

Abstract
The notion that prion-like spreading of misfolded α-synuclein (α-SYN) causes Parkinson's disease (PD) has received a great deal of attention. Although attractive in its simplicity, the hypothesis is difficult to reconcile with postmortem analysis of human brains and connectome-mapping studies. An alternative hypothesis is that PD pathology is governed by regional or cell-autonomous factors. Although these factors provide an explanation for the pattern of neuronal loss in PD, they do not readily explain the apparently staged distribution of Lewy pathology in many PD brains, the feature of the disease that initially motivated the spreading hypothesis by Braak and colleagues. While each hypothesis alone has its shortcomings, a synthesis of the two can explain much of what we know about the etiopathology of PD.
Dual Perspectives Companion Paper: Prying into the Prion Hypothesis for Parkinson's Disease, by Patrik Brundin and Ronald Melki
Keywords: aging, alpha-synuclein, calcium, mitochondria, neurodegeneration, neuron, selective vulnerability, synapse
Introduction
Clinical Parkinson's disease (cPD) is the most common form of a broad class of movement disorders called parkinsonism (Postuma et al., 2015). The cardinal motor manifestations of cPD are attributable to the progressive loss of dopaminergic neurons in the SNc (Hornykiewicz, 2002). In addition to neuronal loss, a defining feature of cPD is the appearance of proteinaceous, α-synuclein (α-SYN) rich inclusions, called Lewy pathology (LP), exclusively in neurons.
While rigorous determination of the regional loss of neurons in postmortem tissue has been difficult, the advent of immunocytochemical techniques allowing the localization of aggregated forms of α-SYN has propelled the study of LP forward. It is always easier to see what is gained than what is lost. Nearly two decades ago, Braak and colleagues used these approaches to compare brains taken from asymptomatic individuals and cPD patients at various times after diagnosis. This exercise led them to hypothesize that LP spreads into the brain from either the olfactory bulb or the dorsal motor nucleus of vagus (DMV) in the caudal medulla, two brain regions with axons extending to body surface (Kosaka et al., 1984; Braak et al., 2003; Beach et al., 2009). It was conjectured that, at these interfaces, LP-inducing, environmental pathogens or infectious agents invaded axon terminals, were retrogradely transported, and then trans-synaptically spread to other neurons (Hawkes et al., 2007). With time, these LP-inducing agents were thought to slowly propagate through the brain connectome, leading to widespread neuronal dysfunction and death (Braak et al., 2004).
This sounds like a neuropathological tsunami, which is essentially the way it is depicted in many reviews (Braak et al., 2004). However, this is misleading. Even in late-stage cPD brains, LP has a discrete, patch-like distribution (Beach et al., 2009; Dijkstra et al., 2014; Surmeier et al., 2017b). Moreover, within each of the nuclei or regions manifesting LP, its distribution is sparse and confined to particular cell types (Braak and Del Tredici, 2009; Dugger and Dickson, 2010). For example, within the DMV, only cholinergic and catecholaminergic neurons ever exhibit LP, whereas GABAergic neurons never do (Kingsbury et al., 2010). A similar discrete distribution of LP is seen in other regions, such as the pedunculopontine nucleus, basal forebrain, and cerebral cortex (Wakabayashi et al., 1995; Hall et al., 2014). It is of some note that GABAergic neurons, regardless of where they are, appear to be resistant to LP. In all of these cases, the percentage of neurons exhibiting LP is small (<15%) and relatively constant over the disease course, even in the absence of neuronal loss (Greffard et al., 2010; Parkkinen et al., 2011; Milber et al., 2012; Dijkstra et al., 2014; Iacono et al., 2015).
The proposition that this distributed pathology evolves over time in a predictable way that is causally related to symptoms clearly is attractive. It was an extraordinary example of inductive reasoning because the human data upon which the hypothesis was built had significant limitations. The most important of these was that the postmortem data did not provide any “hard” longitudinal information; that is, it did not show how LP pathology within individual brains evolved as a function of time and disease state. This relationship had to be inferred from a reasonable, but untested, set of assumptions. As a consequence, it is not surprising that subsequent studies have found that only approximately half of cPD patients have brains with a pattern of LP that is consistent with the Braak staging model (Kalaitzakis et al., 2008; Jellinger, 2009b; Halliday et al., 2012). Some cPD patients have no discernible LP at all (Berg et al., 2014). Moreover, attempts to correlate Braak LP staging with clinical state have been unsuccessful (Jellinger, 2009b).
For the sake of argument, let us set aside those brains that do not conform to Braak staging (we can suppose they are some form of “atypical” cPD for the time being). Is it plausible that the globally and regionally heterogeneous pattern of LP seen in these brains is a consequence of retrograde, trans-synaptic spread of a pathogen from the DMV or olfactory bulb? In principle, what would be needed to evaluate this hypothesis is the retrograde synaptic connectome of neurons in the nuclei from which the pathology is thought to spread. Unfortunately, this type of information is only now being generated in transgenic mice (not humans) using techniques, such as monosynaptic rabies virus mapping (Wall et al., 2010). Nevertheless, there are some data that are relevant if it is assumed that mice and men are approximately similar in the wiring of their brains (an assumption that is generally supported by the experimental literature). Consider the SNc; this is clearly an important node in the network of LP in the cPD brain. If a pathogen is passed from SNc dopaminergic neurons to neurons synapsing upon them, then the probability that this happens should be directly related to the number of synapses formed by the innervating neuron (i.e., the probability of one getting the flu is proportional to the number of times you come into contact with someone who has it). If this is the case, then basal ganglia nuclei (substantia nigra pars reticulata, globus pallidus, subthalamic nucleus, and striatum) should be prominent sites of late stage LP. These nuclei robustly innervate dopaminergic neurons in the SNc (Watabe-Uchida et al., 2012; Ogawa et al., 2014). Yet, none of these regions has any discernible postsynaptic LP, ever. The striatal Lewy neurites seen relatively early in the disease are undoubtedly degenerating dopaminergic axons (Halliday et al., 2011).
Consider the locus ceruleus (LC), another prominent site of LP for which there are connectomic data (Schwarz et al., 2015). Again, there is no correlation between the strength of synaptic connectivity and the probability of manifesting LP at any stage of the disease. The most prominent synaptic inputs to the LC are from the cerebellum and the medial reticular formation. Neither have any significant LP in cPD patients. So, if a pathogen spreads as hypothesized by Braak and others, its spread (or its propensity to induce LP) must be governed by some other factor. It cannot be governed by synaptic connectivity alone.
