry
General Considerations
Standard reagents and solvents were commercially available and used without further purification, unless otherwise noted: sodium chloride, 0.9% USP and sterile water for Injection, USP were purchased from Hospira; Dehydrated Alcohol for Injection, USP was obtained from Akorn Inc.; Ascorbic Acid for Injection, USP was acquired from Bioniche Pharma; molecular sieves were purchased from Alltech; and HPLC columns were acquired from Phenomenex. Other synthesis components were obtained as follows: sterile filters were acquired from Millipore; C18 Sep-Paks were purchased from Waters Corporation; 10 cc sterile vials were obtained from HollisterStier. Sep-Paks were flushed with 10 mL of ethanol followed by 10 mL of sterile water prior to use. [11C]CO2 was produced with a General Electric (GE) PETTrace cyclotron. High purity nitrogen containing 0.5% O2 was irradiated with 16.5 MeV to generate [11C]CO2, which was delivered to a GE TRACERlab FXC-Pro synthesis module via stainless steel lines. Radiochemical syntheses were carried out using GE TRACERLab FXC-Pro synthesis modules housed in Comecer hot-cells. Doses produced were assessed via standard quality control techniques (see below) and were appropriate for rodent studies.
[11C]QZ (1)
A solution of precursor 5 (4 mg) and BEMP (5 μL) dissolved in 0.1 mL acetonitrile was sparged for 4 min with [11C]CO2 (300 mCi) that had been diluted with 1 mL of 12CO2 at 7 mL/min (Ar used for transfer). A solution of POCl3 (0.4 μL in 0.1 mL of acetonitrile) was added and the resulting mixture was stirred for 4 minutes. Then 1 mL of HPLC mobile phase was added and used to transfer the reaction mixture to the HPLC loop. The product was purified by reverse phase HPLC (column: Luna C18 semi-prep, 150 × 10 mm, 10 μ; mobile phase: 25% EtOH, 10 mM NaH2PO4, pH 8.0, 0.50 g/L of L-ascorbic acid; flow rate: 4 mL/min) and the product peak (tR ~ 9 – 10 min) was collected and diluted with USP saline to achieve a 10% ethanol solution suitable for injection. Yield was 2 mCi (1% non-corrected isolated and formulated RCY based upon [11C]CO2; RCP = 100%; pH = 6.5).
[11C]Tideglusib (2)
A solution of naphthalen-1-amine (6) (0.72 mg, 5 μmol) and BEMP (5 μL, 17.5 μmol) dissolved in 0.2 mL acetonitrile was sparged for 6 min with [11C]CO2 (800 mCi) that had been diluted with 0.5 mL of 12CO2 at 5 mL/min (He used for transfer). After this time the reaction mixture was transferred to a vial containing POCl3 (10 μL, 100 μmol) in 100 μL THF (inhibitor-free) and allowed to react for 2 min to generate [11C]7. The solution was then transferred to a vial containing benzylisothiocyanate (8) (3.7 μL, 25 μmol) and N-chlorosuccinimide (13.4 mg, 100 μmol) in 200 μL THF (inhibitor-free) with 20 mL air and allowed to react for 5 min. The crude reaction mixture was diluted with 600 μL of 1:1 ethanol: water and transferred (using 60 mL air) into an 8 mL sterile vial. Analysis of the reaction mixture revealed 14.6 mCi of [11C]titdeglusib, which equated to 2% non-decay corrected RCY based upon [11C]CO2. Total synthesis time was 60 minutes from end of beam. Efforts to purify and reformulate [11C]tideglusib were complicated by radioloysis, but were eventually accomplished using ethanolic solvent systems (see SI for detailed information).
[11C]Ibrutinib (3)
[11CO2] (~3 Ci diluted with 0.5 mL of 12CO2) was bubbled into vinylmagnesium bromide (200 μL of 1.0 M Grignard diluted with 400 μL of THF) at 7 mL/min flow rate for 4 minutes. 250 μL of HCl solution (100 μL of 4.0 M HCl/dioxane diluted with 800 μL of DMF) was then added and the reaction stirred for an additional 1 min. A solution of precursor 11 (3.9 mg) and DIPEA (25 μL) in DMF (150 μL), and a solution of (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) (HATU, 6.5 mg) in DMF (150 μL) were then added and the reaction mixture was stirred at room temperature for 10 min. The reaction was then diluted with 1 mL of HPLC mobile phase and purified by semi-preparative HPLC (column: Prodigy ODS 250×10 mm; mobile phase: 30 mM NH4OAc in 45% MeCN/H2O; flow rate: 4 mL/min). The product peak (tR ~11 – 12 min) was collected into 50 mL of water, and loaded onto a C18 Sep-Pak (Waters, 1cc). The final dose was eluted by 0.5 mL of ethanol followed by 4.5 mL of injectable saline. Total synthesis time was 70 minutes from end of beam and gave 150 mCi of isolated and formulated product (5% non-decay corrected RCY based upon [11C]CO2; RCP >99%; specific activity = 55 mCi/μmol; pH = 6.0). No-carrier-added syntheses were also attempted and further details are provided in the SI).
