

Abstract
The isozymes of monoamine oxidase (MAO-A and MAO-B) are important enzymes involved in the metabolism of numerous biogenic amines, including the neurotransmitters serotonin, dopamine and norepinephrine. Recently, changes in concentrations of MAO-B have been proposed as an in vivo marker of neuroinflammation associated with Alzheimer’s disease. Previous developments of in vivo radiotracers for imaging changes in MAO enzyme expression or activity have utilized the irreversible propargylamine-based suicide inhibitors, or high-affinity reversibly-binding inhibitors. As an alternative approach, we have investigated 1-[11C]methyl-4-aryloxy-1,2,3,6-tetrahydropyridines as metabolic trapping agents for the monoamine oxidases. MAO-mediated oxidation and spontaneous hydrolysis yields 1-[11C]methyl-2,3-dihydro-4-pyridinone as a hydrophilic metabolite that is trapped within brain tissues. Radiotracers with phenyl, biphenyl and 7-coumarinyl ethers were evaluated using microPET imaging in rat and primate brain. No isozyme selectivity for radiotracer trapping was observed in the rat brain for any compound, but in the monkey brain the phenyl ether demonstrated MAO-A selectivity, and the coumarinyl ether showed MAO-B selectivity. These are lead compounds for further development of 1-[11C]methyl-4-aryloxy-1,2,3,6-tetrahydropyridines with optimized brain pharmacokinetics and isozyme selectivity.
Keywords: Alzheimer’s disease, neuroimaging, positron emission tomography, carbon-11, monoamine oxidase
The enzymes monoamine oxidase-A (MAO-A) and –B (MAO-B) are responsible for the oxidation of a wide variety of amines, including the amine neurotransmitters dopamine, norepinephrine, and serotonin. Inhibitors of monoamine oxidases (MAOIs) are important in clinical medicine: inhibition of MAO-A is utilized in the management of depression, and inhibition of MAO-B forms one of the therapeutic approaches to treating Parkinson’s disease1. More recently, the potential for using changes of MAO-B as a marker of astrogliosis in neurodegenerative diseases (such as Alzheimer’s disease) has become of interest2. The importance of the monoamine oxidases in numerous disease processes stimulated interest for in vivo non-invasive imaging of changes of MAO in the human brain, leading to the development of numerous carbon-11 and fluorine-18 labeled radiotracers useful for Positron Emission Tomography (PET) studies3. Prior approaches to isoform-selective MAO radiotracer development have mostly concentrated on (a) irreversibly trapped suicide inhibitors (e.g., [11C]deprenyl and [11C]clorgyline) that form covalent bonds between the radiotracer and the flavin cofactor of the enzymes, and (b) reversibly-binding high-affinity inhibitors (e.g., [11C]befloxatone, [11C]harmine). Several of these have been successfully introduced into human PET studies, including evaluation in Alzheimer’s disease4, but there is continued interest in the development of new MAO radiotracers labeled with carbon-11 and fluorine-18 in the search for optimal in vivo radiotracers3b, 5.
An alternative approach for measuring enzymatic activity in vivo is the use of metabolic trapping, where the product of the catalytic activity of the enzyme is not covalently bound but still retained within the target tissue. This is the mechanism behind such in vivo imaging radiotracers as 2-[18F]fluoro-2-deoxyglucose ([18F]FDG, glucose metabolism), N-[11C]methyl-4-piperidinyl-propionate or acetate ([11C]PMP and [11C]MP4A, acetylcholinesterase)6, and [18F]fluoroDOPA (dopamine synthesis) among other examples. The concept of non-covalent metabolic trapping has been applied to MAO but the effort has been severely limited. The syntheses of radiolabeled forms of N,N-dimethylphenylethylamine were reported, initial biodistribution studies reported in rodents, and even a single human PET image presented7. Preliminary studies in both rat and primate were also done with carbon-11 labeled MPTP8 and a radioiodinated tetrahydropyridine (N-methyl-4-(4′-hydroxy-3′-[125I]iodophenyl)-1,2,3,6-tetrahydropyridine)9, but no additional reports were made for use of either compound. Further development and validation (demonstration of isoform selectivity, pharmacokinetic analyses) of non-covalent metabolic trapping radiotracers for MAO have not been pursued, and there have been no applications of this concept to studies of MAO in human diseases.
We have investigated here the metabolic trapping of 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines, molecules that undergo an initial single oxidation by monoamine oxidases (Fig. 1), forming dihydropyridinium species that are then rapidly hydrolyzed to a phenol and 1-methyl-2,3-dihydro-4-pyridinone10. As that ketone is hydrophilic (cLog P = −0.18), labeling with a carbon-11 at the N-methyl substituent was hypothesized to provide a metabolite that would be trapped within brain tissues. Furthermore, as a second oxidation step of the dihydropyridinium intermediate never occurs, the 4-aryloxy-1,2,3,6-tetrahydropyridines are non-toxic (in contrast to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)). A series of 4-aryloxy substituted tetrahydropyridines were synthesized by Castagnoli and coworkers11 and evaluated as substrates for the monoamine oxidases; those studies demonstrated that a broad range of in vitro kinetic parameters (Km and Kcat) and MAO isoform selectivity are possible by selection of the 4-aryloxy substituent.
