

Abstract
In Parkinson's disease, intracellular α-synuclein inclusions form in neurons. We suggest that prion-like behavior of α-synuclein is a key component in Parkinson's disease pathogenesis. Although multiple molecular changes are involved in the triggering of the disease process, we propose that neuron-to-neuron transfer is a crucial event that is essential for Lewy pathology to spread from one brain region to another. In this review, we describe key findings in human postmortem brains, cultured cells, and animal models of disease that support the idea that α-synuclein can act as a prion. We consider potential triggers of the α-synuclein misfolding and why the aggregates escape cellular degradation under disease conditions. We also discuss whether different strains of α-synuclein fibrils can underlie differences in cellular and regional distribution of aggregates in different synucleinopathies. Our conclusion is that α-synuclein probably acts as a prion in human diseases, and a deeper understanding of this step in the pathogenesis of Parkinson's disease can facilitate the development of disease-modifying therapies in the future.
Dual Perspectives Companion Paper: Parkinson's Disease Is Not Simply a Prion Disorder, by D. James Surmeier, José A. Obeso, and Glenda M. Halliday
Keywords: alpha-synuclein, Lewy body, neurodegenerative disease, propagation, seeding
Introduction
In this brief commentary, we argue the case that prion-like behavior of α-synuclein (α-SYN) plays an important role in the pathogenesis of Parkinson's disease (PD). While we are certain that prion-like mechanisms are crucial, we are also convinced that multiple molecular events play important roles in the pathogenesis of PD and related α-synucleinopathies. Thus, we do not believe that PD pathogenesis is a question of “one mechanism or another.” Instead, we propose that a network of complex and interdependent molecular events play roles in the pathobiology of PD. We also think that the apparent significant heterogeneity between patients regarding clinical features and rate of progression of PD is reflected in individual differences in underlying disease triggers and pathogenetic mechanisms. The identification of causative mutant genes that cause rare, inherited forms of PD (5%–10% of all cases) has led to important insights into diverse molecular pathways that underpin the inherited disease forms. In addition, the facts that increasing age is the greatest single risk factor for sporadic PD, and that certain epidemiological and lifestyle factors modify disease risk, have provided clues to potential triggers and pathogenetic mechanisms underlying idiopathic PD. Together, based on lessons from genetic and sporadic PD, the emerging picture of PD pathogenesis includes protein misfolding, disrupted protein handling, mitochondrial dysfunction, oxidative stress, impaired calcium handling, and inflammation. In this commentary, we will not provide details regarding the contribution of each of these important mechanisms to PD, as they have been reviewed extensively previously (Dias et al., 2013; Rcom-H'cheo-Gauthier et al., 2014; Allen Reish and Standaert, 2015; Bose and Beal, 2016; Ransohoff, 2016; Surmeier et al., 2017b; Wong and Krainc, 2017). Instead, we describe recent advances supporting the idea that a prion-like mechanism operates in PD. Specifically, we propose that propagation of α-SYN pathology from one neuron to another plays an important role in the progressive worsening of symptoms and the gradual involvement of additional brain and autonomic functions as the disease advances. We also suggest that diversity in the molecular structure of α-SYN aggregates, and consequently their capacity to interact with different ensembles of partner proteins and to propagate between different brain regions and in different patients might explain why all PD patients do not follow the same disease course, and could also explain why other synucleinopathies (e.g., Dementia with Lewy bodies [DLB] and multiple system atrophy [MSA]) take a different course to PD. In this commentary, we present evidence to support these views.
What is the prion-like model for PD?
Why did it take so long to embrace the idea that the propagation of protein aggregates is important in PD pathogenesis? Textbooks still classify PD as a movement disorder, and the most conspicuous signs and symptoms of PD involve akinesia, rigidity, and tremor. Most of these motor symptoms are due to reduced striatal dopamine, as a consequence of progressive degeneration of substantia nigra dopaminergic neurons, and dopaminergic drug therapies are initially effective at alleviating these symptoms. Because of the central role of the dopamine deficit in the most conspicuous PD symptoms, most research into underlying pathogenic mechanisms over the past 50 years has focused on the dopamine neurons and their putative selective vulnerability. However, patients also experience a wide range of troublesome nonmotor symptoms (e.g., depression, sleep disorder, hyposmia, constipation, fatigue, cognitive decline), and many of these are not linked to the reduction in striatal dopamine and therefore do not respond well to dopaminergic drug therapies (Chaudhuri and Odin, 2010).