Quality Control
Radiochemical purity of products was assessed using Shimadzu LC-2010A HT system equipped with UV-Vis and radioactivity detectors as follows:
[11C]QZ
Column: Phenomenex Luna C18, 150×4.6 mm; mobile phase: 10 mM NH4OAc in 15 % MeCN; flow rate: 2.0 mL/min; wavelength: 254 nm; room temperature; product tR = 9.9 min.
[11C]Tideglusib
Column: Phenomenex Luna phenyl hexyl, 5 micron, 150×4.6mm; mobile phase: 10 mM KH2PO4 in 70% EtOH, pH 4.6; flow rate: 1 mL/min; wavelength: 254 nm; product tR = 7.2 min.
[11C]Ibrutinib
Column: Kinetex XB-C18 150×4.6 mm; mobile phase: 30 mM NH4OAc in 45% MeCN; flow rate: 1.0 mL/min; wavelength: 254 nm; product tR = 6.9 min.
Supplementary Material
Acknowledgments
PJHS acknowledges US DOE/NIBIB (DE-SC0012484), NIH (T32-EB005172) and the Alzheimer’s Association (NIRP-14-305669) for financial support. Ibrutinib precursor and reference standard were generously provided by Bristol-Myers Squibb.
Footnotes
Supporting Information Available: Experimental procedures for the synthesis of precursors, reference standards and radiotracers, spectral/chromatographic data for all new compounds synthesized, semi-preparative and analytical radio-HPLC for all radiotracers, and details of pre-clinical PET imaging studies. This material is available free of charge via the Internet at http://pubs.acs.org.
LITERATURE CITED
- 1.Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 2.Pither R. PET and the Role of In Vivo molecular Imaging in Personalized Medicine. Expert Rev Mol Diagn. 2003;3:703–713. doi: 10.1586/14737159.3.6.703. [DOI] [PubMed] [Google Scholar]
- 3.Matthews PM, Rabiner EA, Passchier J, Gunn RN. Positron Emission Tomography Molecular Imaging for Drug Development. Br J Clin Pharmacol. 2011;73:175–186. doi: 10.1111/j.1365-2125.2011.04085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N Radiolabels for Positron Emission Tomography. Angew Chem Int Ed. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 5.Scott PJH. Methods for the Incorporation of Carbon-11 To Generate Radiopharmaceuticals for PET Imaging. Angew Chem Int Ed. 2009;48:6001–6004. doi: 10.1002/anie.200901481. [DOI] [PubMed] [Google Scholar]
- 6.Antoni G. Development of Carbon-11 Labelled PET Tracers – Radiochemical and Technological Challenges in a Historic Perspective. J Label Compd Radiopharm. 2015;58:65–72. doi: 10.1002/jlcr.3258. [DOI] [PubMed] [Google Scholar]
- 7.Baumann M, Baxendale IR. An Overview of the Synthetic Routes to the Best-selling Drugs Containing 6-membered Heterocycles. Beilstein J Org Chem. 2013;9:2265–2319. doi: 10.3762/bjoc.9.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Runkle AC, Shao X, Tluczek LJM, Henderson BD, Hockley BG, Scott PJH. Automated Production of [11C]Acetate and [11C]Palmitate using a Modified GE Tracerlab FXC-Pro. Appl Radiat Isot. 2011;69:691–698. doi: 10.1016/j.apradiso.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Riss PJ, Lu S, Telu S, Aigbirhio FI, Pike VW. CuI-Catalyzed 11C Carboxylation of Boronic Acid Esters: a Rapid and Convenient Entry to 11C-Labeled Carboxylic Acids, Esters, and Amides. Angew Chem Int Ed. 2012;51:2698–2702. doi: 10.1002/anie.201107263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rotstein BH, Hooker JM, Woo J, Collier TL, Brady TJ, Liang SH, Vasdev N. Synthesis of [11C]Bexarotene by Cu-Mediated [11C]Carbon Dioxide Fixation and Preliminary PET Imaging. ACS Med Chem Lett. 2014;5:668–672. doi: 10.1021/ml500065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rotstein BH, Liang SH, Holland JP, Collier TL, Hooker JM, Wilson AA, Vasdev N. [11C]CO2 Fixation: a Renaissance in PET Radiochemistry. Chem Commun. 2013;49:5621–5629. doi: 10.1039/c3cc42236d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haji Dheere AK, Bongarzone S, Taddei C, Yan R, Gee AD. Synthesis of 11C-Labelled Symmetrical Ureas via the Rapid Incorporation of [11C]CO2 into Aliphatic Amines. Synlett. 2015;26:2257–2260. [Google Scholar]
