

Abstract
A direct correlation between brain iron and Alzheimer’s disease (AD) raises questions regarding the transport of non-transferrin-bound iron (NTBI), a toxic but less researched pool of circulating iron that is likely to increase due to pathological and/or iatrogenic systemic iron overload. Here, we compared the distribution of radiolabeled-NTBI (59Fe-NTBI) and transferrin-bound iron (59Fe-Tf) in mouse models of iron overload in the absence or presence of inflammation. Following a short pulse, most of the 59Fe-NTBI was taken up by the liver, followed by the kidney, pancreas, and heart. Notably, a strong signal of 59Fe-NTBI was detected in the brain ventricular system after 2 h, and the brain parenchyma after 24 h. 59Fe-Tf accumulated mainly in the femur and spleen, and was transported to the brain at a much slower rate than 59Fe-NTBI. In the kidney, 59Fe-NTBI was detected in the cortex after 2 h, and outer medulla after 24 hours. Most of the 59Fe-NTBI and 59Fe-Tf from the kidney was reabsorbed; negligible amount was excreted in the urine. Acute inflammation increased the uptake of 59Fe-NTBI by the kidney and brain from 2–24 hours. Chronic inflammation, on the other hand, resulted in sequestration of iron in the liver and kidney, reducing its transport to the brain. These observations provide direct evidence for the transport of NTBI to the brain, and reveal a complex interplay between inflammation and brain iron homeostasis. Further studies are necessary to determine whether transient increase in NTBI due to systemic iron overload is a risk factor for AD.
Keywords: Alzheimer’s disease, brain, kidney, inflammation, non-transferrin-bound iron
INTRODUCTION
Iron is an important parameter for proper brain functionality, to the extent that excess iron is linked to neurodegenerative conditions such as Alzheimer’s disease (AD) [1–3], and iron deficiency is implicated in the pathogenesis of restless-leg syndrome and other brain disorders [4, 5]. The correlation between elevated levels of brain iron and AD [6] has led to the use of iron chelators as potential therapeutic agents [7, 8]. The mechanism underlying this change, however, remains controversial. Since the brain is separated from systemic circulation by the blood-brain (BBB) and blood-cerebrospinal fluid (CSF) barriers [9], it has been hypothesized that dyshomeostasis of iron metabolism within the brain is responsible for the increase in brain iron levels. However, limited reports suggest a small but detectable increase in brain iron by systemic iron overload [10], leaving the matter unsettled. Even minor changes in brain iron are likely to have an adverse effect because unlike serum transferrin (Tf), which is only 30–40% saturated with iron, CSF Tf is 100% saturated and has a limited capacity for buffering excess iron [11]. Moreover, iron is likely to accumulate in the brain over time [12], underscoring the relevance of systemic iron overload to AD pathogenesis.
In the blood, the majority of iron circulates as Tf-bound iron (TBI), and is taken up by systemic organs and the brain through the classic Tf/Tf-receptor pathway. A limited amount of iron is associated with citrate and other compounds, and comprises the pool of non-Tf-bound iron (NTBI). The mechanism by which TBI is transported across the blood-brain and blood-CSF barriers has been well characterized [11]. Similar information for NTBI, however, is lacking. Unlike TBI, NTBI requires reduction to the ferrous (Fe2+) form by membrane bound ferrireductases for transport across biological membranes through divalent metal transporters [13]. Although in vitro studies indicate efficient uptake and transport of NTBI by brain micro-vessel endothelial cells [14, 15], it is unclear whether a similar transport mechanism occurs in vivo. Paucity of information on the physiology and pathobiology of NTBI is mainly due to the difficulty in tracking this species experimentally. This question is of particular significance in the pathobiology of AD and other neurodegenerative conditions associated with brain iron dyshomeostasis because unlike TBI that is relatively stable, NTBI has the potential for generating reactive hydroxyl radicals through Fenton chemistry [16]. It is therefore necessary to maintain the concentration of circulating and brain NTBI within safe limits.
It is generally presumed that there is little, if any NTBI in the blood because of the high affinity of Tf for iron. Any NTBI, therefore, is believed to result from a spill-over of Tf saturation. Evidence from several studies, however, suggests otherwise. Such evidence includes: 1) In healthy adults, exogenous iron administered by the oral or parenteral route causes a rapid increase in NTBI despite high Tf iron-binding capacity [17–20]; 2) Conditions unrelated to systemic iron overload such as alcoholic liver disease, diabetes mellitus, and hematological malignancies increase NTBI despite unsaturated Tf [21, 22]; 3) Mice lacking ceruloplasmin have decreased plasma levels of both TBI and NTBI, suggesting that these forms exist in a chemical equilibrium [21, 23]; 4) The rate of iron conjugation with Tf (minutes) is slower than the rate of NTBI uptake by the liver (<3 seconds) [24]; and 5) concentrations of NTBI rise rapidly after iron loading followed by sudden disappearance within seconds, making it unlikely that Tf-saturation is a pre-requisite for the appearance of NTBI [25]. Thus, NTBI is present in the blood under normal conditions and its pool is likely to increase with iron overload. NTBI also contributes substantially to the total pool of iron in the brain [26–29] though it is unclear whether it is generated locally or transported from the blood across the blood-brain and blood-CSF barriers.
To answer this question, we compared the distribution of radiolabeled NTBI (59Fe-NTBI) and Tf (59Fe-Tf) in the brain and systemic organs [30], and the influence of iron overloading in the absence or presence of acute or chronic inflammation on these parameters in a mouse model. We demonstrate that despite adequate sequestration by the liver and kidney, a significant amount of NTBI is transported to the brain parenchyma, and acute and chronic inflammation influences its distribution differentially. These observations have significant implications for AD and other neurodegenerative conditions associated with brain iron imbalance and neuroinflammation.
METHODS
Animal care
Wild-type FVB/NJ mice were obtained from Jackson Laboratories and housed in AAALAC accredited facilities at Case Western Reserve University using standard operating procedures. All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC). Routine veterinary care was provided by technicians under the direction of an attending veterinarian.