Over one hundred years ago, Fritz Heinrich Lewy described intraneuronal hyaline inclusions in cell bodies and neurites in PD, and he mentioned that they could be found in numerous brain regions (Goedert et al., 2013). This revelation, however, did not distract the attention from nigrostriatal degeneration for the 80 years that followed. The seminal discovery in 1997 that the main protein constituent of Lewy pathology (LP) is misfolded α-SYN started a paradigm shift in our thinking about pathogenic mechanisms in PD and other α-synucleinopathies (Spillantini et al., 1997). Using the new tool of α-SYN immunohistochemistry, Braak et al. (2003) described that α-SYN aggregates were not just present in widespread brain areas, but also in peripheral nerves of the autonomic nervous system. They eventually classified six neuropathological stages of PD, with increasing numbers of brain regions exhibiting α-SYN pathology. Taking into consideration that the brain regions that successively exhibited α-SYN aggregates were connected by neural pathways, they proposed that a model where a “causative agent” (e.g., a neurotropic virus) initially affected the olfactory bulb and autonomic nerves innervating the gut, and then progressively involved additional brain regions leading to the gradual spread of α-SYN pathology (Hawkes et al., 2007). It was unclear, and still remains elusive, what the reason is for the disease process possibly being triggered in the olfactory bulb and gut. While this so-called Braak neuropathological staging system for PD is not fully embraced by everyone, and has been suggested to apply to only a subset of patients (Beach et al., 2009), it still had a major impact on PD research. The serendipitous findings in 2008 of LP in fetal neurons grafted to 3 PD patients 11–16 years before their death also significantly influenced the field and dramatically pivoted the research into a new direction (Kordower et al., 2008; Li et al., 2008). Thus, a provocative interpretation of these findings was that α-SYN aggregates had moved from affected cells in the host brain to the grafted neurons and seeded aggregation of endogenous protein in these otherwise healthy and young neurons (Brundin et al., 2008, 2010). What has followed in the wake of the findings in the transplanted patients and the development of the Braak neuropathological staging is an avalanche of studies in experimental models, which we briefly describe in the following sections. Specifically, we briefly review experiments conducted in cell cultures and animal models and describe how they support the idea that a prion-like mechanism is active. We further debate whether there exist different “strains” of α-SYN aggregates that might explain differences between synucleinopathies that occur in humans. We also highlight that the anatomical site (e.g., olfactory system or gut) of the first α-SYN misfolding event can differ between patients, and that the initial triggering event (e.g., genetic predisposition combined with aging, externally induced inflammation, or environmental toxin) is not identical in all patients.
