

Abstract
Purpose
Most patients with pancreatic ductal adenocarcinoma (PDAC) die within 5 years following resection plus adjuvant gemcitabine (Gem) from outgrowth of occult metastases. We hypothesized that inhibition of the KRAS pathway with the MEK inhibitor trametinib would inhibit the outgrowth of occult liver metastases in a preclinical model.
Methods
Liver metastases harvested from two patients with PDAC (Tumors 608, 366) were implanted orthotopically in mice. Tumor cell lines were derived and transduced with lentiviruses encoding luciferase and injected into spleens of mice generating microscopic liver metastases. Growth kinetics of liver metastases were measured with bioluminescent imaging and time-to-progression (TTP), progression-free survival (PFS), and overall survival (OS) were determined.
Results
Trametinib (0.3 mg/kg BID) significantly prolonged OS versus control (Tumor 608: 114 vs. 43 days, p < 0.001; Tumor 366: not reached vs. 167 days, p = 0.0488). In vivo target validation demonstrated trametinib significantly reduced phosphorylated-ERK and expression of the ERK-responsive gene DUSP6. In a randomized, preclinical trial, mice were randomized to: (1) control, (2) adjuvant Gem (100 mg/kg IP, Q3 days) × 7 days followed by surveillance, or (3) adjuvant Gem followed by trametinib. Sequential Gem-trametinib significantly decreased metastatic cell outgrowth and increased TTP and PFS.
Conclusions
Treatment of mice bearing micrometastases with trametinib significantly delayed tumor outgrowth by effectively inhibiting KRAS-MEK-ERK signaling. In a randomized, preclinical, murine trial adjuvant sequential Gem followed by trametinib inhibited occult metastatic cell outgrowth in the liver and increased PFS versus adjuvant Gem alone. An adjuvant trial of sequential Gem-trametinib is being planned in patients with resected PDAC.
Patients who undergo a margin-negative resection of localized pancreatic ductal adenocarcinoma (PDAC) followed by adjuvant chemotherapy have a median survival of only 21 months and 80 % of patients will die within 5 years due to the development of metastases.1,2 Most of these metastases occur in the liver, and thus, patients likely harbor occult metastatic PDAC in the liver at the time of surgery and these occult metastatic cells may be resistant to current adjuvant strategies. The fate of these metastatic cells is dependent on the complex interactions between the liver microenvironment and metastatic niche, local immune factors, and individual PDAC tumor biology.3
Given the prominent role of activating KRAS mutations in PDAC progression, effective inhibition of the RAS-RAF-MEK-ERK signaling cascade is an important target for therapy and has been identified as a priority in pancreatic cancer research.4–8 This signaling is required for epithelial-to-mesenchymal transition (EMT) and increased cancer cellular motility and invasiveness, which are all required for metastasis.9,10 Additionally, cancer cellular-microenvironment interactions are dependent on RAS pathway signaling, including angiogenesis and immune system evasion.9,11,12 However, the role of RAS pathway signaling in cells within the metastatic microenvironment is largely unknown. We previously demonstrated that the MEK inhibitor trametinib significantly inhibited orthotopic tumor growth of patient-derived pancreatic cancer xenografts in mice.13,14 Based on these results and the known role of MEK signaling in processes necessary for metastatic cell growth, we hypothesized that MEK inhibition would inhibit the outgrowth of occult liver metastases from PDAC.
Utilizing a patient-derived splenic injection model of PDAC metastatic to the liver, we demonstrate that trametinib decreased overall liver tumor burden, increased time to proliferative outgrowth of PDAC tumors, and significantly increased survival of mice. Moreover, we designed a rational preclinical trial using this model to reflect an adjuvant clinical trial in patients and show that gemcitabine therapy followed sequentially by maintenance trametinib therapy is superior to standard-of-care gemcitabine.
METHODS
Therapeutics
Trametinib is a potent and selective allosteric inhibitor of MEK1/2 and was obtained from GlaxoSmithKline (Brentford, United Kingdom).15–17 Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), a nucleoside, was obtained from the University of Virginia Clinical Pharmacy.