Figure 1.
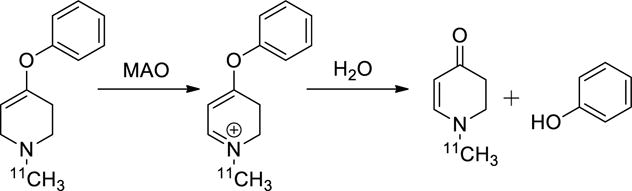
Monoamine oxidase-mediated oxidation of 1-[11C]methyl-4-aryloxy-1,2,3,6-tetrahydropyridines, followed by spontaneous hydrolysis, yields a phenol and 1-[11C]methyl-2,3-dihydro-4-pyridinone.
RESULTS AND DISCUSSION
Chemistry
Using the prior studies of the in vitro kinetics of MAO-mediated hydrolysis as a guide, three target compounds were selected to test the concept of using 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines as isoform-selective MAO-catalyzed metabolic radiotracers (Fig. 2). The simplest molecule, 1-methyl-4-phenyloxy-1,2,3,6-tetrahydropyridine (PHXY, 1a), was reported as a mixed MAO-A/MAO-B substrate with a slight (2-fold) higher reactivity towards MAO-B12. The biphenyl-substituted derivative (BiPHEN, 1c) demonstrated 8-fold higher in vitro reactivity towards MAO-A over MAO-B (Wang and Castagnoli 1995)11a. Finally, the 7-coumarinyl derivative (COU, 1b) was reported by Long and coworkers13 to have a 22-fold selectivity for MAO-B.
Figure 2.
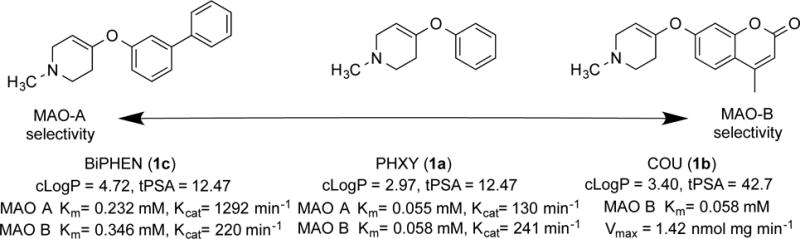
Target carbon-11 radiotracers and reported in vitro enzyme kinetics.
The syntheses of 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines in isotopically unmodified forms were done following modifications of the literature procedure11a, 13 and utilized the reaction of 4-chloro-1-methylpyridin-1-ium triflate with the appropriate phenols (2a–c) to form intermediate 1-methyl-4-aryloxypyridines (3a–c), followed by sodium borohydride reduction of the pyridine ring (Scheme 1). The products 1a–c were isolated in good yields (12–47%) using flash silica gel chromatography. The syntheses of the carbon-11 forms of PHXY, COU and BiPHEN were done using a one-pot procedure of N-[11C]methylation of the 4-aryloxypyridines 4a–c using no-carrier-added [11C]methyl triflate, followed by rapid (5 min) sodium borohydride reduction in ethanolic solution (Scheme 1). The required pyridine precursors (4a–c) were prepared by reaction of 4-chloropyridinium chloride with the corresponding phenols 2a–c. The radiotracers ([11C]PHXY (1a), [11C]COU (1b) and [11C]BiPHEN (1c)) were isolated and purified by HPLC, with overall synthesis times of 30 min. Although isolated radiochemical yields were low (1–5%, not corrected for decay) they were not optimized, and specific activities averaged >55.5 TBq/mmol. Radiotracers were then formulated in isotonic saline for microPET imaging studies in rats and monkey.
Scheme 1.
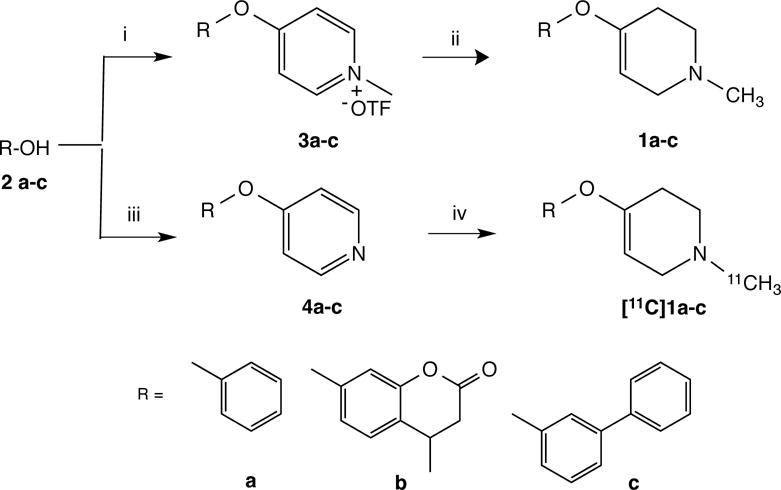
Syntheses of unlabeled and carbon-11 labeled tetrahydropyridine MAO substrates.a
aReagents and Conditions (i) NaOCH3, DMF, 4-chloro-1-methylpyridin-1-ium triflate; (ii) NaBH4, CH3OH; (iii) KOtBu, DMF, 4-chloropyridin-1-ium; (iv) [11C]CH3OTf, EtOH, NaBH4
Biology
The in vivo brain distributions of all three radiotracers were evaluated using microPET imaging, with the initial evaluations of pharmacokinetics and isozyme selectivity performed in the rat brain. However, due to the well-known potential for species differences in the selectivity and rates of substrate oxidations by the monoamine oxidases7a, 14, additional studies were performed in the rhesus monkey.