Radiolabeling and tissue processing
For all experiments, female mice ranging in age from 1.5–2 months on regular laboratory chow were used. In a typical experiment, experimental mice were injected with equal counts of 59FeCl3 (Perkin-Elmer) by the IP route prior to, or 30 min following an injection of 70 μg/22g body weight of FAC (Sigma) dissolved in citrate buffer [30, 31]. To induce acute inflammation, lipopolysaccharide (LPS, 2 mg/kg, E. coli O55:B6) was injected IP followed by FAC and 59FeCl3 as described diagrammatically in the experimental design for each experiment [32]. To induce chronic inflammation, LPS injection was repeated daily for 4 consecutive days [33]. Control animals in each group received the same volume of vehicle.
At the indicated times (2 h, 24 h, and 4d), mice were euthanized as per the approved protocol. Blood was collected by cardiac puncture, and the remaining blood was flushed out by transcardial perfusion of chilled PBS. Major organs were harvested, and uptake of 59Fe in each organ and the remaining carcass was quantified in a γ-counter. Total counts injected and recovered were similar for each set of experiments. Each organ was weighed and fixed in 10% neutral buffered formalin for 24 h. Fixed organs were either embedded in paraffin and processed for immunostaining and Perls’ reaction, or sectioned by a vibratome (Leica VT1000s), dried, and subjected to autoradiography. Control and experimental samples were processed in parallel and exposed to the same X-ray film to allow direct comparison. Scanned autoradiographs were converted to a color gradient image with Adobe Photoshop CC.
SDS-PAGE and western blotting [34], native gel electrophoresis [35], Perls’ staining [36], immunostaining [37], and quantitative RT-PCR were performed essentially as described [38, 39].
RESULTS
Uptake of 59Fe-NTBI and 59Fe-Tf is organ specific, and 59Fe-NTBI is transported faster than 59Fe-Tf to the brain and kidney
To evaluate the kinetics of uptake and distribution of TBI and NTBI in various organs, age and sex-matched mice were injected with equal counts of 59FeCl3 to radiolabel 59Fe-Tf (−FAC) and 59Fe-NTBI (+FAC) [30], and euthanized after 2 and 24 h. The efficacy of radiolabeling was confirmed by fractionating plasma on a native gel followed by autoradiography (Fig. 1A). Uptake of 59Fe by various organs was quantified in a γ-counter (Fig. 1B). The kidney, brain, heart, liver, and spleen were fixed, and thick sections were vacuum-dried and subjected to autoradiography (Fig. 1C–E). The total 59Fe counts recovered from mice in each group were similar (±5%), allowing a direct comparison between the two groups.
Fig. 1.
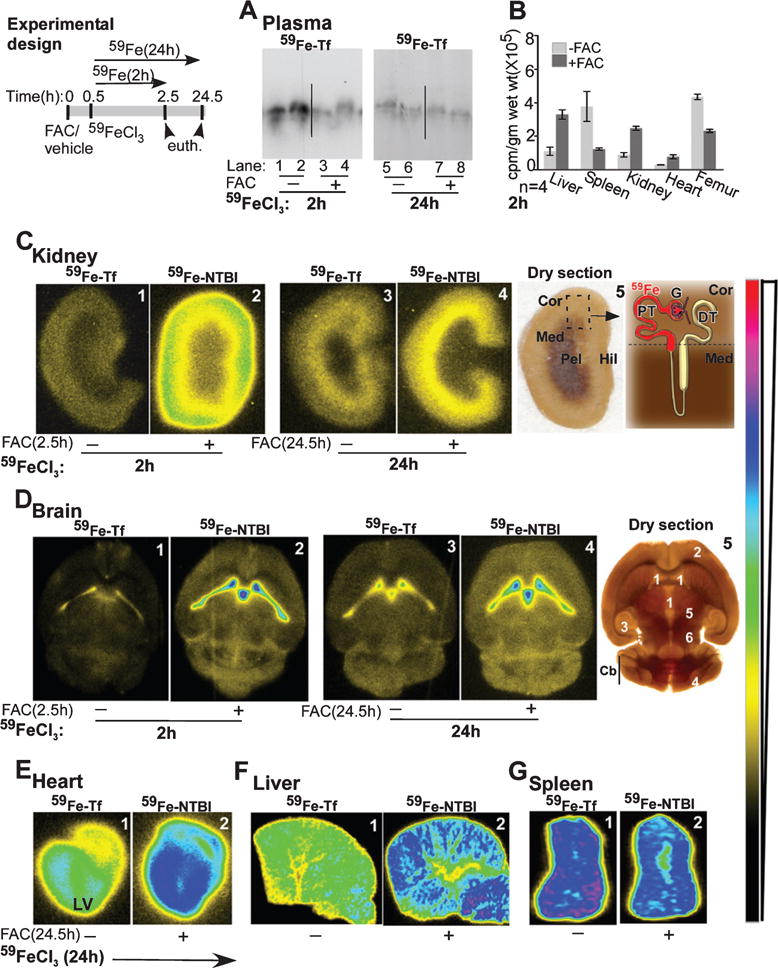
Uptake of 59Fe-NTBI and 59Fe-Tf is organ specific, and 59Fe-NTBI is transported faster than 59Fe-Tf to the brain and the kidney. Experimental design: Time-frame of different treatments for the 2 and 24 h chase experiments in A–G. A) Saturation of plasma Tf with unlabeled FAC results in the distribution of 80% of injected 59Fe to the NTBI pool after 2 h (lanes 1–4). Plasma 59Fe-Tf achieves equilibrium with pools of unlabeled iron in organs by 24 h (lanes 5–8). B) 59Fe-Tf is mainly taken up by the spleen and femur, while 59Fe-NTBI distributes mainly to the liver, kidney, and heart. C) The signal for 59Fe-NTBI in the kidney cortex is 4-fold higher than 59Fe-Tf after 2 h (panels 1 & 2). This difference reduces significantly after 24 h (panels 3 & 4). Both 59Fe-Tf and 59Fe-NTBI show a change in localization from the cortex to the outer medulla after 24 h (panels 3 & 4, panel 5). Panel 5: Dry section used for autoradiography is marked and represented diagrammatically to highlight major sites of iron absorption in the kidney. Cor, cortex; Med, medulla; Pel, pelvis, Hil, hilum; G, glomerulus; PT, proximal tubule; DT, distal tubule. D) The signal from 59Fe-NTBI is significantly higher than 59Fe-Tf in the ventricles and brain parenchyma after 2 h (panels 1 & 2). A significant amount of 59Fe is transported to the brain parenchyma after 24 h, and the signal is higher in 59Fe-NTBI relative to 59Fe-Tf samples (panels 3 & 4, panel 5). Panel 5: Tissue section used for autoradiography is marked to show major sites of iron accumulation in the brain. 1, ventricles; 2, cortex; 3, hippocampus; 4, cerebellar cortex; 5, thalamus; 6, hypothalamus; Cb, cerebellum. E) Accumulation of 59Fe-NTBI in the left ventricular muscle is significantly more than 59Fe-Tf (panels 1 & 2). F) The liver accumulates significantly more 59Fe-NTBI relative to 59Fe-Tf (panels 1 & 2). G) The spleen, on the other hand, accumulates more 59Fe-Tf in comparison to 59Fe-NTBI (panels 1 & 2).