Derivation of Cell Lines and Lentiviral Transduction
Patient-derived cell lines (Tumors 608, 366) were obtained under IRB protocol as previously described and have been extensively characterized.13,14,18,19 Cells were transduced using firefly luciferase lentivirus (KeraFAST, Boston, MA). Cell lines were maintained in RPMI containing 10 % FBS and penicillin/streptomycin. Cells were thawed, propagated, and used for experiments every 4– 6 months (fewer than ten passages).
Splenic Injection Model of PDAC Growth in the Murine Liver
Patient-derived PDAC cell lines (Tumors 608, 366) were harvested by trypsinization and a suspension of 1 × 106 cells/50 μL was prepared in serum-free media for injection. Six-to-eight week old athymic nude mice (NCI, Frederick, MD) were anesthetized with 0.1 cc of ketamine (75 mg/kg) and dexmedetomidine (0.2 mg/kg), intraperitoneal for splenic injection. A 1.5-cm left flank incision was made and the spleen was exteriorized; then, cells were injected into the spleen. Tumor cells were allowed to circulate for 10 min, and then the spleen was resected. The peritoneum and skin were sutured, Atipamezole (2 mg/kg, subcutaneous) was used to reverse the anesthesia, and 100-μL subcutaneous injection of ketoprofen (1 mg/mL, provided by the University of Virginia Department of Comparative Medicine) was administered postoperatively for analgesia.
In Vivo Bioluminescent Imaging
After splenic injection of luciferase-transduced PDAC cell lines, hepatic tumor cell burden was monitored by serial in vivo bioluminescent imaging. Ten minutes before imaging, mice were anesthetized with aerosolized isoflurane and dosed with luciferin substrate (150 mg/kg i.p.). Mice were then imaged using the IVIS 100 imaging system (Xenogen, Alameda, CA). Luminescence was captured and quantified using a region of interest tool, as previously described.20,21 Luminescence data were plotted relative to the initial bioluminescence level to generate tumor growth curves. Time to proliferative outgrowth, or time-to-progression (TTP), was defined as the time to develop relative average hepatic bioluminescence of 2.0. Progression-free survival (PFS) was defined as survival of mice without evidence of tumor progression (i.e., relative average hepatic bioluminescence <2.0).
Histologic Evaluation
Paraffin-embedded sections of murine livers pre- and post-splenic injection of PDAC cell lines were stained with H&E using standard methods. Immunohistochemical staining was performed using antibodies to human EpCAM (CD326) and human phosphorylated ERK1/2. Negative controls were performed omitting the primary antibody.
Quantitative Real-Time PCR
Isolation of tumor cells from the livers of mice was performed utilizing magnetic-activated cell sorting with bead-labeled antibodies to human EpCAM (CD326; Miltenyi Biotec Inc, San Diego, CA). RNA extraction, reverse transcription, and quantitative real-time PCR were performed as previously described.19
Conditions for PCR included 95 °C for 3 min, followed by 40 cycles of 15 s at 95 °C, 30 s at 56.7 °C, and 30 s at 72 °C for GAPDH and DUSP6. Relative expression levels of target sequences were determined by the standard curve method using cDNA from MPanc-96 pancreatic cancer cell line (American Type Culture Collection, Manassas, VA) using serial dilutions. Expression levels of DUSP6 target sequences were normalized to GAPDH expression.
Statistical Analysis
The average hepatic radiance of individual mice during treatment was divided by starting radiance to calculate relative change in radiance. Student’s t test was used to compare continuous variables. All group comparisons were unpaired. Continuous variables were expressed as means ± the standard error of the mean. All p values reported are two-tailed, and statistical significance was indicated by p values <0.05. All experiments were performed in triplicate. GraphPad Prism (Version 5.0b) software (La Jolla, CA) was used for all statistical analyses.