The microPET studies in rat brain demonstrated similar pharmacokinetics for [11C]PHXY and [11C]COU, with rapid initial uptakes peaking at 90–150 seconds and equivalent maximum brain concentrations (1.1 % injected dose/g for both). For both [11C]PHXY and [11C]COU, there followed a rapid washout of radioactivity until constant levels of trapped radioactivity were reached by 20–30 min, with trapping fractions (plateau/peak) of 55–60% for [11C]PHXY and 10–15% for [11C]COU. [11C]BiPHEN showed different pharmacokinetics, with a significantly lower and broader initial uptake (0.43 % injected dose/g) and a more gradual washout that did not quite reach a constant level by the end of the 60 min imaging period.
To test for isozyme selectivity, studies of trapping efficiencies were performed after pretreatments with deprenyl (selective MAO-B inhibitor, 10 mg/kg i.p. 90 min prior to scan) or clorgyline (selective MAO-A inhibitor, 10 mg/kg i.p. 90 min prior to scan) or both irreversible inhibitors. The in vivo trapping of [11C]PHXY and [11C]BiPHEN were more sensitive to MAO-A inhibition, and [11C]COU more sensitive to MAO-B inhibition, but none of the three radiotracers examined here exhibited specificity for either isozyme in the rat brain. The pharmacological blocking studies support little non-specific binding of [11C]PHXY and [11C]COU, as residual trapped radioactivity levels are very low after enzyme inhibition. The lower brain uptake and slower kinetics of [11C]BiPHEN suggests the radiotracer might exhibit higher non-specific distribution, consistent with its higher lipophilicity.
The rat studies were encouraging and supported the hypothesis that MAO-mediated oxidation of 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines could be imaged in the mammalian brain. Metabolite studies of rat brain extracts at 10 minutes after injection of [11C]PHXY confirmed the formation of a single polar radioactive metabolite, consistent with formation of 1-[11C]methyl-2,3-dihydro-4-pyridinone. The failure to achieve isozyme selectivity (or specificity) for any of the radiotracers in the rat brain was however not discouraging. The prior studies11a, 12 of 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines had utilized MAO isolated from bovine or human tissues (or both), and numerous studies have demonstrated significant species variability in the behavior of both inhibitors and substrates towards the two isozymes of MAO7a, 14. This variability supported the notion that our new radiotracers needed to be evaluated in a second species.
The imaging studies in the rhesus monkey brain showed pharmacokinetics of all three radiotracers that were different than seen in the rat brain, with more encouraging isozyme selectivity for [11C]PHXY and [11C]COU. [11C]PHXY, the lowest molecular weight and least lipophilic compound, exhibited the highest uptake and trapping, with clear heterogeneity between the major regions of the brain (striatum > thalamus > cerebellum > cortex) (Fig. 4A). The coumarin derivative [11C]COU showed similar pharmacokinetics, with slightly lower trapping of radioactivity, and little or no distinction between levels in the striatum and thalamus (Fig 4D). Finally, the kinetics of [11C]BiPHEN were significantly different, with much lower brain uptake and trapping, and less heterogeneity in the regional brain distribution (Fig. 4G). No attempts have been made to correlate the observed regional radiotracer trapping concentrations with in vitro values for protein concentrations or enzyme activities of MAO-A or MAO-B, as there is very limited data available for rhesus monkey brain15. The rank order of striatum > cerebellar cortex > cortex for [11C]COU is however consistent with the in vitro studies of Riachi and Harik15a using either binding of [3H]pargyline or oxidation of substrates (MPTP, benzylamine).
Fig. 4.