The other major caveat of the Braak hypothesis is that the relationship between LP, neuronal dysfunction, and neuronal death was, and remains, uncertain. Braak's conjecture was that LP was a harbinger of death and dysfunction. In contrast to LP, there have been relatively few rigorous studies of neuronal death in PD. This is hard to do. In brains with LP confined to the caudal medulla, there is a significant loss of SNc DA neurons in the ventral tier of the SNc (Milber et al., 2012; Dijkstra et al., 2014). There is not any substantial neurodegeneration in the other regions, most notably those that had LP. In the brains of recently diagnosed cPD patients, DA neurons in the ventral tier of the SNc are nearly gone (Halliday et al., 1996; Damier et al., 1999), and neuronal loss is apparent in a handful of other regions. For example, cholinergic neurons in the pedunculopontine nucleus are lost, but not glutamatergic or GABAergic pedunculopontine nucleus neurons (Halliday et al., 1990b). There also is modest loss of glutamatergic neurons in the intralaminar nuclei of the thalamus and the basolateral amygdala (Henderson et al., 2000; Harding et al., 2002). Thus, in the early stages of PD, there is not a compelling correlation between LP and neuronal loss.
With clinical progression, neuronal death is found in other regions, particularly those with LP (Halliday et al., 1990b; Kremer and Bots, 1993; Thannickal et al., 2007; Fronczek et al., 2008; Jellinger, 2009a). But there are plenty of exceptions (Halliday et al., 1990a; Ansorge et al., 1997; MacDonald and Halliday, 2002; Pedersen et al., 2005). Thus, both early and late in the disease, the correlation between LP and neuronal loss is poor (Fig. 1).
Figure 1.
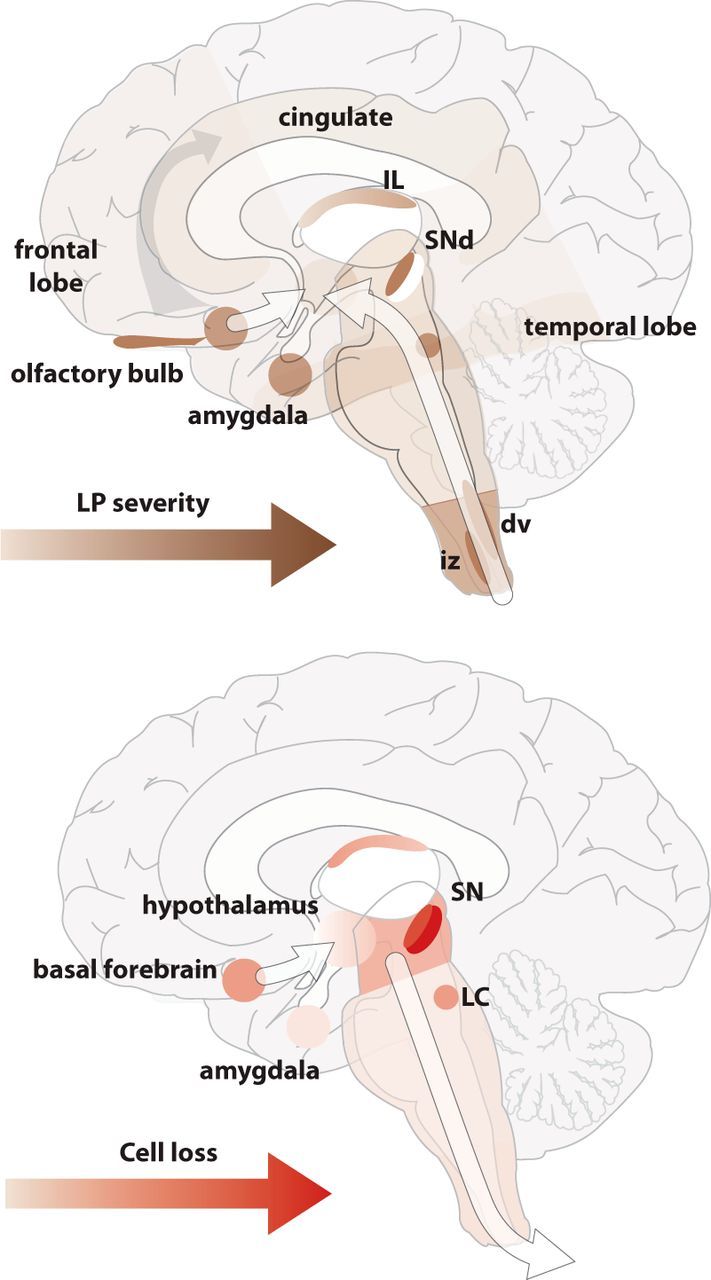
Top, The selective regions containing LP and the severity and conceptual progression based on cross-sectional postmortem data from patients at different stages of Parkinson's disease. Bottom, The selective regions with neuronal cell loss, and the severity and conceptual progression based on cross-sectional postmortem data from patients at different stages of PD. Although there is some overlap in the regions identified with LP and neuronal loss, the severity and regions affected over the disease course indicate different progression patterns, and these patterns are independent of the major projections of the regions affected, suggesting that prion propagation through neuronal connections is unlikely as a simplistic mechanism. IL, Intralaminar thalamus; SNd, dorsal tier of the substantia nigra pars compacta; dv, dorsal motor nucleus of the vagus nerve; iz, intermediate reticular zone.
Braak redux
The recognition that the pathology in PD is distributed and not restricted to the SNc by Braak and others fundamentally changed thinking about cPD pathogenesis. But the proposition that LP spreads through the brain connectome from well-defined nuclei and is solely responsible for the pathophysiology underlying cPD symptoms was inconsistent with much of the literature, so it languished, until recently.