- 13.Haji Dheere AK, Yusuf N, Gee A. Rapid and Efficient Synthesis of [11C]Ureas via the Incorporation of [11C]CO2 into Aliphatic and Aromatic Amines. Chem Commun. 2013;49:8193–8195. doi: 10.1039/c3cc44046j. [DOI] [PubMed] [Google Scholar]
- 14.Hicks JW, Parkes J, Tong J, Houle S, Vasdev N, Wilson AA. Radiosynthesis and Ex Vivo Evaluation of [11C-Carbonyl]carbamate- and Urea-based Monoacylglycerol Lipase Inhibitors. Nucl Med Biol. 2014;41:688–694. doi: 10.1016/j.nucmedbio.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N, Wilson AA. Synthesis and Preclinical Evaluation of [11C-Carbonyl]PF-04457845 for Neuroimaging of Fatty Acid Amide Hydrolase. Nucl Med Biol. 2013;40:740–746. doi: 10.1016/j.nucmedbio.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Towards the Preparation of Radiolabeled 1-Aryl-3-benzyl ureas: Radiosynthesis of [11C-Carbonyl] AR-A014418 by [11C]CO2 Fixation. Bioorg Med Chem Lett. 2012;22:2099–2101. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 17.Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N. Synthesis and Application of Isocyanates Radiolabeled with Carbon-11. Chem Eur J. 2011;17:259–264. doi: 10.1002/chem.201002345. [DOI] [PubMed] [Google Scholar]
- 18.Hooker JM, Reibel AT, Hill SM, Schueller MJ, Fowler JF. One-Pot, Direct Incorporation of [11C]CO2 into Carbamates. Angew Chem Int Ed. 2009;48:3482–3485. doi: 10.1002/anie.200900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, Fowler CJ, Vasdev N. Radiosynthesis and Evaluation of [11C-Carbonyl]-Labeled Carbamates as Fatty Acid Amide Hydrolase Radiotracers for Positron Emission Tomography. J Med Chem. 2013;56:201–209. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasdev N, Sadovski O, Garcia A, Dollé F, Meyer J, Houle S, Wilson AA. Radiosynthesis of [11C]SL25.1188 via [11C]CO2 Fixation for Imaging Monoamine Oxidase B. J Label Compd Radiopharm. 2011;54:678–680. [Google Scholar]
- 21.Wilson AA, Garcia A, Houle S, Vasdev N. Direct Fixation of [11C]-CO2 by Amines: Formation of [11C-Carbonyl]-methylcarbamates. Org Biomol Chem. 2010;8:428–432. doi: 10.1039/b916419g. [DOI] [PubMed] [Google Scholar]
- 22.Miller PW, Bender D. [11C]Carbon Disulfide: A Versatile Reagent for PET Radiolabelling. Chem Eur J. 2012;18:433–436. doi: 10.1002/chem.201103128. [DOI] [PubMed] [Google Scholar]
- 23.Tran PT, Hoang VH, Thorat SA, Kim SE, Ann J, Chang YJ, Nam DW, Song H, Mook-Jung I, Lee J, Lee J. Structure-activity Relationship of Human Glutaminyl Cyclase Inhibitors having an N-(5-methyl-1H-imidazol-1-yl)propyl Thiourea Template. Bioorg Med Chem. 2013;21:3821–3830. doi: 10.1016/j.bmc.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J Biol Chem. 2012;287:893–904. doi: 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davids MS, Brown JR. Ibrutinib: a First in Class Covalent Inhibitor of Bruton’s Tyrosine Kinase. Future Oncol. 2014;10:957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao X, Hoareau R, Runkle AC, Tluczek LJM, Hockley BG, Henderson BD, Scott PJH. Highlighting the versatility of the Tracerlab synthesis modules. Part 2: fully automated production of [11C]-labeled radiopharmaceuticals using a Tracerlab FXC-Pro. J Label Compd Radiopharm. 2011;54:819–838. doi: 10.1002/jlcr.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, et al. Glutaminyl Cyclase Inhibition Attenuates Pyroglutamate Aβ and Alzheimer’s Disease-like Pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 28.Morawski M, Schilling S, Kreuzberger M, Waniek A, Jäger C, Koch B, Cynis H, Kehlen A, Arendt T, Hartlage-Rübsamen M, et al. Glutaminyl Cyclase in Human Cortex: Correlation with (pGlu)-amyloid-β Load and Cognitive Decline in Alzheimer’s Disease. J Alzheimer’s Dis. 2014;39:385–400. doi: 10.3233/JAD-131535. [DOI] [PubMed] [Google Scholar]