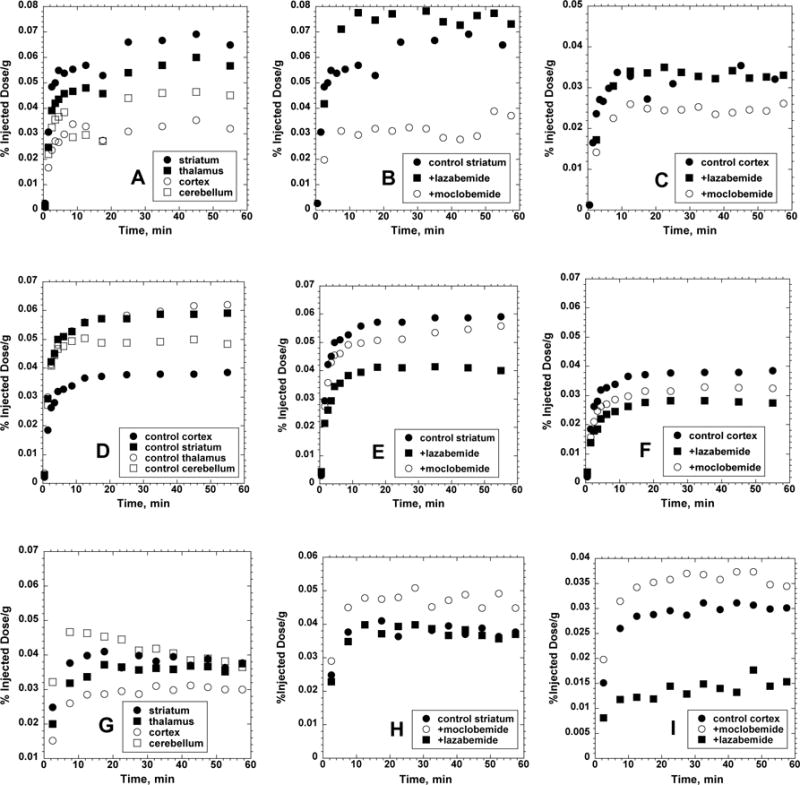
Representative curves (single scans) for in vivo trapping of [11C]PHXY (panels A–C), [11C]COU (panel D–F) and [11C]BiPHEN (panels G–I) in striatum, thalamus, cerebral cortex and cerebellum of rhesus monkey brain. Pharmacological interventions were done using pretreatment with MAO-A inhibitor (moclobemide) or MAO-B inhibitor (lazabemide).
Pharmacological studies for isozyme selectivity in the monkey were done using the reversible MAO inhibitors moclobemide (MAO-A, 1.0 mg/kg i.v.) and lazabemide (MAO-B, 0.5 mg/kg i.v.), to avoid the long-lasting irreversible inactivation of the enzymes known to occur with deprenyl and clorgyline16. The doses were administered as a 10 min infusion prior to radiotracer injection. The doses of inhibitors were limited to amounts supported by prior safe administration to monkeys, based on literature reports; without independent evidence that the doses were sufficient for complete inhibition of enzyme activities, or that the radiotracers themselves do not effectively displace these reversibly-bound inhibitors. These studies with moclobemide and lazabemide are considered here as pharmacological intervention studies but not necessarily full blocking studies (in the sense the enzymes can be fully and irreversibly inhibited, as was achieved with deprenyl and clogyline in the rat brain).
Radiotracer trapping of the simplest substrate [11C]PHXY was significantly reduced in all brain regions by inhibition of MAO-A by moclobemide, with data for striatum and cortex shown in Figs 4B–C. In contrast, administration of the MAO-B inhibitor lazabemide produced no change in the cortex and a small and perhaps insignificant increase in the striatum. These preliminary results suggest that, contrary to the in vitro assays which used a combination of enzymes from human (MAO-A) and bovine (MAO-B) tissue sources and contrary to the rat imaging studies, [11C]PHXY exhibits MAO-A selectivity in the monkey brain.
As was hoped based on the reported in vitro selectivity of [11C]COU, where human tissues provided the sources for both MAO-A and MAO-B, the in vivo blocking studies in the monkey brain showed a significant reduction in trapping in striatum and cortex after inhibition of MAO-B (Fig. 4E–F), with lesser effects effects for inhibition of MAO-A. Again, the monkey imaging studies are more encouraging than those of the rat where significant effect of both MAO-A and MAO-B inhibition were observed for [11C]COU. These studies emphasize the importance of species selection when evaluating MAO substrates or inhibitors.
Finally, the results for [11C]BiPHEN, a compound which was more selective for MAO-A in vitro (using human and bovine sources of MAO), were less clear. Inhibition of MAO-B had mixed effects on [11C]BiPHEN pharmacokinetics, and the inhibition with the MAO-A inhibitor moclobemide produced an unexpected increase in trapping of radioactivity in all brain regions (Figs. 4 H–I). One possible explanation is that moclobemide inhibition of peripheral MAO-A activity increased the available [11C]BiPHEN in the blood, resulting in increased radiotracer delivery to the brain. We had observed such changes in brain pharmacokinetics in previous studies of radiolabeled trapping agents for the enzyme acetylcholinesterase, where inhibition of blood enzymes significantly increased radiotracer delivery to the brain. Studies of [11C]BiPHEN employing metabolite-corrected blood input data would allow us to verify this hypothesis, however, the lower initial brain uptake, poor regional heterogeneity, mixed pharmacological selectivity and potential for significant non-specific distribution due to its high lipophilicity (clog P = 4.72) make [11C]BiPHEN at this point a less interesting candidate MAO-A radiotracer than [11C]PHXY.
SUMMARY
These preliminary studies have demonstrated the feasibility of using 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines as substrates for in vivo PET imaging studies of the enzymatic activity of brain monoamine oxidases. Two of the initial radiotracers examined here, [11C]PHXY and [11C]COU, already show encouraging isozyme selectivity in the monkey brain, and are of immediate interest for further evaluation. The general synthetic strategy shown in Scheme I can be used to prepare a wide variety of substituted tetrahydropyridines in the continued search for radiotracers having the optimal combinations of lipophilicity, affinity for the enzyme (Km) and rate of enzymatic hydrolysis (Kcat). Additional modifications of the rate of MAO-mediated oxidation of 1-methyl-4-aryloxy-1,2,3,6-tetrahydropyridines should also be possible by selective incorporation of deuterium at the 6-position of the tetrahydropyridine ring, as demonstrated to be effective for MPTP17.