Two set of observations have resurrected Braak's conjecture. One piece comes from histological analysis of fetal transplants into the striatum of PD patients. Most (but not all) of these studies revealed that, after only a decade or so, a few dopaminergic neurons exhibited proteinaceous inclusions that strongly resembled LP (Kordower et al., 2008; Li et al., 2008; compare Mendez et al., 2008). This was interpreted as spread of LP from the host to the graft. Although there is no doubt that α-SYN can be taken up by neurons from the extracellular space (Desplats et al., 2009; Hansen et al., 2011), the proposition that LP per se spread from the host to the graft was quite speculative and left several basic questions unanswered. For example, from where in the host did the seeding pathology spread? In late-stage PD patients, there is not any discernible LP in the host striatum, meaning it would have had to spread from some distant, nonsynaptically coupled site. Another question was why the Lewy-like pathology was restricted to dopaminergic neurons in the graft (compare Ahn et al., 2012), sparing neighboring GABAergc neurons? And why does the fraction of neurons displaying α-SYN pathology not increase with the duration the graft is in the host (Cooper et al., 2009; Kurowska et al., 2011; Li et al., 2016)? Could it not simply be the case that living in a graft is stressful and that the cumulative effect of this environment on already stressed (see below) dopaminergic neurons is proteostatic dysfunction and α-SYN accumulation?
A more compelling argument for spreading comes from experiments where α-SYN fibrils have been directly injected into the brain of mice and monkeys. In contrast to monomeric α-SYN (Kirik et al., 2003; Maingay et al., 2006; Ulusoy et al., 2013), synthetic, preformed α-SYN fibrils (PFFs) injected into the mouse striatum can propagate to synaptically connected, neighboring structures, creating Lewy-like pathology (Luk et al., 2012; Masuda-Suzukake et al., 2013; Peelaerts et al., 2015). In monkeys, proteins extracted from human brains with LP (that would contain α-SYN fibrils and other LP proteins) also can retrogradely propagate from the striatum after injection (Recasens et al., 2014). More recent work has shown spreading from the olfactory bulb (Rey et al., 2016). Although there are a lot of questions about these experiments and their interpretation (Sacino et al., 2016; Uchihara and Giasson, 2016; Walsh and Selkoe, 2016), they do demonstrate that extracellular α-SYN fibrils can be taken up, retrogradely (and possibly anterogradely) transported, and induce Lewy-like pathology (and cell death). Moreover, because endogenous α-SYN is recruited to the intracellular PFF aggregates and is necessary for the spreading of pathology (Volpicelli-Daley et al., 2011, 2014; Luk et al., 2012; Masuda-Suzukake et al., 2013; Peelaerts et al., 2015; compare Helwig et al., 2016), the PFFs have been likened to prions (Olanow and Brundin, 2013; Brettschneider et al., 2015). The hypothesis that prion-like fibrillary α-SYN drive pathogenesis in PD is attractive in many ways, as it posits a conceptually simple mechanism that, taken at face value, explains the evolution of LP in patients and captures the essence of the original Braak idea.
But is this what happens in PD? In principle, a prion-like process should follow one of two rules: a nearest neighbor rule or a synaptic connectivity rule. The nearest neighbor rule, whereby the probability of manifesting LP is directly related to the physical proximity to an initial seeding site, clearly is not consistent with the pattern of LP in PD patients. LP does not simply fill up the brain; it has a discrete distribution. Does the spread follow a simple synaptic connectivity rule? As described above, the available data argue that it does not. Hence, if LP spreads in PD, then there must be some other determinant of spreading in addition to just connectivity.
Neuronal phenotype and PD pathogenesis
An alternative to the Braak hypothesis is that neuronal death and LP are driven by cell-autonomous or regionally autonomous mechanisms, not a propagated pathogen. One of the features of the brain that distinguishes it from other organs is its incredible cellular diversity. Neurons, in particular, vary enormously in their size, shape, and function. The neurons at risk in PD may have a phenotype that renders them particularly vulnerable to factors known to cause the disease, age, genetic mutations, and environmental toxins.
What do we know about the phenotype of neurons that are at risk in PD? Almost all of the work on this topic has focused on SNc dopaminergic neurons whose loss is responsible for the core motor features of PD. While the SNc dopaminergic neuron may be the “poster child” for the disease, Braak and colleagues have shown that PD stretches well beyond them. Any general theory of PD pathogenesis must explain this feature.
Many (if not all) of the neurons that degenerate or manifest profound LP in PD seem to have a set of shared traits. The most notable and best characterized of these shared traits is a long and highly branched axon with an extraordinary number of transmitter release sites. This diffuse axonal arbor helps them coordinate the activity in large networks, such as the basal ganglia or the spinal cord. For example, SNc DA neurons in the rodent have axons that branch profusely in the striatum and possess as many as 200,000 vesicular release sites (Matsuda et al., 2009). This branching is similar in primates (Parent and Parent, 2006). Why might a long and highly branched axon be problematic? There are several hypotheses that have been proposed (Venda et al., 2010; Bolam and Pissadaki, 2012; Hunn et al., 2015), but only one has compelling experimental support at this point. This hypothesis posits that the bioenergetic demands of sustaining electrical excitability in a highly branched axon leads to mitochondrial oxidant stress. Indeed, it has been shown that in vitro mitochondrial oxidant stress is higher in SNc DA neuron axons than in the axons of less vulnerable VTA DA neurons and that reducing the size of the arbor (by manipulating axon guidance signals) decreases this stress (Pacelli et al., 2015). That said, it is puzzling that neurons, such as striatal cholinergic interneurons (Zhou et al., 2002), which have axons that are similar in complexity to those of an SNc dopaminergic neuron, seem to be resistant to whatever is going on in PD.
In addition to having a long axon, many vulnerable neurons share a set of physiological traits (Surmeier et al., 2017b). In vivo, at-risk neurons that have been studied are tonically active (Surmeier et al., 2012). Typically, the action potentials of these neurons are slow and broad, which maximizes Ca2+ entry and promotes slow rhythmic activity (Bean, 2007). In that subset of neurons studied in depth, the slow, rhythmic activity (2–10 Hz) is autonomously generated and accompanied by slow oscillations in intracellular Ca2+ concentration that are triggered by the opening of plasma membrane Cav1 and Cav3 Ca2+ channels and release of Ca2+ from intracellular, ER stores (Nedergaard et al., 1993; Wolfart and Roeper, 2002; Puopolo et al., 2007; Guzman et al., 2010; Morikawa and Paladini, 2011; Goldberg et al., 2012; Sanchez-Padilla et al., 2014; Matschke et al., 2015). In these cells, the diffusion of Ca2+ in the cytosol is unimpeded by the expression of Ca2+ buffering proteins, such as calbindin (Foehring et al., 2009; Goldberg et al., 2012; Sanchez-Padilla et al., 2014). This combination of features, broad spikes, pacemaking, low intrinsic Ca2+ buffering, and cytosolic Ca2+ oscillations (not any one) is what appears to distinguish vulnerable neurons.