RESULTS
Splenic Injection of PDX Cell Lines
After splenic injection, both patient-derived cell lines developed liver metastases and followed reproducible phases of growth kinetics (Fig. 1a, b). Both cell lines experienced an initial rapid clearance phase corresponding with decay in hepatic bioluminescence, which led to a cell survival phase (the minimum measured hepatic bioluminescence). This phase of cellular survival was followed by proliferative outgrowth and the rapid and robust increase in bioluminescent signal from the murine livers (Fig. 1a, b).
FIG. 1.
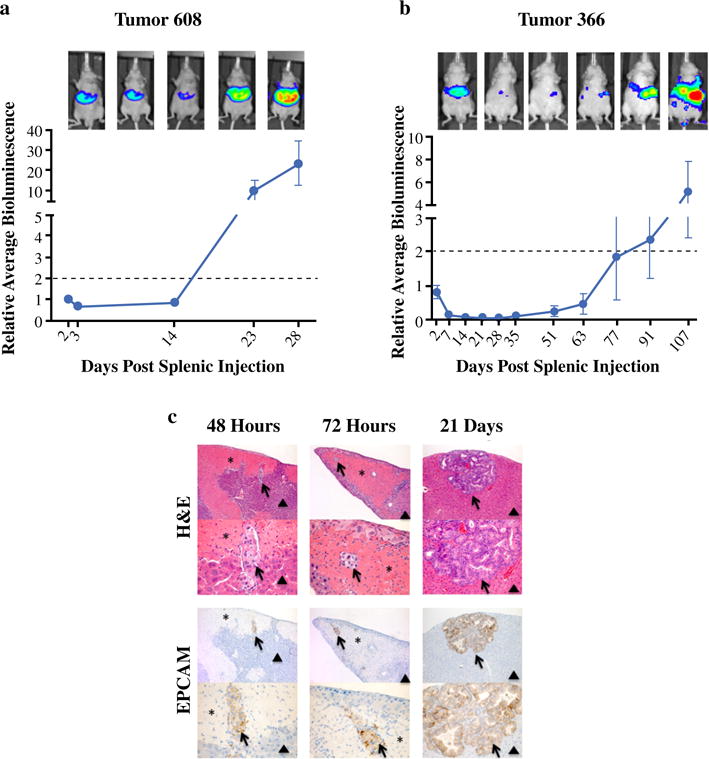
Patient-derived PDAC cell lines exhibit characteristic and reproducible growth kinetics in the murine liver microenvironment following splenic injection. Sequential in vivo bioluminescent imaging following splenic injection of 106 tumor cells reveals both Tumor 608 (a) and Tumor 366 (b) cell lines share conserved phases of growth but have differential growth kinetics. Dotted line signifies a twofold increase in average hepatic bioluminescence from 48 h post-splenic injection and represents time to proliferative outgrowth (days). Patient-derived PDAC cell lines develop into mature PDAC tumors (c) and recapitulate metastatic disease in the liver of patients. Livers were harvested from mice harboring Tumor 608 cells 48, 72 h, and 21 days post-splenic injection and prepared for histologic review. Hematoxylin and eosin staining reveals tumor architecture and corresponding areas were prepared for IHC utilizing an anti-EpCAM (CD326) antibody recognizing human epithelial cells and, thus, patient-derived PDAC cells. Arrows, metastatic tumor cells. Asterisks, areas of peritumoral hepatic injury and necrosis. Arrow heads, normal liver
The phases of rapid clearance and cellular survival were temporally different between Tumor 608 and 366, which led to a time to proliferative outgrowth (twofold increase in relative hepatic bioluminescence) that was characteristic and reproducible for each cell line examined (Tumor 608: 18 days, Fig. 1a vs. Tumor 366: 80 days, Fig. 1b).