Following a pulse of 2 h, plasma 59Fe-Tf was reduced by ∼80% in +FAC samples relative to −FAC controls, confirming successful radiolabeling of NTBI and TBI, respectively (Fig. 1A, lanes 1 & 2 versus 3 & 4). Levels of 59Fe-Tf were similar in both sets of mice after 24 h, indicating equilibration of iron pools in different organs (Fig. 1A, lanes 5–8). Quantitation of 59Fe counts in different organs revealed preferential uptake of 59Fe-NTBI by the liver, kidney, heart, and 59Fe-Tf by the spleen and femur (Fig. 1B) [30].
Examination of kidney sections revealed significantly more 59Fe-NTBI relative to 59Fe-Tf after 2 and 24 h, and a change in the localization of both forms from the cortex at 2 h to the outer medulla after 24 h (Fig. 1C, panels 1–4). Notably, 59Fe-Tf increased, while 59Fe-NTBI decreased from 2 to 24 h (Fig. 1C, panel 1 versus 3 & 2 versus 4), indicating that uptake and efflux of 59Fe-NTBI in the kidney is faster than 59Fe-Tf, and the total amount of 59Fe-NTBI passing through is significantly higher than 59Fe-Tf. A dry section of the kidney used for fluorography indicates the anatomical regions of 59Fe accumulation (Fig. 1C, panel 5).
As in the kidney, significantly more 59Fe-NTBI accumulated in the brain after 2 h relative to 59Fe-Tf, especially in the lateral ventricles (Fig. 1D, panels 1 & 2). Unlike the kidney, both 59Fe-Tf and 59Fe-NTBI continued to accumulate after 24 h, and a significant amount was detected in the brain parenchyma in addition to the ventricles (Fig. 1D, panels 1 versus 3 & 2 versus 4). A section of the brain used for fluorography indicates major areas that accumulate 59Fe (Fig. 1D, panel 5).
Sections of the heart displayed significantly more 59Fe-NTBI relative to 59Fe-Tf, most of which accumulated in the left ventricular muscle after 24 h (Fig. 1E). The liver revealed a strong signal for 59Fe-NTBI, and the spleen for 59Fe-Tf as expected from the 59Fe counts in Fig. 1B (Fig. 1F, G). Sections of the heart, liver, and spleen (Fig. 1E–G) were exposed to X-ray film for variable times, and cannot be compared with each other or with the kidney and brain sections above.
These observations demonstrate that 1) transport of 59Fe-NTBI to the kidney is faster than 59Fe-Tf, 2) 59Fe from 59Fe-NTBI is transported across the blood-brain and blood-CSF barriers to the brain parenchyma faster than 59Fe from 59Fe-Tf, and 3) a significant amount of 59Fe-NTBI accumulates in the heart.
Endothelial and epithelial cells of the brain and kidney accumulate iron and upregulate ferritin during systemic iron overload
To evaluate whether the 59Fe signal in kidney cortex and outer medulla represents transient passage or accumulation of iron in these regions, sections of kidney from control (−FAC) and FAC treated mice were stained with Perls’ reagent (Fig. 2A). Surprisingly, prominent Perls’ positive deposits were visible in epithelial cells lining the proximal tubules and Bowman’s capsule of mice treated with FAC (Fig. 2A, panels 2 & 4). Minimal reactivity was detected in control sections (Fig. 2A, panels 1 & 3).
Fig. 2.