The slow Ca2+ oscillations in at-risk neurons subserve two complementary functions. First, they help maintain the slow tonic spiking in these neurons (Nedergaard et al., 1993; Puopolo et al., 2007; Putzier et al., 2009). Second, they promote Ca2+ entry into mitochondria, oxidative phosphorylation (OXPHOS), and the production of ATP (Guzman et al., 2010; Sanchez-Padilla et al., 2014; Llorente-Folch et al., 2015). In principle, this feedforward control of OXPHOS helps to ensure that bioenergetic needs are met (Budd and Nicholls, 1998; Balaban, 2009) and that intracellular ATP levels do not fall into a range that would trigger protective activation of K-ATP channels and cessation of ongoing activity (Dragicevic et al., 2015). Even temporary cessation of activity in neuronal networks necessary to mobilize sensory and motor systems directing escape or attack behavior would lessen the chances of survival in an unpredictable environment. As a consequence, there should have been strong evolutionary pressure to design neurons in these “too important to fail” networks with this type of feedforward control mechanism.
There are two obvious downsides of this design. First, stimulating OXPHOS in the absence of strong ATP demand increases the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Votyakova and Reynolds, 2001; Guzman et al., 2010; Goldberg et al., 2012; Sanchez-Padilla et al., 2014). ROS and RNS damage proteins, lipids, and DNA, particularly in mitochondria. Sustained oxidant stress could be a major factor underlying declining mitochondrial function in at-risk neurons with age (Reeve et al., 2014). ROS and RNS also exacerbate the impact of genetic mutations and environmental toxins affecting mitochondria (Gegg and Schapira, 2016), as well as increase the propensity of α-SYN to aggregate (Gupta et al., 2008). The second downside is that it results in sustained elevations in cytosolic Ca2+ concentration. Ca2+ promotes α-SYN aggregation both directly (Rcom-H'cheo-Gauthier et al., 2014) and indirectly through activation of calpain and calcineurin (Dufty et al., 2007; Caraveo et al., 2014; Diepenbroek et al., 2014). Elevated cytosolic Ca2+ also impairs lysosomal motility and turnover of misfolded proteins (Gómez-Sintes et al., 2016), potentially synergizing with other defects in proteasomal/autophagic function to increase the likelihood of LP (Wong and Cuervo, 2010). Thus, by design, these vulnerable neurons appear to reside close to mitochondrial and degradative “tipping points.”
But do all of the neurons at risk in PD conform to this model? It is unclear. In-depth analysis has only been performed in SNc, LC, and DMV neurons. While much of the brainstem data are consistent with a shared phenotype, more in-depth phenotyping needs to be done. However, healthy, young telencephalic neurons are not phenocopies of SNc dopaminergic neurons. That said, many of the telencephalic regions at-risk in PD (and AD) are part of a “default” network, which manifests high resting activity, albeit of synaptic origin (Andrews-Hanna et al., 2007). It is possible that, in aged, late-stage PD patients, network dysfunction (Hammond et al., 2007; Ko et al., 2013) triggers adaptations that bring these neurons and networks phenotypically closer to other at-risk neurons. Cav1 Ca2+ channels, which are key determinants of the SNc phenotype, could be a major factor in this process. Sustained Ca2+ entry through Cav1 channels in forebrain neurons has long been associated with aging-related cognitive decline and AD (Disterhoft et al., 1994; Thibault et al., 2007). Moreover, in PD patients, Cav1 Ca2+ channels are upregulated in limbic and motor cortices (Hurley et al., 2013, 2014).
Can the phenotype of at-risk neurons account for LP staging? The simple answer is no, at least at this point in time. From what we currently know about cell-autonomous risk factors, LP should appear in the SNc before it does in the DMV. Barring the emergence of some other cell-autonomous factor that drives LP, the most parsimonious explanation of the LP pattern in PD is that there is spreading of α-SYN pathology, as posited by Braak and colleagues and the proponents of the prion model, but that spreading is limited to a subset of neurons whose phenotype renders them susceptible to spreading, a proposition that is very consistent with the phenotype outlined above.
What is better explained by cell-autonomous factors is the sequence of cell death in PD (Fig. 2). The earliest known loss of neurons in PD is the SNc. These neurons are at one extreme of the anatomical, physiological, and molecular spectrum of vulnerable neurons as we currently understand it (Sulzer and Surmeier, 2013; Poulin et al., 2014; Anderegg et al., 2015; Brichta et al., 2015; Surmeier et al., 2017a), exhibiting the highest basal levels of mitochondrial oxidant stress and free cytosolic Ca2+ of any cell examined. Mitochondria and intracellular Ca2+ are linchpins of all three major death cascades (apoptotic, autophagic, and necrotic) (Nagley et al., 2010). In human SNc, there are telltale signs of sustained mitochondrial oxidant stress with aging and PD, such as mitochondrial DNA deletions (Bender et al., 2006, 2008). Against this backdrop, it makes sense that genetic mutations that compromise mitochondrial oxidant defenses, biogenesis, or quality control cause the preferential loss of SNc dopaminergic neurons and early onset forms of PD (Lin and Farrer, 2014; Kumaran and Cookson, 2015; Mullin and Schapira, 2015; Beilina and Cookson, 2016). The tipping point for these neurons also could be reached by other genetic mutations that indirectly compromise mitochondrial function (McCoy and Cookson, 2012; Mullin and Schapira, 2013; Brini et al., 2014; Guardia-Laguarta et al., 2015; Beilina and Cookson, 2016; Gegg and Schapira, 2016).
Figure 2.

Schematic summary of the factors potentially driving LP and neurodegeneration in PD. The vulnerable neuronal phenotype has a long, highly branched axon, which could lead to elevated expression of α-SYN, as well as increase transmission sites for misfolded α-SYN. Both of these factors could promote α-SYN aggregation, oligomer formation, LP, and possibly neurodegeneration. In parallel, pacemaking, elevated cytosolic Ca2+, and mitochondrial oxidant stress could put vulnerable neurons at risk, both by promoting mitochondrial and lysosomal dysfunction with aging as well as by promoting α-SYN aggregation (through elevated ROS/RNS, Ca2+, and calpain activation, proteostatic deficits). Other potential factors, such as a reactive neurotransmitter (e.g., dopamine), also could contribute.