Histologic Evaluation of Liver Microenvironment After Splenic Injection
Hematoxylin and eosin staining of livers at 48 h after injection of Tumor 608 cells demonstrated intact tumor cells within the distal hepatic portal venous structures with corresponding well-demarcated areas of regional hepatic injury and necrosis (Fig. 1c). The areas of hepatic injury were devoid of intact hepatic architecture and associated cells. Higher magnification revealed intact embolized PDAC tumor cells filling the hepatic portal venous structures within the area of injury. After 72 h post-injection, tumor cells were still evident within the vasculature and there was an increase of immune response cells surrounding the perimeter of injury and beginning to infiltrate the remodeling liver (Fig. 1c). Intact disordered glandular structures characteristic of adenocarcinoma were clearly developed by 21 days with associated stroma reflective of PDAC’s characteristic desmoplastic reaction (Fig. 1c). Similar results were seen for Tumor 366 (data not shown).
Role of RAS-MEK-ERK Signaling on PDX Cell Proliferation in the Liver
Mice underwent splenic injection of either Tumor 608 or Tumor 366 cells then were randomized to either trametinib (0.3 mg/kg, oral gavage, daily) or vehicle control beginning 48 h post-splenic injection (Fig. 2). Mice harboring Tumor 608 and treated with control vehicle exhibited a steady decay of bioluminescence to a nadir at approximately 7–10 days post-splenic injection, whereas mice treated with trametinib reached a nadir in bioluminescence at approximately 14 days and at a level lower than that of the bioluminescence in the livers of control mice (Fig. 2a, b). Mice bearing Tumor 366 cells and treated with vehicle control reached a nadir in hepatic bioluminescence at approximately 10 days (vs. 17 days for trametinib, Fig. 2c). Control mice harboring Tumor 608 cells reached proliferative outgrowth at approximately 16 days post-splenic injection (vs. 28 days with trametinib treatment, Fig. 2b). For mice harboring Tumor 366 cells in the liver, time to proliferative outgrowth with trametinib treatment was over 170 days, compared with approximately 60 days for control mice (Fig. 2c). Thus, trametinib therapy resulted in a more than twofold increase in time to proliferative outgrowth for both patient-derived PDAC cell lines.
FIG. 2.
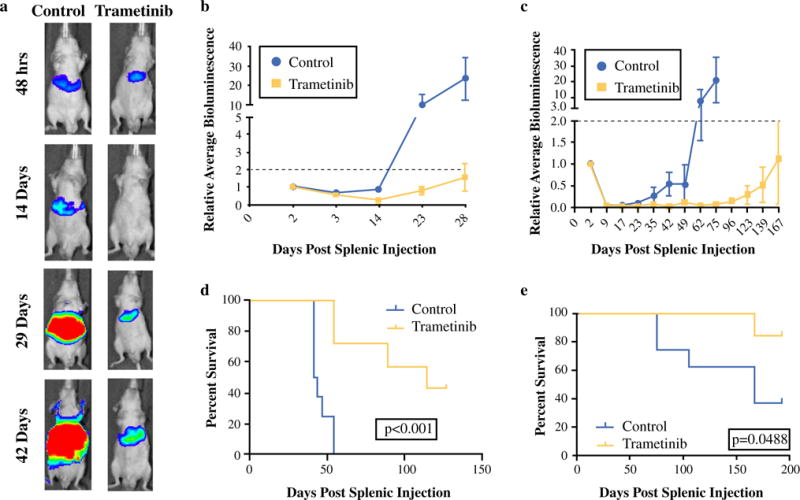
Blocking the RAS-RAF-MEK-ERK signaling pathway results in significant inhibition of PDAC growth in the liver and prolongs survival of mice harboring hepatic PDAC. Mice received either the MEK inhibitor trametinib (0.3 mg/kg, oral gavage, daily) or vehicle control beginning 48 h post-splenic injection. a Representative images of hepatic bioluminescence from mice harboring Tumor 608 over the course of therapy (quantified linearly in b). Daily trametinib beginning 48 h post-splenic injection significantly decreased hepatic tumor burden (a) and prolonged time to proliferative outgrowth for both Tumor 608 (b) and Tumor 366 (c). Moreover, trametinib significantly prolonged survival of mice harboring hepatic Tumor 608 (d) and Tumor 366 (e)
In a survival study, mice with hepatic Tumor 608 treated with trametinib at 48 h post-injection had a median overall survival of 114 vs. 43 days for control (p < 0.001; Fig. 2d). Control mice harboring Tumor 366 had a median overall survival of 167 days. Trametinib significantly prolonged survival and median survival was not reached in the trametinib group by the end of the study as very few mice died (p = 0.0488; Fig. 2e). Trametinib therapy resulted in a dramatic increase in overall survival for mice harboring hepatic patient-derived PDAC tumors.