These studies also demonstrate some of the difficulties in developing new radiotracers for in vivo imaging studies of the monoamine oxidases. The potential for significant species differences is well recognized for the isozymes of MAO, and were reflected in the relatively little selectivity of [11C]PHXY and [11C]COU in the rat brain. A correlation of regional in vivo brain trapping of our new radiotracers with in vitro values for enzyme activities is essentially impossible given the extreme paucity of available data on the regional distributions of enzymatic activities in the monkey brain. Finally, pharmacological studies in the monkey have to be planned carefully, as the use of irreversible inhibitors (e.g., deprenyl and clogyline) that unequivocally block enzyme action poses a challenge given the now recognized long-lasting effects of such inhibitors on the concentrations of active enzyme molecules16.
EXPERIMENTAL SECTION
Chemistry
General Considerations
All solvents and reagents were commercially available and used without further purification unless otherwise stated. 7-Methylumbeliferone and 3-phenylphenol were obtained from Sigma-Aldrich; 4-Phenoxypyridine was purchased from TCI America and used directly as a precursor for radiolabeling. NMR spectra were recorded with a Varian 400 MHz instrument at room temperature with tetramethylsilane (TMS) as an internal standard. Mass spectra were performed on a Micromass LCT Time-of-Flight mass spectrometer or an Agilent Q-TOF HPLC-MS employing the electrospray ionization (ESI) method. High performance liquid chromatography (HPLC) was performed using a Shimadzu LC-2010A HT system equipped with a Bioscan B-FC-1000 radiation detector.
General Procedure for preparation of 1-methyl-4-aryloxy-1,2,3,4-tetrahydropyridines (1a–c)
Phenol starting material (0.72 mmol) was added to sodium methoxide (0.87 mmol) dissolved in DMF (3 mL) and stirred for 10 minutes. 4-Chloro-1-methylpyridin-1-ium triflate (0.73 mmol) was added and the reaction was stirred for 18 h. The solvent was removed in vacuo and the resulting intermediate was suspended in methanol (3 mL). The reaction mixture was cooled to 0 °C in a water ice bath, and NaBH4 (2.9 mmol) was added slowly. After 1 h the solvent was removed in vacuo. Water and ethyl acetate were added to the residue and the mixture was transfer to a separatory funnel. The product was extracted with ethyl acetate (3X), dried over sodium sulfate, filtered, and concentrated in vacuo. The product was purified by flash silica gel chromatography (dichloromethane, methanol gradient).
1-Methyl-4-phenoxy-1,2,3,6-tetrahydropyridine (PHXY, 1a)
Starting from 4-phenoxypyridine, the reduction step reaction yielded 0.056 g (41 % yield) of 1a as a white solid. 1H NMR (400 MHz; CH3OD-d4)/δ (ppm): 7.32 (2H, t, J= 7.8, 2H), 7.08 (1H, t, J=7.4), 7.00 (2H, d, J= 7.8), 4.77 (1H, t, J= 3.2), 2.98 (2H, d J=3.1), 2.70 (2H, t, J=5.9), 2.38 (2H, m), 2.37 (3H, s); HRMS: calculated for [M+H]+(M = C12H15NO), 190.1226, found 190.1230.
4-Methyl-7-((1-methyl-1,2,3,6-tetrahydropyridin-4-yl)oxy)-2H-chromen-2-one (COU, 1b)
Starting with 4-methylumbeliferone, the reaction sequence yielded 0.091 g (47 % yield) of 1b as an off-white solid. 1H NMR (400 MHz; CH3OD-d4)/δ (ppm): 7.74 (1H, d, J=8.7), 7.06 (1H, dd, J=2.4, 8.7), 7.00(1H, d, J=2.4), 6.23(1H, d, J=1.1), 5.21 (1H, t, J=3.4); 3.17 (2H, d, J=3.4) 2.84 (2H, t, J=5.9), 2.48 (3H, s), 2.46 (3H, s), 2.42 (2H, m); HRMS: calculated for [M+H]+(M = C16H17NO3), 272.1281, found 272.184.
4-([1,1′-Biphenyl]-3-yloxy)-1-methyl-1,2,3,6-tetrahydropyridine (BiPHEN, 1c)
Starting from 3-hydroxybiphenyl, the reaction sequence yielded 0.0743 g (39 % yield) of 1c as a pale yellow oil. 1H NMR (400 MHz; CH3OD-d4)/δ (ppm): 7.57 (2H, d, J=7.2), 7.42-7.30 (5H, m), 7.27 (1H, t, J=1.9), 6.98 (1H, dq J=7.8, 1.2), 4.86 (1H, t, J=3.5), 2.96 (2H, dd, J=5.8, 2.5), 2.67 (2H, t, J=5.9) 2.39 (2H, m), 2.34 (3H, s); HRMS: calculated for [M+H]+(M = C18H19NO), 266.1539, found 266.1538.