It also is important to acknowledge that other forms of α-SYN may be more toxic than LP and contribute to pathogenesis and cell death in PD (Ingelsson, 2016). Soluble, oligomeric forms of α-SYN clearly can induce cell death when present in sufficient quantities. Given that Ca2+ and ROS/RNS promote α-SYN aggregation (see above), toxic oligomers, and proto-fibrils could be more likely to form in nominally vulnerable neurons, effectively synergizing with mitochondrial and lysosomal dysfunction to trigger cell death (Fig. 2). New strategies for visualization of oligomeric forms of α-SYN (Roberts et al., 2015) should allow this possibility to be tested.
Another factor that has long been hypothesized to put SNc neurons specifically at risk is DA (Sulzer, 2007; Zucca et al., 2017). Elevated cytosolic Ca2+, α-SYN, and DA in SNc DA neurons could be a particularly toxic combination, especially in axon terminals and dendrites (Mosharov et al., 2009; Dryanovski et al., 2013; Caraveo et al., 2014; Brimblecombe et al., 2015). Indeed, striatal DA axon terminals appear to be lost early in the development of PD, preceding the loss of DA cell bodies (Kordower et al., 2013). In this regard, the inference that levodopa therapy does not accelerate disease progression (Fahn, 2005) might be wrong if the primary site of DA toxicity is the axon terminal, terminals that are largely gone by the time levodopa therapy is usually started.
If cell-autonomous factors are critical to the evolution of PD, then “normalizing” one or more of these factors should slow disease progression. As outlined above, Ca2+ entry through Cav1 Ca2+ channels appears to be a major driver of mitochondrial oxidant stress in all of the at-risk neurons examined to date. Moreover, these channels can be targeted. Dihydropyridines are FDA-approved, selective negative allosteric modulators of Cav1 channels that have good brain bioavailability (Striessnig et al., 1998; Anekonda et al., 2011; Surmeier et al., 2017a). Because dihydropyridines are voltage-dependent negative allosteric modulators that bind to and inhibit channels only when the plasma membrane is depolarized for sustained periods of time (as in pacemaking neurons), they should effectively blunt Ca2+ entry only in a small subset of healthy neurons, precisely the pacemaking neurons at risk in PD. Moreover, at FDA-approved doses, the inhibition of Cav1 channels is decidedly partial (Ilijic et al., 2011). Epidemiological studies have consistently found that the use of dihydropyridines is associated with a decreased risk of developing PD (Becker et al., 2008; Ritz et al., 2010; Pasternak et al., 2012; Lee et al., 2014; Gudala et al., 2015); their use even seems to slow progression after diagnosis (Marras et al., 2012). The combination of preclinical and clinical data implicating Cav1 channels in PD pathogenesis motivated the National Institutes of Health to mount a 5 year, Phase III, disease modification clinical trial in early stage PD patients with the dihydropyridine isradipine that will be completed in 2018.
In conclusion, the prevailing view of PD etiology is that LP spreads in the brain through synaptically coupled networks, driving cell death, and clinical manifestations. However, the distribution of pathology in PD brains and recent connectomics are not consistent with this simple model. Moreover, the relationship between LP, neuronal dysfunction, and death remains uncertain. If LP spreads trans-synaptically in PD, the processes must be gated by cell- or region-autonomous mechanisms. Indeed, at-risk neurons appear to share a set of traits that would not only make them more vulnerable to α-SYN pathology, but would make them more vulnerable to age, as well as toxins and genetic mutations associated with the disease. Although the relative roles of neuronal design and propagated pathology in the etiology of PD remain to be determined, it is clear that both factors need to be considered.
Footnotes
This work was supported by National Institutes of Health Grant NS047085 and the JPB and IDP Foundations to D.J.S. G.M.H. is a National Health and Medical Research Council of Australia Senior Principal Research Fellow (Grant 1079679). J.A.O. was supported by Plan Nacional, Ministerio de Economía y Competitividad Grants SAF2012-40216 and SAF 2015-67239-P.
We thank Heidi Cartwright for help with the figures.
The authors declare no competing financial interests.
References
- Ahn TB, Langston JW, Aachi VR, Dickson DW (2012) Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson's disease. Am J Neurodegener Dis 1:49–59. [PMC free article] [PubMed] [Google Scholar]
- Anderegg A, Poulin JF, Awatramani R (2015) Molecular heterogeneity of midbrain dopaminergic neurons: moving toward single cell resolution. FEBS Lett 589:3714–3726. 10.1016/j.febslet.2015.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL (2007) Disruption of large-scale brain systems in advanced aging. Neuron 56:924–935. 10.1016/j.neuron.2007.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL (2011) L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer's disease. Neurobiol Dis 41:62–70. 10.1016/j.nbd.2010.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge O, Daniel SE, Pearce RK (1997) Neuronal loss and plasticity in the supraoptic nucleus in Parkinson's disease. Neurology 49:610–613. 10.1212/WNL.49.2.610 [DOI] [PubMed] [Google Scholar]
- Balaban RS. (2009) The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta 1787:1334–1341. 10.1016/j.bbabio.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. 10.1007/s00401-009-0538-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8:451–465. 10.1038/nrn2148 [DOI] [PubMed] [Google Scholar]
- Becker C, Jick SS, Meier CR (2008) Use of antihypertensives and the risk of Parkinson disease. Neurology 70:1438–1444. 10.1212/01.wnl.0000303818.38960.44 [DOI] [PubMed] [Google Scholar]
- Beilina A, Cookson MR (2016) Genes associated with Parkinson's disease: regulation of autophagy and beyond. J Neurochem 139 [Suppl 1]:91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. 10.1038/ng1769 [DOI] [PubMed] [Google Scholar]
- Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, Elstner M, Turnbull DM, Klopstock T (2008) Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol 255:1231–1235. 10.1007/s00415-008-0892-9 [DOI] [PubMed] [Google Scholar]
- Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, Goetz CG, Halliday GM, Hardy J, Lang AE, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2014) Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson's disease. Mov Disord 29:454–462. 10.1002/mds.25844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord 27:1478–1483. 10.1002/mds.25135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K (2009) Neuroanatomy and pathology of sporadic Parkinson's disease. Adv Anat Embryol Cell Biol 201:1–119. [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. 10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 318:121–134. 10.1007/s00441-004-0956-9 [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. 10.1038/nrn3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, Zhang J, Roussarie JP, Alvarez MJ, Califano A, Przedborski S, Greengard P (2015) Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat Neurosci 18:1325–1333. 10.1038/nn.4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, Gracie CJ, Platt NJ, Cragg SJ (2015) Gating of dopamine transmission by calcium and axonal N-, Q-, T- and L-type voltage-gated calcium channels differs between striatal domains. J Physiol 593:929–946. 10.1113/jphysiol.2014.285890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Calì T, Ottolini D, Carafoli E (2014) Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci 71:2787–2814. 10.1007/s00018-013-1550-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG (1998) Mitochondria in the life and death of neurons. Essays Biochem 33:43–52. 10.1042/bse0330043 [DOI] [PubMed] [Google Scholar]
- Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny M, Soste M, Picotti P, Kim H, Caldwell KA, Caldwell GA, Sulzer D, Yue DT, Lindquist S (2014) Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc Natl Acad Sci U S A 111:E3544–E3552. 10.1073/pnas.1413201111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Astradsson A, Hallett P, Robertson H, Mendez I, Isacson O (2009) Lack of functional relevance of isolated cell damage in transplants of Parkinson's disease patients. J Neurol 256 [Suppl 3]:310–316. [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain 122:1437–1448. 10.1093/brain/122.8.1437 [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) From the Cover: Inclusion formation and neuronal cell death through neuron-to-neuron transmission of -synuclein. Proc Natl Acad Sci U S A 106:13010–13015. 10.1073/pnas.0903691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepenbroek M, Casadei N, Esmer H, Saido TC, Takano J, Kahle PJ, Nixon RA, Rao MV, Melki R, Pieri L, Helling S, Marcus K, Krueger R, Masliah E, Riess O, Nuber S (2014) Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum Mol Genet 23:3975–3989. 10.1093/hmg/ddu112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra AA, Voorn P, Berendse HW, Groenewegen HJ, Rozemuller AJ, van de Berg WD (2014) Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson's disease. Mov Disord 29:1244–1251. 10.1002/mds.25952 [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Moyer JR, Thompson LT (1994) The calcium rationale in aging and Alzheimer's disease: evidence from an animal model of normal aging. Ann N Y Acad Sci 747:382–406. 10.1111/j.1749-6632.1994.tb44424.x [DOI] [PubMed] [Google Scholar]
- Dragicevic E, Schiemann J, Liss B (2015) Dopamine midbrain neurons in health and Parkinson's disease: emerging roles of voltage-gated calcium channels and ATP-sensitive potassium channels. Neuroscience 284C:798–814. 10.1016/j.neuroscience.2014.10.037 [DOI] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ (2013) Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci 33:10154–10164. 10.1523/JNEUROSCI.5311-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT (2007) Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 170:1725–1738. 10.2353/ajpath.2007.061232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger BN, Dickson DW (2010) Cell type specific sequestration of choline acetyltransferase and tyrosine hydroxylase within Lewy bodies. Acta Neuropathol 120:633–639. 10.1007/s00401-010-0739-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. (2005) Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol 252:iv37–iv42. 10.1007/s00415-005-4008-5 [DOI] [PubMed] [Google Scholar]
- Foehring RC, Zhang XF, Lee JC, Callaway JC (2009) Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J Neurophysiol 102:2326–2333. 10.1152/jn.00038.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronczek R, Overeem S, Lee SY, Hegeman IM, van Pelt J, van Duinen SG, Lammers GJ, Swaab DF (2008) Hypocretin (orexin) loss and sleep disturbances in Parkinson's disease. Brain 131:e88. 10.1093/brain/awm222 [DOI] [PubMed] [Google Scholar]
- Gegg ME, Schapira AH (2016) Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol Dis 90:43–50. 10.1016/j.nbd.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, Surmeier DJ (2012) Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci 15:1414–1421. 10.1038/nn.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sintes R, Ledesma MD, Boya P (2016) Lysosomal cell death mechanisms in aging. Ageing Res Rev 32:150–168. 10.1016/j.arr.2016.02.009 [DOI] [PubMed] [Google Scholar]
- Greffard S, Verny M, Bonnet AM, Seilhean D, Hauw JJ, Duyckaerts C (2010) A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol Aging 31:99–103. 10.1016/j.neurobiolaging.2008.03.015 [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Schon EA, Przedborski S (2015) A new role for α-synuclein in Parkinson's disease: alteration of ER-mitochondrial communication. Mov Disord 30:1026–1033. 10.1002/mds.26239 [DOI] [PubMed] [Google Scholar]
- Gudala K, Kanukula R, Bansal D (2015) Reduced risk of Parkinson's disease in users of calcium channel blockers: a meta-analysis. Int J Chronic Dis 2015:697404. 10.1155/2015/697404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Dawson VL, Dawson TM (2008) What causes cell death in Parkinson's disease? Ann Neurol 64:S3–S15. 10.1002/ana.21573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468:696–700. 10.1038/nature09536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D (2014) Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain 137:2493–2508. 10.1093/brain/awu193 [DOI] [PubMed] [Google Scholar]
- Halliday GM, Blumbergs PC, Cotton RG, Blessing WW, Geffen LB (1990a) Loss of brainstem serotonin- and substance P-containing neurons in Parkinson's disease. Brain Res 510:104–107. 10.1016/0006-8993(90)90733-R [DOI] [PubMed] [Google Scholar]
- Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW, Geffen LB (1990b) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol 27:373–385. 10.1002/ana.410270405 [DOI] [PubMed] [Google Scholar]
- Halliday GM, McRitchie DA, Cartwright H, Pamphlett R, Hely MA, Morris JG (1996) Midbrain neuropathology in idiopathic Parkinson's disease and diffuse Lewy body disease. J Clin Neurosci 3:52–60. 10.1016/S0967-5868(96)90083-1 [DOI] [PubMed] [Google Scholar]
- Halliday GM, Song YJ, Harding AJ (2011) Striatal β-amyloid in dementia with Lewy bodies but not Parkinson's disease. J Neural Transm 118:713–719. 10.1007/s00702-011-0641-6 [DOI] [PubMed] [Google Scholar]
- Halliday G, McCann H, Shepherd C (2012) Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson's disease? Expert Rev Neurother 12:673–686. 10.1586/ern.12.47 [DOI] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P (2007) Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci 30:357–364. 10.1016/j.tins.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725. 10.1172/JCI43366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Stimson E, Henderson JM, Halliday GM (2002) Clinical correlates of selective pathology in the amygdala of patients with Parkinson's disease. Brain 125:2431–2445. 10.1093/brain/awf251 [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. 10.1111/j.1365-2990.2007.00874.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwig M, Klinkenberg M, Rusconi R, Musgrove RE, Majbour NK, El-Agnaf OM, Ulusoy A, Di Monte DA (2016) Brain propagation of transduced α-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 139:856–870. 10.1093/brain/awv376 [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM (2000) Degeneration of the centré median-parafascicular complex in Parkinson's disease. Ann Neurol 47:345–352. 10.1002/1531-8249(200003)47:3%3C345::AID-ANA10%3E3.0.CO%3B2-V [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. (2002) Dopamine miracle: from brain homogenate to dopamine replacement. Mov Disord 17:501–508. 10.1002/mds.10115 [DOI] [PubMed] [Google Scholar]