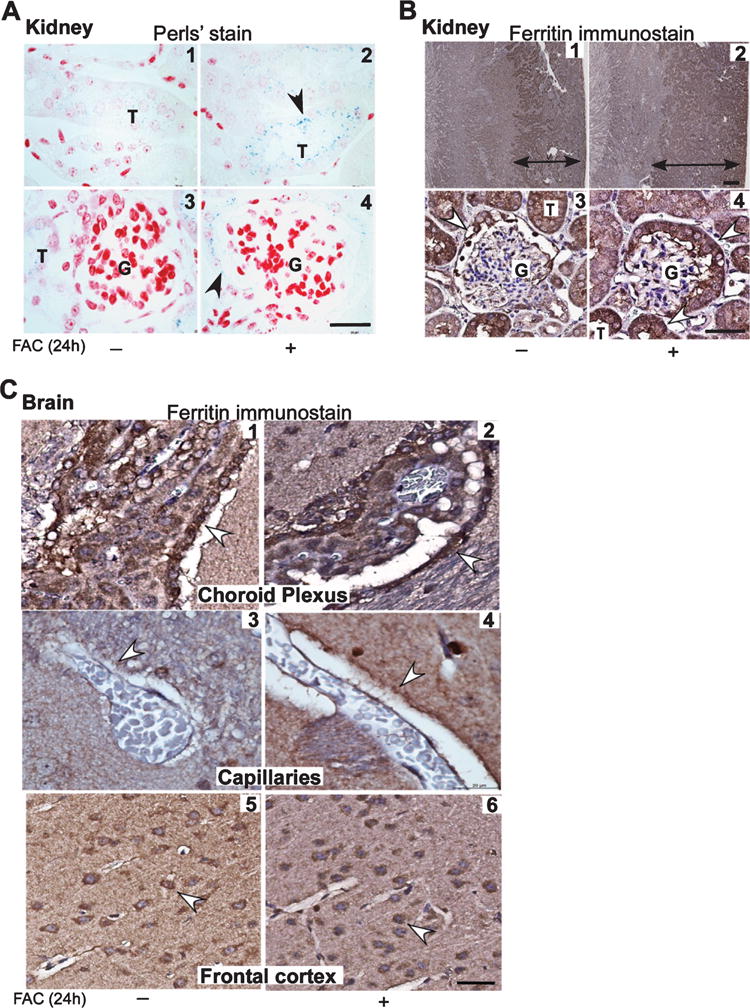
Endothelial and epithelial cells of the brain and kidney upregulate ferritin in iron overloaded mice. A) Perls’ staining of kidney sections shows a blue reaction product of iron in epithelial cells lining the proximal tubules and Bowman’s capsule in FAC treated samples (panels 2 & 4). A weak reaction is also noted in control sections (panels 1 & 3). B) Immunostaining for ferritin followed by enhancement of the reaction with diaminobenzidine shows a much larger area of positivity in the cortex and outer and inner medulla in FAC treated samples relative to controls where the reactivity is limited to the cortex (panels 1 & 2). Glomeruli show a strong reaction for ferritin in cells lining the Bowman’s capsule of FAC treated samples relative to controls (panels 3 & 4). C) Reactivity for ferritin is stronger in brain sections from mice exposed to FAC relative to controls in epithelial cells of the choroid plexus (panels 1 & 2), endothelial cells lining blood capillaries (panels 3 & 4), and neuronal cell bodies in the frontal cortex (panels 5 & 6). Scale bar, 10 μm.
Immunostaining for ferritin revealed a much larger area of reactivity in the cortex of FAC treated samples relative to controls (Fig. 2B, panels 1 & 2). Higher magnification revealed that ferritin was localized to epithelial cells lining the Bowman’s capsule and proximal tubules (Fig. 2B, panels 3 & 4).
Likewise, brain sections from FAC treated mice showed a strong reaction for ferritin in epithelial cells of the choroid plexus and capillary endothelial cells (Fig. 2C, panels 1–4). Neuronal cells in the frontal cortex also reacted strongly for ferritin (Fig. 2C, panels 5 & 6), indicating that despite intact blood-brain and blood-CSF barriers, iron overloading resulted in the transport of iron to the brain parenchyma and uptake by neuronal cells.
Combined with the 59Fe results in Fig. 1, these observations suggest that transient iron overload results in the accumulation of iron and consequent upregulation of ferritin in epithelial lining kidney proximal tubules and Bowman’s capsule. Likewise, brain capillary endothelial and choroid plexus epithelial cells upregulate ferritin, indicating a protective and regulatory response to excess iron.
Inflammation influences the uptake of 59Fe-NTBI by the brain and kidney
Systemic inflammation is known to sequester iron in macrophages, creating functional iron deficiency and anemia of chronic disease [40]. The effect of neuroinflammation that accompanies AD [41] on brain iron homeostasis, however, is less clear. To evaluate whether inflammation alters the uptake of NTBI by the brain, mice were injected with a single or multiple doses of LPS, an agent widely used to induce acute and chronic systemic and neuroinflammation [42, 43]. This was followed by vehicle or FAC after 2 h, and 30 min later, 59FeCl3 to radiolabel TBI and NTBI respectively (Fig. 3, experimental design).
Fig. 3.
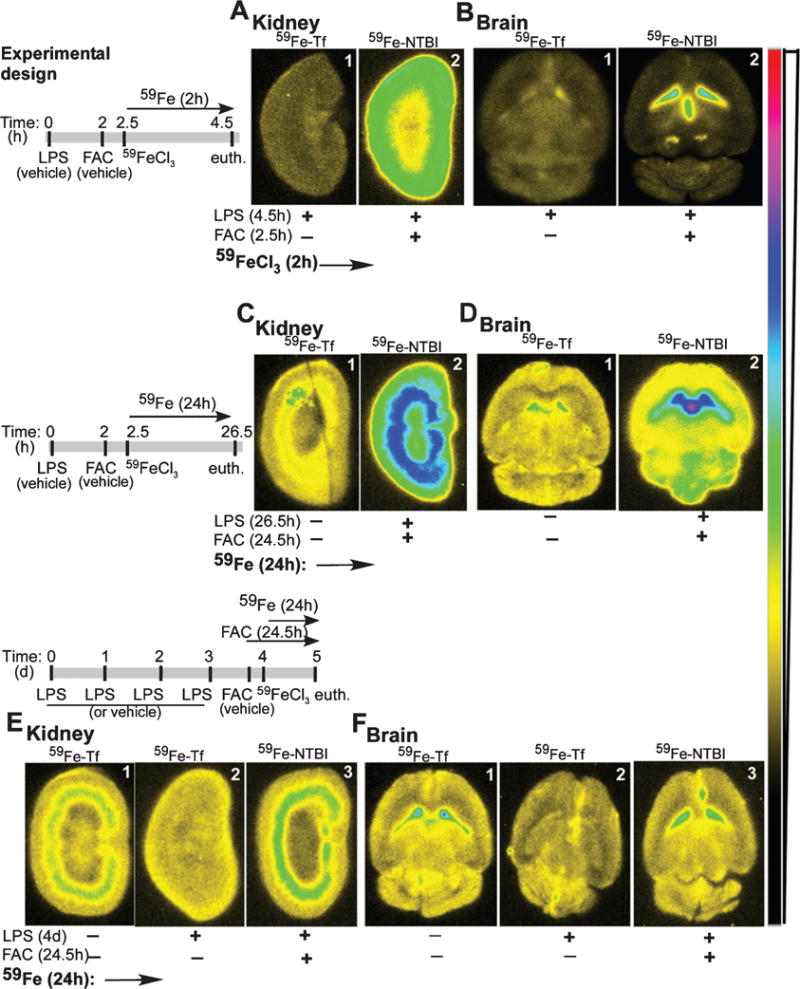
Inflammation alters the distribution of NTBI in the brain and kidney. Experimental design: Time-frame of experimental manipulations in A–F. A) Acute inflammation shows increased transport of 59Fe-NTBI to the kidney cortex relative to 59Fe-Tf after a chase of 2 h (panels 1 & 2), and when compared with non-inflamed samples in Fig. 1C, panels 1 & 2. B) Likewise, significantly more 59Fe-NTBI accumulates in the ventricles in the presence of acute inflammation relative to 59Fe-Tf (panels 1 & 2). However, transport of 59Fe from 59Fe-NTBI to the brain parenchyma is reduced in comparison with 59Fe-Tf (panels 1 & 2) and non-inflamed samples in Fig. 1D, panels 1 & 2. C) A longer chase of 24 h in the