In Vivo Target Validation
We next investigated the degree that trametinib inhibits the RAS-RAF-MEK-ERK signaling axis in vivo. Mice harboring Tumor 608 and treated with either control or trametinib starting at 48 h post-injection were sacrificed after 22 days and histologic analyses were performed. Immunohistochemical staining of tumors for phosphorylated ERK (pERK1/2) revealed a significant decrease of this activated protein with trametinib treatment (Fig. 3a). RT-PCR for DUSP6 from tumor cells harvested from the livers demonstrated decreased levels of transcripts of DUSP6 in trametinib treated mice (Fig. 3b), consistent with DUSP6’s role as a negative feedback regulator of the RAS pathway and a biomarker of RAS pathway output.22 These results demonstrated in vivo target validation that trametinib decreases RAS-RAF-MEK-ERK signaling.
FIG. 3.
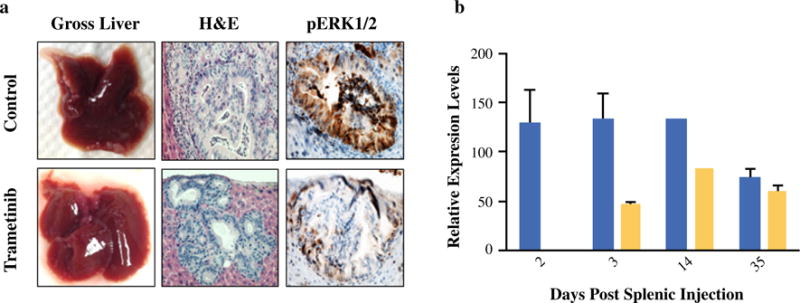
Trametinib decreases RAS pathway signaling output in PDAC tumors in the liver microenvironment. a Gross and histologic review of livers harvested from mice harboring Tumor 608 21 days post-splenic injection. Mice were treated with either vehicle control or trametinib beginning at 48 h post-splenic injection. Immunohistochemistry utilizing an antibody recognizing phosphorylated human ERK1/2 revealed decreased levels of activated ERK1/2 in mice receiving trametinib. b RT-PCR for DUSP6 from RNA isolated from PDAC cells isolated from the livers of mice harboring Tumor 608 at various time points after splenic injection revealed decreased levels of transcripts of DUSP6 in mice treated with trametinib (yellow bars) versus mice that received vehicle control (blue bars). Expression levels of DUSP6 were normalized to expression levels of a housekeeping gene, GAPDH
Preclinical Trial of Gemcitabine Followed by Trametinib for Occult Metastatic PDAC
We next designed a preclinical adjuvant trial utilizing patient-derived PDAC cells in our splenic injection model. Currently, most patients with PDAC receive adjuvant gemcitabine following surgical resection.23 We chose a study design of sequential therapy with gemcitabine followed by trametinib as in vivo studies demonstrated that simultaneous therapy was not superior to trametinib alone (data not shown). Given that both agents target proliferating cells, these results were not surprising and were consistent with results in human clinical trials.24,25 Mice underwent splenic injection of patient-derived PDAC cells (Tumors 608, 366) and were then randomized to receive placebo (vehicle control), gemcitabine (100 mg/kg, IP, q3 days) for 7 days followed by placebo, or gemcitabine for 7 days followed by trametinib (0.3 mg/kg, oral, daily). A total of 7 days of gemcitabine was chosen to allow modest response to gemcitabine and initiation of trametinib during a period of occult-only disease to model an adjuvant setting in humans in whom there is occult-only disease.