General Procedure for preparation of 4-aryloxypyridines (4b,c)
Phenol starting material (1.6 mmol) was added to potassium tert-butoxide (2.9 mmol) dissolved in DMF (7 mL). The reaction mixture was heated to 140 °C, 4-chloropyridin-1-ium (1.33 mmol) was added, and the reaction was stirred for 18 h. The reaction was cooled to room temperature and quenched with aqueous saturated NH4Cl. The product was extracted with ethyl acetate (3X), dried over sodium sulfate, filtered, and concentrated in vacuo. The product was purified by flash silica gel chromatography (hexanes, ethyl acetate gradient).
4-Methyl-7-(pyridin-4-yloxy)-2H-chromen-2-one (4b)
Starting with 4-methylumbeliferone the reaction yielded 0.040 g (12 % yield) of 4b as an off-white solid. 1H NMR (400 MHz; CH3OD-d4)/δ (ppm): 8.48 (2H, d, J=6.1), 7.88 (1H, d, J=9.3), 7.170-7.155 (2H, m), 7.08 (2H, d, J=6.1), 6.33 (1H, s), 2.50 (3H, s); HRMS: calculated for [M+H]+(M = C15H11NO3), 254.0812, found 254.0815.
4-([1,1′-Biphenyl]-3-yloxy)pyridine (4c)
Starting with 3-hydroxybiphenyl the reaction yielded 0.1831 g (56 % yield) of 4c as a yellow solid. 1H NMR (400 MHz; CH3OD-d4)/δ (ppm): 8.41 (2H, d, J=4.9, 1.5), 7.62 (2H, d, J=7.5), 7.55 (2H, m), 7.44 (2H, t, J=7.5), 7.40-7.34 (2H, m), 7.13 (1H, dt, J=7.1, 2.2), 7.00 (2H, dd, J=4.9, 1.5); HRMS: calculated for [M+H]+(M = C17H13NO), 248.1070, found 248.1068.
Radiochemistry
General Considerations
Reagents and solvents were commercially available and used without further purification, unless otherwise noted: sodium chloride (0.9% USP) and sterile water for Injection (USP) were purchased from Hospira; Dehydrated Alcohol for Injection (USP) was obtained from Akorn Inc. Shimalite-Nickle was purchased from Shimadzu; iodine was obtained from EMD; phosphorus pentoxide was acquired from Fluka; molecular sieves were purchased from Alltech; and HPLC columns were acquired from Phenomenex. Other synthesis components were obtained as follows: sterile filters were acquired from Millipore; C18-light Sep-Paks and Porapak Q were purchased from Waters Corporation; 10 cc sterile vials were obtained from HollisterStier. Sep-Paks were flushed with 10 mL of ethanol followed by 10 mL of sterile water prior to use.
General Procedure for Radiochemical Syntheses
Production of carbon-11 labeled radiotracers was carried out using a TracerLab FXC-Pro automated radiochemistry synthesis module (General Electric, GE). [11C]Carbon dioxide was produced using a GE PETTrace cyclotron (40 μA beam for 20 min) and converted by standard procedures into carbon-11 labeled methyl triflate ([11C]CH3OTf). The [11C]CH3OTf in helium carrier gas was bubbled into a vial containing a solution of precursor (1 mg) dissolved in ethanol (0.2 mL). At completion of transfer of radioactivity into the reaction vial, the ethanol solution was then transferred to a second conical vial containing sodium borohydride (2 mg) in ethanol (0.3 mL). The resulting mixture was stirred for 5 min at room temperature and then the reaction was quenched by addition of HPLC buffer. The crude product was loaded onto a semi-preparative HPLC loop. The product was purified by reverse phase chromatography (Prodigy ODS prep, 250 × 10 mm, 10μ, 4 mL/min), collected and diluted into H2O (40 mL) and reformulated using a C-18 extraction disk into a final 5 mL total volume of 10 % ethanol in saline. The doses produced were assessed via standard quality control techniques and were appropriate for rodent and non-human primate studies. Average specific activity was 69116 GBq/mmol (range of 23236-178081 GBq/mmol). Overall synthesis times were 30 min from end-of-bombardment.
[11C]1-Methyl-4-phenoxy-1,2,3,6-tetrahydropyridine ([11C]PHXY, [11C]1a)
In non-optimized yields, 270 ± 173 mBq of the phenyl ether (0.8 % yield from [11C]CH3OTf, not decay corrected; n=8) were collected. The product was purified by semi-preparative reverse phase chromatography (250 × 10, 10μ, ODS prep column eluted with 40 % CH3CN, 60 % H2O, 10 mM NH4OAc).
[11C]4-Methyl-7-(pyridin-4-yloxy)-2H-chromen-2-one ([11C]COU, [11C]1b)
In non-optimized yields, 577 ± 141 MBq of the coumarin ether (1.7 % yield from [11C]CH3OTf, not decay corrected; n=10) were collected. The product was purified by semi-preparative reverse phase chromatography (250 × 10, 10μ, ODS prep column eluted with 30 % CH3CN, 70 % H2O, 10 mM NH4OAc).