absence or presence of acute inflammation shows significantly more 59Fe-NTBI relative to 59Fe-Tf, and their accumulation in the outer and inner medulla (panels 1 & 2). D) Brain sections show increased accumulation of 59Fe-NTBI in the ventricles, and significantly more transport of 59Fe to the parenchyma relative to 59Fe-Tf in non-inflamed samples (panels 1 & 2). E) Chronic inflammation reduces the transport of 59Fe-Tf to the kidney, but has minimal effect on the uptake of 59Fe-NTBI (panels 1–3). Notably, uptake of 59Fe-NTBI remains higher than 59Fe-Tf during chronic inflammation (panels 2 & 3). F) The transport of 59Fe-Tf and 59Fe-NTBI to the ventricles and brain parenchyma is reduced during chronic inflammation, though uptake of 59Fe-NTBI is significantly more than 59Fe-Tf (panels 1–3).
Following a pulse of 4.5 h, accumulation of 59Fe-NTBI in cortical region of the kidney was significantly more than 59Fe-Tf (Fig. 3A, panels 1 & 2). The brain showed significantly more 59Fe-NTBI in the ventricles relative to 59Fe-Tf. However, the transport of 59Fe-NTBI to the brain parenchyma was significantly less than 59Fe-Tf (Fig. 3B, panels 1 & 2).
After a longer chase of 26.5 h, LPS treated samples continued to show significantly more 59Fe-NTBI relative to 59Fe-Tf in the kidney cortex (Fig. 3C, panels 1 & 2). Brain sections showed a strong signal for 59Fe-NTBI in the ventricles, and in contrast to the 4.5 h time-point, significantly more transport to the parenchyma relative to 59Fe-Tf (Fig. 3D, panels 1 & 2). Chronic inflammation, on the other hand, caused a distinct decrease in 59Fe-Tf in the kidney and brain relative to the control (Fig. 3E, F, panels 1 & 2). Uptake of 59Fe-NTBI by the kidney and brain, however, was similar to controls (Fig. 3E. F, panel 1 versus 3).
Chronic inflammation increases Perls’ positive iron in the kidney and liver
Reduced uptake of 59Fe-NTBI and 59Fe-Tf by the brain and kidney following chronic inflammation indicated sequestration in the liver, thus reducing circulating levels of radiolabeled iron. To confirm this, liver and kidney sections from mice injected with LPS were stained with the Perls’ reagent [33, 44]. Surprisingly, Perls’ positive iron deposits were detected in epithelial cells lining the kidney proximal tubules and Bowman’s capsule (Fig. 4A, panels 1 & 2). Similar reactivity was also detected in hepatocytes and Kupffer cells (Fig. 4A, panels 3 & 4). Western blot analysis of liver homogenates revealed increased levels of ferritin in LPS treated samples, a significant amount of which fractionated in the detergent insoluble fraction (Fig. 4B, lanes 1–6, Fig. 4C), indicating a change in the biophysical characteristics of ferritin.
Fig. 4.
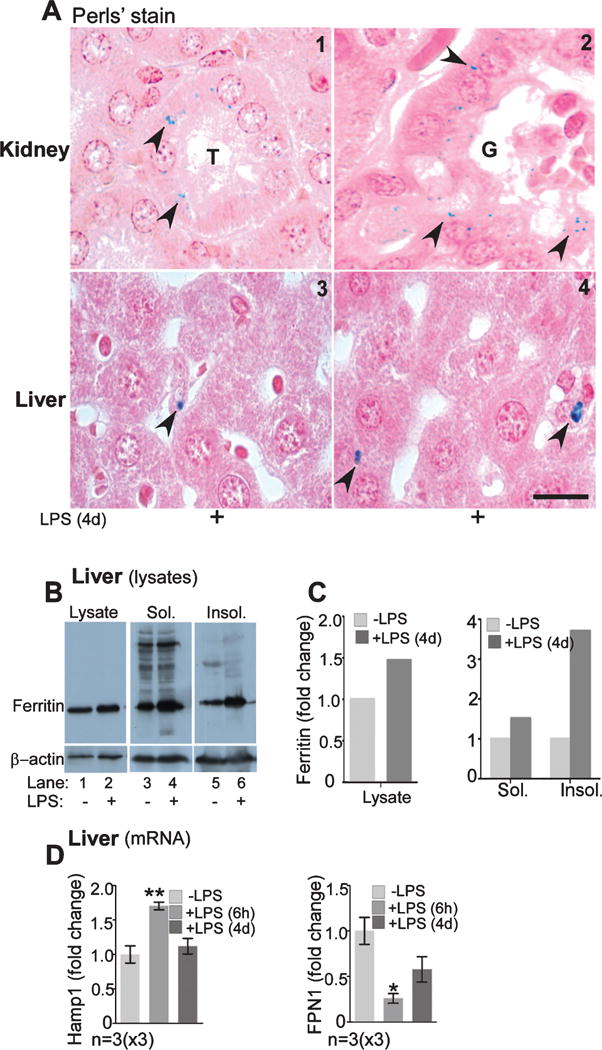
Chronic inflammation increases sequestration of iron in the liver and kidney. A) Perls’ reaction is positive in the epithelial cells lining the kidney proximal tubules and Bowman’s capsule (panels 1 & 2). Perls’ positive granules are also evident in the hepatocytes and Kupffer cells (panels 3 & 4). Scale bar, 10 μm. B) Western blot of liver homogenates shows upregulation of ferritin by LPS (lanes 1 & 2), a significant amount of which partitions in the detergent-insoluble fraction (lanes 3 & 4). C) Quantification shows 1.3-fold increase in ferritin in the total lysate, and 1.2 and 3-fold increase in ferritin in the soluble and insoluble fractions respectively by LPS. D) Quantitative analysis of mRNA levels shows a significant increase in Hamp1 (hepcidin) mRNA after 6 h, and a fall to near control levels after 4 days of LPS (p < 0.01). mRNA levels for Fpn, on the other hand, remain low after 6 h and 4 days of LPS treatment (p < 0.05).