The results of the preclinical trials are depicted in Fig. 4. Adjuvant gemcitabine followed sequentially by daily trametinib significantly prolonged TTP for Tumor 608 (Fig. 4a; 39 vs. 30 days, p = 0.0384) and for Tumor 366 (Fig. 4b; >132 vs. 53 days, p = 0.00013). The growth inhibition of occult liver metastases by sequential gemcitabine-trametinib translated to a significant increase in PFS in Tumor 608 (Fig. 4c) and Tumor 366 (Fig. 4d).
FIG. 4.
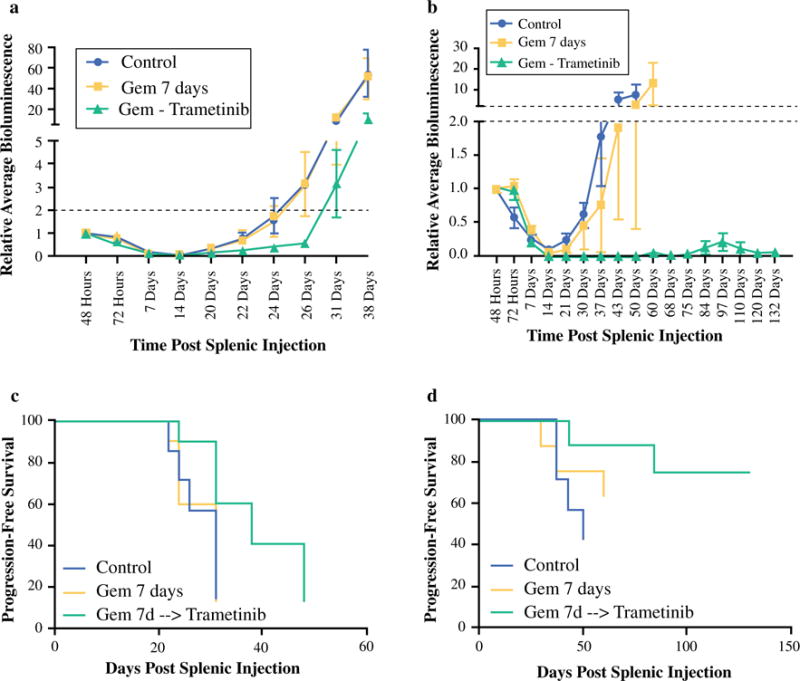
Preclinical trial of gemcitabine followed by trametinib for occult metastatic PDAC. Adjuvant gemcitabine followed sequentially by daily trametinib therapy resulted in significantly increased time to proliferative outgrowth and decreased metastatic tumor burden at conclusion of the trial in the livers of mice harboring Tumor 608 (a, p = 0.0262) and Tumor 366 (b, p = 0.0007) compared with vehicle control or gemcitabine alone. This adjuvant regimen resulted in increased 35-day PFS (p = 0.0108) in Tumor 608 (c) and improved median PFS (p = 0.0367) and 100-day PFS (p = 0.007) in tumor 366 (d)
DISCUSSION
Patients diagnosed with PDAC have a prognosis that is among the worst of any solid organ malignancy and current therapy unfortunately leads to meager increases in survival after surgery.1,2,26 The lack of significant improvement in patient survival following complete resection of localized PDAC can be attributed to the limited efficacy of current adjuvant therapies. This may be due in part to a lack of efficient, reproducible preclinical models of metastatic PDAC that are necessary to foster therapeutic development.27 We have developed a reproducible patient-derived xenograft model that recapitulates many of the steps of metastatic PDAC to the liver and this model has been extensively internally validated (data not shown). The patient-derived cell lines used are from untreated patients (e.g., drug-naïve) and are sensitive to gemcitabine in vitro and in vivo.