- 29.Brooks AF, Jackson IM, Shao X, Kropog GW, Sherman P, Quesada CA, Scott PJH. Synthesis and Evaluation of [11C]PBD150, a Radiolabeled Glutaminyl Cyclase Inhibitor for the Potential Detection of Alzheimer’s Disease Prior to Amyloid β Aggregation. Med Chem Commun. 2015;6:1065–1068. doi: 10.1039/C5MD00148J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchholz M, Heiser U, Schilling S, Niestroj AJ, Zunkel K, Demuth HU. The First Potent Inhibitors for Human Glutaminyl Cyclase: Synthesis and Structure-activity Relationship. J Med Chem. 2006;49:664–677. doi: 10.1021/jm050756e. [DOI] [PubMed] [Google Scholar]
- 31.Woodget JR. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hooper C, Killick R, Lovestone S. The GSK3 Hypothesis of Alzheimer’s Disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avila J, Wandosell F, Hernańdez F. Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease Pathogenesis and Glycogen Synthase Kinase-3 Inhibitors. Expert Rev Neurother. 2010;10:703–710. doi: 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- 34.Cole EL, Shao X, Sherman P, Quesada C, Fawaz MV, Desmond TJ, Scott PJH. Synthesis and evaluation of [11C]PyrATP-1, a Novel Radiotracer for PET Imaging of Glycogen Synthase Kinase-3β (GSK-3β) Nucl Med Biol. 2014;41:507–512. doi: 10.1016/j.nucmedbio.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Shao X, Ohnmacht SA, Ferrari V, Hong YT, Williamson DJ, Fryer TD, Quesada CA, Sherman P, Riss PJ, Scott PJH, Aigbirhio FI. Synthesis and Initial In Vivo Studies with [11C]SB-216763: The First Radiolabeled Brain Penetrative Inhibitor of GSK-3. ACS Med Chem Lett. 2015;6:548–552. doi: 10.1021/acsmedchemlett.5b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolosa E, Litvan I, Höglinger GU, Burn D, Lees A, Andrés MV, Gómez-Carrillo B, León T, Del Ser T, the TAUROS Investigators A Phase 2 Trial of the GSK-3 Inhibitor Tideglusib in Progressive Supranuclear Palsy. Mov Disord. 2014;29:470–478. doi: 10.1002/mds.25824. [DOI] [PubMed] [Google Scholar]
- 37.Lovestone S, Boada M, Dubois B, Huell M, Rinne JO, Huppertz HJ, Calero M, Andres MV, Gomez-Carrillo B, Leon T, del Ser T. A Phase II Trial of Tideglusib in Alzheimer’s Disease. Journal of Alzheimer’s Disease. 2015;45:75–88. doi: 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- 38.Medina Padilla M, Alonso Cascon M, Dorronsoro Diaz I, Martinez Gil A, Panizo Del Pliego G, Fuertes Huerta A, Perez Puerto MJ. GSK-3 Inhibitors. 2005 US20050222220. [Google Scholar]
- 39.Nasim S, Crooks PA. N-Chlorosuccinimide is a Convenient Oxidant for the Synthesis of 2,4-Disubstituted 1,2,4-Thiadiazolidine-3,5-diones. Tetrahedron Lett. 2009;50:257–259. [Google Scholar]
- 40.Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J Biol Chem. 2012;287:893–904. doi: 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van der Veldt AAM, Lubberink M, Mathijssen RHJ, Loos WJ, Herder GJM, Greuter HN, Comans EFI, Rutten HB, Eriksson J, Windhorst AD, Hendrikse NH, Postmus PE, Smit EF, Lammertsma AA. Toward Prediction of Efficacy of Chemotherapy: a Proof of Concept Study in Lung Cancer Patients using [11C]Docetaxel and Positron Emission Tomography. Clin Cancer Res. 2013;19:4163–4173. doi: 10.1158/1078-0432.CCR-12-3779. [DOI] [PubMed] [Google Scholar]
- 42.Mishani E, Levitzki A, Ortu G, Ben-David I, Rozen Y. Radiolabeled Anilinoquinazolines and their use in Radioimaging and Radiotherapy. PCT Int Appl. 2005 WO 2005023315. [Google Scholar]
- 43.http://www.imbruvica.com; accessed March 2016
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.