In response to inflammation, the liver releases Hepcidin, a peptide hormone that downregulates the iron export protein ferroportin (Fpn), thereby reducing uptake from the intestine and release from macrophages [45]. To confirm efficient induction of both acute and chronic inflammation by LPS, quantitative RT-PCR was used to determine levels of hepcidin (Hamp1) and Fpn that show upregulation and downregulation, respectively, in response to inflammatory stimuli. As expected, there was significant upregulation of Hamp1 and down-regulation of Fpn 6 h after the first injection of LPS (Fig. 4D). Levels of Hamp1 fell to control levels 24 h after the 4th injection (Fig. 4D), though Fpn remained below control levels under both experimental conditions, partially explaining Perls’ positive iron deposits in the inflamed kidney and liver.
DISCUSSION
Our data demonstrate that a significant amount of NTBI is transported to the brain of iron-overloaded, disease-free mice with intact blood-brain and blood-CSF barriers. The uptake of 59Fe-NTBI by the brain parenchyma continued for up to 24 h even though a significant amount was sequestered in the liver and transported to the kidney within 2 h of administration. Increased reactivity of endothelial and epithelial cells of the blood-brain and blood-CSF barriers for ferritin 24 h after iron administration indicated accumulation of access iron in these cells, a phenotype expected to downregulate iron uptake proteins and reduce further uptake of iron [46]. Acute inflammation increased transport of 59Fe-NTBI to the brain, perhaps due to disruption of the blood-brain and blood-CSF barriers. Chronic inflammation, on the other hand, resulted in the accumulation of iron in the liver and kidney, thereby reducing circulating levels of NTBI and transport to the brain.
Faster accumulation of NTBI in the ventricles and brain parenchyma relative to TBI despite similar 59Fe counts injected and recovered could be explained by the faster kinetics of NTBI, which is cleared from the circulation within seconds relative to the conjugation of iron with Tf, which takes minutes [24]. However, the continued transport and accumulation of 59Fe-NTBI after 24 h, despite systemic iron overload, is difficult to explain, and suggests the following possibilities: 1) transport of 59Fe-NTBI to the brain is not as stringent as 59Fe-Tf, and continues despite adequate brain iron and upregulation of ferritin in endothelial and epithelial cells of the blood-brain and blood-CSF barriers; 2) 59Fe-NTBI transported to the brain is taken up by astrocytes and microglia, and accumulates in ferritin; and 3) drainage of brain interstitial fluid and CSF into the venous system is slower than the kinetics of 59Fe-NTBI uptake, resulting in transient accumulation in the brain parenchyma. Accumulation of relatively less 59Fe-Tf in the brain after 2 and 24 h reflects the uptake and distribution of TBI under normal conditions, and suggests utilization by brain cells and accumulation of excess in ferritin. Alternately, the increase in 59Fe-Tf signal with time could reflect slower kinetics of export relative to import. Further studies are necessary to understand the kinetics of NTBI and TBI into, and out of the brain.
The kidneys accumulated mainly 59Fe-NTBI as expected [31]. However, unlike the brain, a significant amount of 59Fe-NTBI was transported out of the kidney within 24 h, indicating reabsorption by the blood. Minimal 59Fe was lost in the urine [47]. The transport of 59Fe-Tf to and from the kidney was less dramatic than 59Fe-NTBI, reinforcing the conclusion that the kinetics and distribution of 59Fe-NTBI differ from those of 59Fe-Tf, and are organ-specific. The accumulation of iron and upregulation of ferritin in epithelial cells lining the glomeruli and proximal tubules of iron overloaded mice, anatomical sites susceptible to iron-induced toxicity, is of concern specially in cases of chronic kidney disease where intravenous iron therapy is the norm, and is likely to damage the already compromised kidney [47]. Moreover, significant accumulation of 59Fe-NTBI in the heart suggests that in addition to the brain, the kidney and heart are also sensitive to iron-induced toxicity due to systemic iron overload.
Acute inflammation increased the transport of 59Fe-NTBI to the brain and the kidney, a surprising observation since the majority of NTBI is expected to accumulate in the liver due to upregulation of divalent metal transporters DMT1 and ZIP14 [48, 49] and downregulation of transferrin receptor and Fpn due to inflammation-induced up regulation of hepcidin [32, 50]. It is likely that altered expression of iron transporters combined with compromised integrity of the brain barriers due to inflammatory cytokines contribute to this phenotype [51, 52]. Chronic inflammation, on the other hand, reduced the transport of 59Fe-NTBI and 59Fe-Tf to the brain and kidney because the majority of circulating iron accumulated in the liver and kidney glomerular and proximal tubule epithelial cells as Perls’ positive deposits. Thus, chronic inflammation played a protective role by reducing iron deposition in the brain while increasing the susceptibility of kidney tubules to iron-induced damage. Interestingly, knock-out of Zip14 in normal mice or models of hemochromatosis decreased uptake of NTBI by the liver and increased its transport to the kidney [53–55], suggesting that excess iron that escapes the liver is diverted to the kidney. It is likely that shedding of ferritin-rich proximal tubule epithelial cells in the urine helps in ridding the body of excess iron and is therefore a protective mechanism. The aggregation of ferritin within the chronically inflamed liver, however, was surprising, and suggested a change in its biophysical characteristics as a result of inflammation. This observation has implications for neurodegenerative diseases such as AD in which neuroinflammation plays a dominant role, and could account for the increase in brain iron as noted in brains affected by a prion disease [35, 56].