This preclinical model has limitations in that nude mice lack T cells, and thus, any contribution of these cells to tumor growth and response to MEK inhibition cannot be assessed. However, nude mice do have an intact innate immunity (fibroblasts, macrophages, natural killer cells). Another limitation of this model, like any preclinical model, is the difficulty in translating drug dosing and scheduling from humans to mice. In these studies, we used previously published drug doses and schedules.
The RAS-RAF-MEK-ERK signaling cascade has been shown to be integral in the development and proliferation of cancers harboring activating RAF or RAS mutations, such as PDAC.11,28 Inhibiting this growth and differentiation pathway via BRAF inhibitors has significantly improved the outcomes of patients with melanoma, and clinical trials have demonstrated the efficacy of combination therapy with BRAF inhibitor plus MEK inhibitor.29,30 Recently, a phase II trial of concurrent gemcitabine plus trametinib in patients with previously untreated, radio-graphically detectable (non-occult) metastatic PDAC did not translate into any significant improvement in survival.31 Our preclinical model of advanced PDAC (unresectable primary tumor and grossly visible metastatic disease) using patient-derived tumors corroborated the findings of this phase II trial, demonstrating that concurrent trametinib plus gemcitabine is ineffective in the setting of advanced PDAC (data not shown). In contrast, the studies described here demonstrate that micrometastatic PDAC cells in the liver are susceptible to RAS pathway signaling inhibition. Trametinib treatment significantly delayed time to metastatic cell outgrowth and this led to significant increase in survival. Sequential, rather than simultaneous, treatment with gemcitabine and trametinib may prove superior in patients because of the competing mechanisms of action of these drugs—gemcitabine is a nucleoside-analog that targets dividing cancer cells and similarly trametinib has been shown to induce cell-cycle arrest, thus inhibiting cell division. The archived tumor tissues generated from this work will allow for future investigation of the mechanism of resistance to MEK inhibition with trametinib (i.e., trametinib-treated tumors in Figs. 2 and 4 that eventually grew out). This could lead to novel combination therapies employing MEK inhibition for PDAC.
A randomized, phase II, clinical trial is being planned to evaluate the efficacy and tolerability of adjuvant sequential gemcitabine followed by trametinib for patients with resected PDAC (Fig. 5). Following surgical resection of pathologically confirmed PDAC patients will be randomized to adjuvant gemcitabine followed by placebo versus gemcitabine followed by trametinib. Patients will be monitored for disease progression with CT every 3 months. The primary endpoint of the study will be DFS, and the secondary endpoint will be OS. Correlative studies on pancreatic resection specimens will include KRAS status and staining for ERK and phosphorylated-ERK.
FIG. 5.
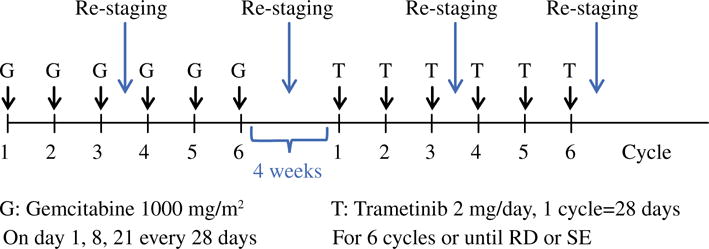
Planned clinical trial schema. A randomized, phase II, clinical trial is being planned to evaluate the efficacy and tolerability of adjuvant sequential gemcitabine followed by trametinib for patients with resected PDAC. RD recurrent disease, SE side effects
CONCLUSIONS
In a preclinical model we have shown that the MEK inhibitor trametinib is effective at increasing TTP and survival of mice harboring occult patient-derived PDAC in the liver. Furthermore, we demonstrated that sequential treatment with adjuvant gemcitabine followed by trametinib is superior to gemcitabine alone in a preclinical trial of PDAC metastatic to the liver. These promising preclinical results support the conduct of a clinical trial (currently in development) of a MEK inhibitor following standard-of-care gemcitabine for patients with resected PDAC.