[11C]4-([1,1′-Biphenyl]-3-yloxy)-1-methyl-1,2,3,6-tetrahydropyridine ([11C]BiPHEN, [11C]1c)
In non-optimized yields, 551 ± 126 MBq of the biphenyl ether (1.7 % yield from [11C]CH3OTf, non-decay corrected; n=7). The product was purified by semi-preparative reverse phase chromatography (250 × 10, 10μ, ODS prep column eluted with 50 % CH3CN, 50% H2O, 10 mM NH4OAc, pH 4.5).
Fig. 3.
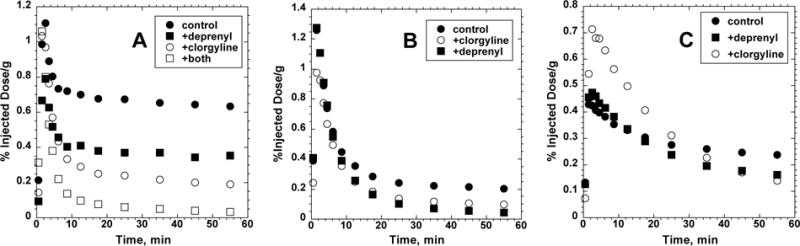
Representative curves (single scans) for in vivo trapping of [11C]PHXY (panel A), [11C]COU (panel B) and [11C]BiPHEN (panel C) in rat brain cerebrum. Data is expressed as percent injected dose per gram for rat brain cerebrum. Pharmacological interventions were done using pretreatment with MAO-A inhibitor (clorgyline) or MAO-B inhibitor (deprenyl) or both together.
Acknowledgments
Funding
Financial support for this work from the National Institutes of Health (National Institute of Neurological Diseases and Stroke, Award Number 1R21 NS075553 (MRK), and National Institute of Biomedical Imaging and Bioengineering, Award Number T32-EB005172 (PJHS, MRK) is gratefully acknowledged. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional funding for this research from the Alzheimer’s Association (NIRP-14-305669 (PJHS)) is also acknowledged.
Footnotes
Spectra for all novel compounds synthesized, chromatographic data for radiotracer purifications, and procedures for microPET imaging. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Finberg JPM. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: Focus on modulation of CNS monoamine neurotransmitter release. Pharmacology & Therapeutics. 2014;143(2):133–152. doi: 10.1016/j.pharmthera.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs AH, Tavitian B. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32(7):1393–1415. doi: 10.1038/jcbfm.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Kersemans K, Van Laeken N, De Vos F. Radiochemistry devoted to the production of monoamine oxidase (MAO-A and MAO-B) ligands for brain imaging with positron emission tomography. Journal of Labelled Compounds and Radiopharmaceuticals. 2013;56(3–4):78–88. doi: 10.1002/jlcr.3007. [DOI] [PubMed] [Google Scholar]; (b) Fowler JS, Logan J, Shumay E, Alia-Klein N, Wang GJ, Volkow ND. Monoamine oxidase: radiotracer chemistry and human studies. Journal of Labelled Compounds and Radiopharmaceuticals. 2015;58(3):51–64. doi: 10.1002/jlcr.3247. [DOI] [PubMed] [Google Scholar]
- 4.(a) Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, Nordberg A. Evidence for Astrocytosis in Prodromal Alzheimer Disease Provided by 11C-Deuterium-L-Deprenyl: A Multitracer PET Paradigm Combining 11C-Pittsburgh Compound B and 18F-FDG. Journal of Nuclear Medicine. 2012;53(1):37–46. doi: 10.2967/jnumed.110.087031. [DOI] [PubMed] [Google Scholar]; (b) Choo I, Carter S, Schöll M, Nordberg A. Astrocytosis measured by 11C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur J Nucl Med Mol Imaging. 2014;41(11):2120–2126. doi: 10.1007/s00259-014-2859-7. [DOI] [PubMed] [Google Scholar]
- 5.Tong J, Meyer JH, Furukawa Y, Boileau I, Chang LJ, Wilson AA, Houle S, Kish SJ. Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab. 2013;33(6):863–871. doi: 10.1038/jcbfm.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kikuchi T, Okamura T, Zhang MR, Irie T. PET probes for imaging brain acetylcholinesterase. Journal of Labelled Compounds and Radiopharmaceuticals. 2013;56(3–4):172–179. doi: 10.1002/jlcr.3002. [DOI] [PubMed] [Google Scholar]
- 7.(a) Inoue H, Castagnoli K, Van der Schyf C, Mabic S, Igarashi K, Castagnoli N. Species-Dependent Differences in Monoamine Oxidase A and B-Catalyzed Oxidation of Various C4 Substituted 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridinyl Derivatives. Journal of Pharmacology and Experimental Therapeutics. 1999;291(2):856–864. [PubMed] [Google Scholar]; (b) Shinotoh H, Inoue O, Suzuki K, Yamasaki T, Iyo M, Hashimoto K, Tominaga T, Itoh T, Tateno Y, Ikehira H. Kinetics of [11C]N,N-Dimethylphenylethylamine in Mice and Humans: Potential for Measurement of Brain MAO-B Activity. Journal of Nuclear Medicine. 1987;28(6):1006–1011. [PubMed] [Google Scholar]; (c) Halldin C, Bjurling P, Stalnacke CG, Jossan SS, Oreland L, Langstrom B. 11C-labelling of dimethylphenethylamine in two different positions and biodistribution studies. International journal of radiation applications and instrumentation Part A, Applied radiation and isotopes. 1989;40(7):557–60. doi: 10.1016/0883-2889(89)90108-1. [DOI] [PubMed] [Google Scholar]