In conclusion, this study demonstrates that NTBI is transported to the brain in addition to the liver, kidney, and heart. Acute inflammation increases iron uptake by the brain while chronic inflammation is relatively neuroprotective due to sequestration of iron in the liver and kidney. Further studies are necessary to evaluate the concentration of NTBI in the brain under normal and diseased conditions, and the correlation between circulating NTBI and AD.
Acknowledgments
This study was supported by grants NS077438 and NS092145 from the National Institutes of Health, USA, to NS.
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0097r1).
References
- 1.Bartzokis G, Lu PH, Mintz J. Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 2007;3:122–125. doi: 10.1016/j.jalz.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartzokis G, Sultzer D, Cummings J, Holt LE, Hance DB, Henderson VW, Mintz J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch Gen Psychiatry. 2000;57:47–53. doi: 10.1001/archpsyc.57.1.47. [DOI] [PubMed] [Google Scholar]
- 3.Ayton S, Faux NG, Bush AI, Alzheimer’s Disease Neuroimaging I Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun. 2015;6:6760. doi: 10.1038/ncomms7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen RP, Earley CJ. The role of iron in restless legs syndrome. Mov Disord. 2007;22(Suppl 18):S440–S448. doi: 10.1002/mds.21607. [DOI] [PubMed] [Google Scholar]
- 5.Youdim MB. Brain iron deficiency and excess; cognitive impairment and neurodegeneration with involvement of striatum and hippocampus. Neurotox Res. 2008;14:45–56. doi: 10.1007/BF03033574. [DOI] [PubMed] [Google Scholar]
- 6.Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J Neurochem. 2016;139(Suppl 1):179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 8.Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A, Gabbiani C, Messori L, Moneti G, Casamenti F. Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2009;17:423–440. doi: 10.3233/JAD-2009-1063. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- 10.Milward E, Acikyol B, Bassett B, Williams E, Graham R, Delima R, Trinder D, Olynyk J, Johnstone D. Brain changes in iron loading disorders. In: Linert W, Kozlowski H, editors. Metal Ions in Neurological Systems. Springer; 2012. pp. 17–29. [Google Scholar]
- 11.Bradbury MW. Transport of iron in the blood-brain-cerebrospinal fluid system. J Neurochem. 1997;69:443–454. doi: 10.1046/j.1471-4159.1997.69020443.x. [DOI] [PubMed] [Google Scholar]
- 12.Pirpamer L, Hofer E, Gesierich B, De Guio F, Freudenberger P, Seiler S, Duering M, Jouvent E, Duchesnay E, Dichgans M, Ropele S, Schmidt R. Determinants of iron accumulation in the normal aging brain. Neurobiol Aging. 2016;43:149–155. doi: 10.1016/j.neurobiolaging.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 13.McKie AT, Latunde-Dada GO, Miret S, McGregor JA, Anderson GJ, Vulpe CD, Wrigglesworth JM, Simpson RJ. Molecular evidence for the role of a ferric reductase in iron transport. Biochem Soc Trans. 2002;30:722–724. doi: 10.1042/bst0300722. [DOI] [PubMed] [Google Scholar]
- 14.McCarthy RC, Kosman DJ. Mechanistic analysis of iron accumulation by endothelial cells of the BBB. Biometals. 2012;25:665–675. doi: 10.1007/s10534-012-9538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy RC, Kosman DJ. Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front Mol Neurosci. 2015;8:31. doi: 10.3389/fnmol.2015.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop GM, Dang TN, Dringen R, Robinson SR. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox Res. 2011;19:443–451. doi: 10.1007/s12640-010-9195-x. [DOI] [PubMed] [Google Scholar]
- 17.Rooyakkers TM, Stroes ES, Kooistra MP, van Faassen EE, Hider RC, Rabelink TJ, Marx JJ. Ferric saccharate induces oxygen radical stress and endothelial dysfunction in vivo. Eur J Clin Invest. 2002;32(Suppl 1):9–16. doi: 10.1046/j.1365-2362.2002.0320s1009.x. [DOI] [PubMed] [Google Scholar]
- 18.Stefansson BV, Haraldsson B, Nilsson U. Ascorbyl free radical reflects catalytically active iron after intravenous iron saccharate injection. Free Radic Biol Med. 2008;45:1302–1307. doi: 10.1016/j.freeradbiomed.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Dresow B, Petersen D, Fischer R, Nielsen P. Non-transferrin-bound iron in plasma following administration of oral iron drugs. Biometals. 2008;21:273–276. doi: 10.1007/s10534-007-9116-5. [DOI] [PubMed] [Google Scholar]
- 20.Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med. 2013;65:1174–1194. doi: 10.1016/j.freeradbiomed.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820:403–410. doi: 10.1016/j.bbagen.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 22.Sebastiani G, Pantopoulos K. NTBI unveiled by chelatable fluorescent beads. Biochem J. 2014;463:e7–e9. doi: 10.1042/BJ20141083. [DOI] [PubMed] [Google Scholar]
- 23.Loreal O, Turlin B, Pigeon C, Moisan A, Ropert M, Morice P, Gandon Y, Jouanolle AM, Verin M, Hider RC, Yoshida K, Brissot P. Aceruloplasminemia: New clinical, pathophysiological and therapeutic insights. J Hepatol. 2002;36:851–856. doi: 10.1016/s0168-8278(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 24.Chakrabarti M, Barlas MN, McCormick SP, Lindahl LS, Lindahl PA. Kinetics of iron import into developing mouse organs determined by a pup-swapping method. J Biol Chem. 2015;290:520–528. doi: 10.1074/jbc.M114.606731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito S, Ikuta K, Kato D, Lynda A, Shibusa K, Niizeki N, Toki Y, Hatayama M, Yamamoto M, Shindo M, Iizuka N, Kohgo Y, Fujiya M. In vivo behavior of NTBI revealed by automated quantification system. Int J Hematol. 2016;104:175–181. doi: 10.1007/s12185-016-2002-6. [DOI] [PubMed] [Google Scholar]