Acknowledgments
Financial Support: NIH T32 CA163177 (TEN, JML), Virginia M. Kincaid Foundation Grant (TWB). The data described were gathered on Caliper IVIS Spectrum funded by the National Institutes of Health Grant (1S10RR025694-01).
Footnotes
DISCLOSURE The authors disclose no potential conflicts of interest.
References
- 1.Herman JM, Swartz MJ, Hsu CC, et al. Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. J Clin Oncol. 2008;26(21):3503–10. doi: 10.1200/JCO.2007.15.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–10. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 3.Paez D, Labonte MJ, Bohanes P, et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res. 2012;18(3):645–53. doi: 10.1158/1078-0432.CCR-11-2186. [DOI] [PubMed] [Google Scholar]
- 4.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18(1):1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 5.Shibata D, Capella G, Perucho M. Mutational activation of the c-K-ras gene in human pancreatic carcinoma. Baillieres Clin Gastroenterol. 1990;4(1):151–69. doi: 10.1016/0950-3528(90)90044-h. [DOI] [PubMed] [Google Scholar]
- 6.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16(16):7773–82. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Philip PA, Mooney M, Jaffe D, et al. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009;27(33):5660–9. doi: 10.1200/JCO.2009.21.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30(32):3477–88. doi: 10.1038/onc.2011.160. [DOI] [PubMed] [Google Scholar]
- 10.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochimica et biophysica acta. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 11.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/ MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et Biophysica Acta. 2007;1773(8):1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer R, Sers C. RAS oncogene-mediated deregulation of the transcriptome: from molecular signature to function. Adv Enzym Regul. 2011;51(1):126–36. doi: 10.1016/j.advenzreg.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Walters DM, Lindberg JM, Adair SJ, et al. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013;15(2):143–55. doi: 10.1593/neo.121712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindberg JM, Newhook TE, Adair SJ, et al. Co-treatment with panitumumab and trastuzumab augments response to the MEK inhibitor trametinib in a patient-derived xenograft model of pancreatic cancer. Neoplasia. 2014;16(7):562–71. doi: 10.1016/j.neo.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi T, Yoshida T, Kurachi R, et al. Identification of JTP-70902, a p15(INK4b)-inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci. 2007;98(11):1809–16. doi: 10.1111/j.1349-7006.2007.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stokes JB, Adair SJ, Slack-Davis JK, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–45. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walters DM, Stokes JB, Adair SJ, et al. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PLoS One. 2013;8(10):e77065. doi: 10.1371/journal.pone.0077065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajendran S, Salwa S, Gao X, et al. Murine bioluminescent hepatic tumour model. J Vis Exp. 2010;(41) doi: 10.3791/1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edinger M, Cao YA, Hornig YS, et al. Advancing animal models of neoplasia through in vivo bioluminescence imaging. Eur J Cancer. 2002;38(16):2128–36. doi: 10.1016/s0959-8049(02)00410-0. [DOI] [PubMed] [Google Scholar]
- 22.Jing J, Greshock J, Holbrook JD, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11(3):720–9. doi: 10.1158/1535-7163.MCT-11-0505. [DOI] [PubMed] [Google Scholar]
- 23.Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA. 2013;310(14):1473–81. doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 24.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 25.Bodoky G, Timcheva C, Spigel DR, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- 26.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 27.Little EC, Wang C, Watson PM, Watson DK, Cole DJ, Camp ER. Novel immunocompetent murine models representing advanced local and metastatic pancreatic cancer. J Surg Res. 2012;176(2):359–66. doi: 10.1016/j.jss.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 29.Neuzillet C, Hammel P, Tijeras-Raballand A, Couvelard A, Raymond E. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32(1–2):147–62. doi: 10.1007/s10555-012-9396-2. [DOI] [PubMed] [Google Scholar]
- 30.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Infante JR, Somer BG, Park JO, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]