- Hunn BH, Cragg SJ, Bolam JP, Spillantini MG, Wade-Martins R (2015) Impaired intracellular trafficking defines early Parkinson's disease. Trends Neurosci 38:178–188. 10.1016/j.tins.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley MJ, Brandon B, Gentleman SM, Dexter DT (2013) Parkinson's disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 136:2077–2097. 10.1093/brain/awt134 [DOI] [PubMed] [Google Scholar]
- Hurley MJ, Gentleman SM, Dexter DT (2014) Calcium CaV1 channel subtype mRNA expression in Parkinson's disease examined by in situ hybridization. J Mol Neurosci 55:1–10. 10.1007/s12031-014-0410-8 [DOI] [PubMed] [Google Scholar]
- Iacono D, Geraci-Erck M, Rabin ML, Adler CH, Serrano G, Beach TG, Kurlan R (2015) Parkinson disease and incidental Lewy body disease: just a question of time? Neurology 85:1670–1679. 10.1212/WNL.0000000000002102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 43:364–371. 10.1016/j.nbd.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M. (2016) Alpha-synuclein oligomers-neurotoxic molecules in Parkinson's disease and other Lewy body disorders. Front Neurosci 10:408. 10.3389/fnins.2016.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. (2009a) Formation and development of Lewy pathology: a critical update. J Neurol 256:270–279. 10.1007/s00415-009-5243-y [DOI] [PubMed] [Google Scholar]
- Jellinger KA. (2009b) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740. 10.1016/j.bbadis.2008.07.006 [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease: a critical analysis of α-synuclein staging. Neuropathol Appl Neurobiol 34:284–295. 10.1111/j.1365-2990.2007.00923.x [DOI] [PubMed] [Google Scholar]
- Kingsbury AE, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, Maeir H, Poewe W, Lees AJ (2010) Brain stem pathology in Parkinson's disease: an evaluation of the Braak staging model. Mov Disord 25:2508–2515. 10.1002/mds.23305 [DOI] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A (2003) Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci U S A 100:2884–2889. 10.1073/pnas.0536383100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JH, Mure H, Tang CC, Ma Y, Dhawan V, Spetsieris P, Eidelberg D (2013) Parkinson's disease: increased motor network activity in the absence of movement. J Neurosci 33:4540–4549. 10.1523/JNEUROSCI.5024-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504–506. 10.1038/nm1747 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136:2419–2431. 10.1093/brain/awt192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka K, Yoshimura M, Ikeda K, Budka H (1984) Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree—a new disease? Clin Neuropathol 3:185–192. [PubMed] [Google Scholar]
- Kremer HP, Bots GT (1993) Lewy bodies in the lateral hypothalamus: do they imply neuronal loss? Mov Disord 8:315–320. 10.1002/mds.870080310 [DOI] [PubMed] [Google Scholar]
- Kumaran R, Cookson MR (2015) Pathways to Parkinsonism redux: convergent pathobiological mechanisms in genetics of Parkinson's disease. Hum Mol Genet 24:R32–R44. 10.1093/hmg/ddv236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P (2011) Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson's disease. J Parkinsons Dis 1:83–92. 10.3233/JPD-2011-11004 [DOI] [PubMed] [Google Scholar]
- Lee YC, Lin CH, Wu RM, Lin JW, Chang CH, Lai MS (2014) Antihypertensive agents and risk of Parkinson's disease: a nationwide cohort study. PLoS ONE 9:e98961. 10.1371/journal.pone.0098961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14:501–503. 10.1038/nm1746 [DOI] [PubMed] [Google Scholar]
- Li W, Englund E, Widner H, Mattsson B, van Westen D, Lätt J, Rehncrona S, Brundin P, Björklund A, Lindvall O, Li JY (2016) Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A 113:6544–6549. 10.1073/pnas.1605245113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MK, Farrer MJ (2014) Genetics and genomics of Parkinson's disease. Genome Med 6:48. 10.1186/gm566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J (2015) The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 593:3447–3462. 10.1113/JP270254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald V, Halliday GM (2002) Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson's disease. Mov Disord 17:1166–1173. 10.1002/mds.10258 [DOI] [PubMed] [Google Scholar]
- Maingay M, Romero-Ramos M, Carta M, Kirik D (2006) Ventral tegmental area dopamine neurons are resistant to human mutant alpha-synuclein overexpression. Neurobiol Dis 23:522–532. 10.1016/j.nbd.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Marras C, Gruneir A, Rochon P, Wang X, Anderson G, Brotchie J, Bell CM, Fox S, Austin PC (2012) Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann Neurol 71:362–369. 10.1002/ana.22616 [DOI] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M (2013) Prion-like spreading of pathological α-synuclein in brain. Brain 136:1128–1138. 10.1093/brain/awt037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschke LA, Bertoune M, Roeper J, Snutch TP, Oertel WH, Rinné S, Decher N (2015) A concerted action of L- and T-type Ca(2+) channels regulates locus coeruleus pacemaking. Mol Cell Neurosci 68:293–302. 10.1016/j.mcn.2015.08.012 [DOI] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci 29:444–453. 10.1523/JNEUROSCI.4029-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Cookson MR (2012) Mitochondrial quality control and dynamics in Parkinson's disease. Antioxid Redox Signal 16:869–882. 10.1089/ars.2011.4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez I, Viñuela A, Astradsson A, Mukhida K, Hallett P, Robertson H, Tierney T, Holness R, Dagher A, Trojanowski JQ, Isacson O (2008) Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat Med 14:507–509. 10.1038/nm1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, Duda JE (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79:2307–2314. 10.1212/WNL.0b013e318278fe32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Paladini CA (2011) Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience 198:95–111. 10.1016/j.neuroscience.2011.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D (2009) Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62:218–229. 10.1016/j.neuron.2009.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullin S, Schapira A (2013) α-Synuclein and mitochondrial dysfunction in Parkinson's disease. Mol Neurobiol 47:587–597. 10.1007/s12035-013-8394-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullin S, Schapira A (2015) The genetics of Parkinson's disease. Br Med Bull 114:39–52. 10.1093/bmb/ldv022 [DOI] [PubMed] [Google Scholar]