- 26.Molina JA, Jimenez-Jimenez FJ, Aguilar MV, Meseguer I, Mateos-Vega CJ, Gonzalez-Munoz MJ, de Bustos F, Porta J, Orti-Pareja M, Zurdo M, Barrios E, Martinez-Para MC. Cerebrospinal fluid levels of transition metals in patients with Alzheimer’s disease. J Neural Transm (Vienna) 1998;105:479–488. doi: 10.1007/s007020050071. [DOI] [PubMed] [Google Scholar]
- 27.Moos T, Morgan EH. Evidence for low molecular weight, non-transferrin-bound iron in rat brain and cerebrospinal fluid. J Neurosci Res. 1998;54:486–494. doi: 10.1002/(SICI)1097-4547(19981115)54:4<486::AID-JNR6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno S, Mihara T, Miyaoka T, Inagaki T, Horiguchi J. CSF iron, ferritin and transferrin levels in restless legs syndrome. J Sleep Res. 2005;14:43–47. doi: 10.1111/j.1365-2869.2004.00403.x. [DOI] [PubMed] [Google Scholar]
- 29.Hozumi I, Hasegawa T, Honda A, Ozawa K, Hayashi Y, Hashimoto K, Yamada M, Koumura A, Sakurai T, Kimura A, Tanaka Y, Satoh M, Inuzuka T. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J Neurol Sci. 2011;303:95–99. doi: 10.1016/j.jns.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis. Proc Natl Acad Sci U S A. 1987;84:3457–3461. doi: 10.1073/pnas.84.10.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haldar S, Tripathi A, Qian J, Beserra A, Suda S, McElwee M, Turner J, Hopfer U, Singh N. Prion protein promotes kidney iron uptake via its ferrireductase activity. J Biol Chem. 2015;290:5512–5522. doi: 10.1074/jbc.M114.607507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aydemir TB, Chang SM, Guthrie GJ, Maki AB, Ryu MS, Karabiyik A, Cousins RJ. Zinc transporter ZIP14 functions in hepatic zinc, iron and glucose homeostasis during the innate immune response (endotoxemia) PLoS One. 2012;7:e48679. doi: 10.1371/journal.pone.0048679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathi AK, Singh N. Prion protein-hemin interaction upregulates hemoglobin synthesis: Implications for cerebral hemorrhage and sporadic Creutzfeldt-Jakob disease. J Alzheimers Dis. 2016;51:107–121. doi: 10.3233/JAD-151039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh A, Mohan ML, Isaac AO, Luo X, Petrak J, Vyoral D, Singh N. Prion protein modulates cellular iron uptake: A novel function with implications for prion disease pathogenesis. PLoS One. 2009;4:e4468. doi: 10.1371/journal.pone.0004468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Grigoryan MM, Vasilevko V, Sumbria RK, Paganini-Hill A, Cribbs DH, Fisher MJ. Comparative analysis of H&E and Prussian blue staining in a mouse model of cerebral microbleeds. J Histochem Cytochem. 2014;62:767–773. doi: 10.1369/0022155414546692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baksi S, Tripathi AK, Singh N. Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: Implications for visual manifestations of Parkinson’s disease. Free Radic Biol Med. 2016;97:292–306. doi: 10.1016/j.freeradbiomed.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tripathi AK, Haldar S, Qian J, Beserra A, Suda S, Singh A, Hopfer U, Chen SG, Garrick MD, Turner JR, Knutson MD, Singh N. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol Med. 2015;84:322–330. doi: 10.1016/j.freeradbiomed.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010;30:105–122. doi: 10.1146/annurev.nutr.012809.104804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, Balling R, Neumann H. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34:8546–8556. doi: 10.1523/JNEUROSCI.5002-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–510. doi: 10.1038/nri3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simpson IA, Ponnuru P, Klinger ME, Myers RL, Devraj K, Coe CL, Lubach GR, Carruthers A, Connor JR. A novel model for brain iron uptake: Introducing the concept of regulation. J Cereb Blood Flow Metab. 2015;35:48–57. doi: 10.1038/jcbfm.2014.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martines AM, Masereeuw R, Tjalsma H, Hoenderop JG, Wetzels JF, Swinkels DW. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nat Rev Nephrol. 2013;9:385–398. doi: 10.1038/nrneph.2013.98. [DOI] [PubMed] [Google Scholar]
- 48.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006;103:13612–13617. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, Ganz T, Cousins RJ. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843–6848. doi: 10.1073/pnas.0502257102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersen HH, Johnsen KB, Moos T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol Life Sci. 2014;71:1607–1622. doi: 10.1007/s00018-013-1509-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, Gonzalez-Billault C, Nunez MT. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. 2013;126:541–549. doi: 10.1111/jnc.12244. [DOI] [PubMed] [Google Scholar]
- 52.Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12. doi: 10.1016/j.bbi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Canonne-Hergaux F, Gros P. Expression of the iron transporter DMT1 in kidney from normal and anemic mk mice. Kidney Int. 2002;62:147–156. doi: 10.1046/j.1523-1755.2002.00405.x. [DOI] [PubMed] [Google Scholar]
- 54.Fujishiro H, Yano Y, Takada Y, Tanihara M, Himeno S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics. 2012;4:700–708. doi: 10.1039/c2mt20024d. [DOI] [PubMed] [Google Scholar]
- 55.Jenkitkasemwong S, Wang CY, Coffey R, Zhang W, Chan A, Biel T, Kim JS, Hojyo S, Fukada T, Knutson MD. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 2015;22:138–150. doi: 10.1016/j.cmet.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, Kong Q, Bartz J, Singh N. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. 2009;5:e1000336. doi: 10.1371/journal.ppat.1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]