- 8.Livni E, Spellman JP, Correia JA, Alpert NM, Brownell GL, Strauss HW, Elmaleh DR. [11C]MPTP: A Potential Tracer for Parkinson’s Disease Research in Laboratory Animals. Journal of Nuclear Medicine. 1986;27(10):1600–1603. [PubMed] [Google Scholar]
- 9.(a) Efange SM, Mash D, Hefti F, Kung HF, Billings J. Selective visualization of rodent locus ceruleus by a radiolabeled N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine analog. Journal of neurochemistry. 1989;53(2):459–64. doi: 10.1111/j.1471-4159.1989.tb07356.x. [DOI] [PubMed] [Google Scholar]; (b) Efange SM, Kung HF, Mash DC, Jabir M, Billings J, Pablo J, Dutta A, Freshler A. Pargyline-sensitive selective accumulation of a radiolabeled MPTP analog in the primate cerebral cortex and basal ganglia. Synapse (New York, NY) 1990;5(3):207–12. doi: 10.1002/syn.890050306. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Z, Dalvie D, Naiman N, Castagnoli K, Castagnoli N. Design, synthesis, and biological evaluation of novel 4-substituted 1-methyl-1,2,3,6-tetrahydropyridine analogs of MPTP. Journal of Medicinal Chemistry. 1992;35(23):4473–4478. doi: 10.1021/jm00101a026. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wang YX, Castagnoli N. Studies on the Monoamine Oxidase (MAO)-Catalyzed Oxidation of Phenyl-Substituted 1-Methyl-4-phenoxy-1,2,3,6-tetrahydropyridine Derivatives: Factors Contributing to MAO-A and MAO-B Selectivity. Journal of Medicinal Chemistry. 1995;38(11):1904–1910. doi: 10.1021/jm00011a010. [DOI] [PubMed] [Google Scholar]; (b) Yu J, Castagnoli N., Jr Synthesis and MAO-B substrate properties of 1-methyl-4-heteroaryl-1,2,3,6-tetrahydropyridines. Bioorganic & medicinal chemistry. 1999;7(2):231–9. doi: 10.1016/s0968-0896(98)00201-6. [DOI] [PubMed] [Google Scholar]
- 12.Kalgutkar AS, Castagnoli K, Hall A, Castagnoli N., Jr Novel 4-(aryloxy)tetrahydropyridine analogs of MPTP as monoamine oxidase A and B substrates. J Med Chem. 1994;37(7):944–9. doi: 10.1021/jm00033a012. [DOI] [PubMed] [Google Scholar]
- 13.Long S, Chen L, Xiang Y, Song M, Zheng Y, Zhu Q. An activity-based fluorogenic probe for sensitive and selective monoamine oxidase-B detection. Chemical communications (Cambridge, England) 2012;48(57):7164–6. doi: 10.1039/c2cc33089j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fowler JS, Ding YS, Logan J, MacGregor RR, Shea C, Garza V, Gimi R, Volkow ND, Wang GJ, Schlyer D, Ferrieri R, Gatley SJ, Alexoff D, Carter P, King P, Pappas N, Arnett CD. Species differences in [11C]clorgyline binding in brain. Nuclear Medicine and Biology. 2001;28(7):779–785. doi: 10.1016/s0969-8051(01)00245-1. [DOI] [PubMed] [Google Scholar]; (b) Novaroli L, Daina A, Favre E, Bravo J, Carotti A, Leonetti F, Catto M, Carrupt PA, Reist M. Impact of species-dependent differences on screening, design, and development of MAO B inhibitors. J Med Chem. 2006;49(21):6264–72. doi: 10.1021/jm060441e. [DOI] [PubMed] [Google Scholar]
- 15.(a) Riachi NJ, Harik SI. Monoamine oxidases of the brains and livers of macaque and cercopithecus monkeys. Experimental neurology. 1992;115(2):212–7. doi: 10.1016/0014-4886(92)90055-u. [DOI] [PubMed] [Google Scholar]; (b) Campbell IC, Marangos PJ, Parma A, Garrick NA, Murphy DL. Localization of monoamine oxidases A and B in primate brains relative to neuron-specific and non-neuronal enolases. Neurochemical research. 1982;7(6):657–66. doi: 10.1007/BF00965519. [DOI] [PubMed] [Google Scholar]
- 16.Fowler JS, Volkow ND, Logan J, Wang GJ, MacGregor RR, Schyler D, Wolf AP, Pappas N, Alexoff D, Shea C, et al. Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse (New York, NY) 1994;18(2):86–93. doi: 10.1002/syn.890180203. [DOI] [PubMed] [Google Scholar]
- 17.Ottoboni S, Caldera P, Trevor A, Castagnoli N., Jr Deuterium isotope effect measurements on the interactions of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine with monoamine oxidase B. The Journal of biological chemistry. 1989;264(23):13684–8. [PubMed] [Google Scholar]