- Nagley P, Higgins GC, Atkin JD, Beart PM (2010) Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta 1802:167–185. 10.1016/j.bbadis.2009.09.004 [DOI] [PubMed] [Google Scholar]
- Nedergaard S, Flatman JA, Engberg I (1993) Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 466:727–747. 10.1113/jphysiol.1993.sp019742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa SK, Cohen JY, Hwang D, Uchida N, Watabe-Uchida M (2014) Organization of monosynaptic inputs to the serotonin and dopamine neuromodulatory systems. Cell Rep 8:1105–1118. 10.1016/j.celrep.2014.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Brundin P (2013) Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov Disord 28:31–40. 10.1002/mds.25373 [DOI] [PubMed] [Google Scholar]
- Pacelli C, Giguère N, Bourque MJ, Lévesque M, Slack RS, Trudeau LÉ (2015) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25:2349–2360. 10.1016/j.cub.2015.07.050 [DOI] [PubMed] [Google Scholar]
- Parent M, Parent A (2006) Relationship between axonal collateralization and neuronal degeneration in basal ganglia. J Neural Transm Suppl 70:85–88. 10.1007/978-3-211-45295-0_14 [DOI] [PubMed] [Google Scholar]
- Parkkinen L, O'Sullivan SS, Collins C, Petrie A, Holton JL, Revesz T, Lees AJ (2011) Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson's disease. J Parkinsons Dis 1:277–286. 10.3233/JPD-2011-11046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A (2012) Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol 175:627–635. 10.1093/aje/kwr362 [DOI] [PubMed] [Google Scholar]
- Pedersen KM, Marner L, Pakkenberg H, Pakkenberg B (2005) No global loss of neocortical neurons in Parkinson's disease: a quantitative stereological study. Mov Disord 20:164–171. 10.1002/mds.20289 [DOI] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 30:1591–1601. 10.1002/mds.26424 [DOI] [PubMed] [Google Scholar]
- Poulin JF, Zou J, Drouin-Ouellet J, Kim KY, Cicchetti F, Awatramani RB (2014) Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep 9:930–943. 10.1016/j.celrep.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27:645–656. 10.1523/JNEUROSCI.4341-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzier I, Kullmann PH, Horn JP, Levitan ES (2009) Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci 29:15414–15419. 10.1523/JNEUROSCI.4742-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rcom-H'cheo-Gauthier A, Goodwin J, Pountney DL (2014) Interactions between calcium and alpha-synuclein in neurodegeneration. Biomolecules 4:795–811. 10.3390/biom4030795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. 10.1002/ana.24066 [DOI] [PubMed] [Google Scholar]
- Reeve A, Simcox E, Turnbull D (2014) Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev 14:19–30. 10.1016/j.arr.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. 213:1759–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S (2010) L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 67:600–606. 10.1002/ana.21937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain. Brain 138:1642–1657. 10.1093/brain/awv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Ayers JI, Brooks MM, Chakrabarty P, Hudson VJ 3rd, Howard JK, Golde TE, Giasson BI, Borchelt DR (2016) Non-prion-type transmission in A53T α-synuclein transgenic mice: a normal component of spinal homogenates from naive non-transgenic mice induces robust α-synuclein pathology. Acta Neuropathol 131:151–154. 10.1007/s00401-015-1505-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, Schieber S, Oertel W, Wokosin D, Schumacker PT, Surmeier DJ (2014) Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci 17:832–840. 10.1038/nn.3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, Luo L (2015) Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524:88–92. 10.1038/nature14600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H (1998) Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol Sci 19:108–115. 10.1016/S0165-6147(98)01171-7 [DOI] [PubMed] [Google Scholar]
- Sulzer D. (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci 30:244–250. 10.1016/j.tins.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Sulzer D, Surmeier DJ (2013) Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord 28:41–50. 10.1002/mds.25095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT (2012) Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb Perspect Med 2:a009290. 10.1101/cshperspect.a009290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E (2017a) Calcium and Parkinson's disease. 483:1013–1019. 10.1016/j.bbrc.2016.08.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM (2017b) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113. 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Lai YY, Siegel JM (2007) Hypocretin (orexin) cell loss in Parkinson's disease. Brain 130:1586–1595. 10.1093/brain/awm097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell 6:307–317. 10.1111/j.1474-9726.2007.00295.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, Giasson BI (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Ata Neuropathol 131:49–73. 10.1007/s00401-015-1485-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Di Monte DA (2013) Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 5:1119–1127. 10.1002/emmm.201302475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venda LL, Cragg SJ, Buchman VL, Wade-Martins R (2010) α-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci 33:559–568. 10.1016/j.tins.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Lee VM (2014) Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9:2135–2146. 10.1038/nprot.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ (2001) DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem 79:266–277. 10.1046/j.1471-4159.2001.00548.x [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Hansen LA, Masliah E (1995) Cortical Lewy body-containing neurons are pyramidal cells: laser confocal imaging of double-immunolabeled sections with anti-ubiquitin and SMI32. Acta Neuropathol 89:404–408. 10.1007/BF00307643 [DOI] [PubMed] [Google Scholar]
- Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM (2010) Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc Natl Acad Sci U S A 107:21848–21853. 10.1073/pnas.1011756107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ (2016) A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci 17:251–260. 10.1038/nrn.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N (2012) Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74:858–873. 10.1016/j.neuron.2012.03.017 [DOI] [PubMed] [Google Scholar]
- Wolfart J, Roeper J (2002) Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci 22:3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811. 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA (2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53:590–605. 10.1002/neu.10150 [DOI] [PubMed] [Google Scholar]
- Zucca FA, Segura-Aguilar J, Ferrari E, Muñoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2017) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol 155:96–119. 10.1016/j.pneurobio.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]