Evidence for cell-to-cell transfer of α-SYN obtained in cell culture
A key premise for the hypothesis that α-SYN acts as a prion-like protein in PD is that α-SYN assemblies, which can act as seeds for further aggregation, can be taken up by neurons, then undergo transport along axons, and finally transfer to another neuron (e.g., by being released into the extracellular space). Numerous cell culture studies have addressed different aspects of this complex series of events (Lee et al., 2008a; Danzer et al., 2009; Desplats et al., 2009; Luk et al., 2009; Hansen et al., 2011; Volpicelli-Daley et al., 2011; Freundt et al., 2012; Bousset et al., 2013; Aulić et al., 2014; Reyes et al., 2015). For example, it has been demonstrated repeatedly that the key α-SYN assemblies can bind to the surfaces of a variety of cultured cells, ranging from cell lines to primary neurons. The binding of assemblies to the cell surface, which can occur both if they are free in solution or associated within extracellular vesicles, is a key step for subsequent events. Heparan sulfate proteoglycans are reported to bind α-SYN assemblies and facilitate uptake via endocytosis (Holmes et al., 2013). Naked α-SYN assemblies also interact with membranous proteins. Some of these were recently identified (Shrivastava et al., 2015; Mao et al., 2016) and, in some cases, are reported to promote the uptake of α-SYN from the extracellular space (Mao et al., 2016). After binding to membranes (e.g., via the specific membrane proteins), naked α-SYN assemblies are taken up by neurons and are directed, at least in part, to the lysosomal compartment (Lee et al., 2008a). They can also undergo anterograde and retrograde transport, which has been monitored in primary neurons grown in microfluidic devices (Freundt et al., 2012; Tran et al., 2014; Brahic et al., 2016), and some of the movement of α-SYN assemblies occurs at a velocity consistent with fast axonal transport. As a crucial step for the prion hypothesis for PD, intraneuronal α-SYN assemblies can be exported into the extracellular space and then be taken up by neighboring neurons (Hansen et al., 2011; Freundt et al., 2012; Tran et al., 2014; Reyes et al., 2015), microglia (Lee et al., 2008b), or astrocytes (Lee et al., 2010). If the lysosomal or proteosomal systems are inhibited, the excretion of α-SYN increases (Lee et al., 2005, 2013; Alvarez-Erviti et al., 2011; Fernandes et al., 2016), and some of the exported α-SYN is associated with microvesicles that can display markers found on exosomes (Emmanouilidou et al., 2010; Danzer et al., 2012). When α-SYN assemblies are encapsulated within extracellular vesicles, export is dependent on the cell packaging machinery and uptake is certainly the consequence of membrane fusion events (Martens and McMahon, 2008; Traub, 2009). Cell contacts are not required for the passage of α-SYN aggregates from cell to cell (Brahic et al., 2016). Nonetheless, recent evidence suggests that tunneling nanotubes can act as conduits for α-SYN assemblies that transfer from one cell to another (Abounit et al., 2016).
Once inside the naive cell, α-SYN assemblies derived from the extracellular space can reach the cytosol of neurons where they amplify by triggering the aggregation of endogenous cytosolic α-SYN, through a yet unknown process. It is discussed how α-SYN that is enclosed inside an endosome can gain access to the cytosol. Although this debate is not resolved, one suggestion is that α-SYN might penetrate and lyse the lysosomal membrane in a fashion akin to that used by viral proteins (Freeman et al., 2013). The accumulation of endogenous α-SYN following exposure of cells to exogenous fibrils, as has been demonstrated by many independent groups, is unlikely to be simply due to perturbed cellular proteostasis. Indeed, not only do exogenous fluorescently labeled α-SYN fibrils colocalize with newly aggregated endogenous α-SYN, but structurally distinct α-SYN assemblies imprint their intrinsic structural characteristics on the endogenous α-SYN (Danzer et al., 2009; Desplats et al., 2009; Hansen et al., 2011; Volpicelli-Daley et al., 2011; Bousset et al., 2013). This strongly suggests that endogenous α-SYN aggregates through a seeded process where the imported α-SYN acts as a template. The α-SYN molecules in one given conformation within the α-SYN fibrils that are derived from the extracellular space recruit monomers of α-SYN that are produced endogenously by the cell (Bousset et al., 2013). The intrinsic structure of the seeds is preserved by structurally well-defined and specific longitudinal and lateral interactions between newly recruited α-SYN monomers and the ends of the seeds (Fig. 1). Together, we think the evidence is overwhelming that α-SYN can be taken up from the extracellular space by neurons and seed aggregation, as well as undergo long-distance axonal transport. Further, we think it has been clearly demonstrated that neurons can excrete misfolded α-SYN, allowing the vicious cycle to be repeated in a new set of neurons. Therefore, some of the fundamental processes required for the prion hypothesis are evident in the culture dish of a laboratory. The next question is: can we demonstrate in laboratory animals that α-SYN can spread from one neuron to another, trigger misfolding, and start a cascade of spreading neuropathology in brain regions that are distant from each other? This is the topic of the next section.
Figure 1.
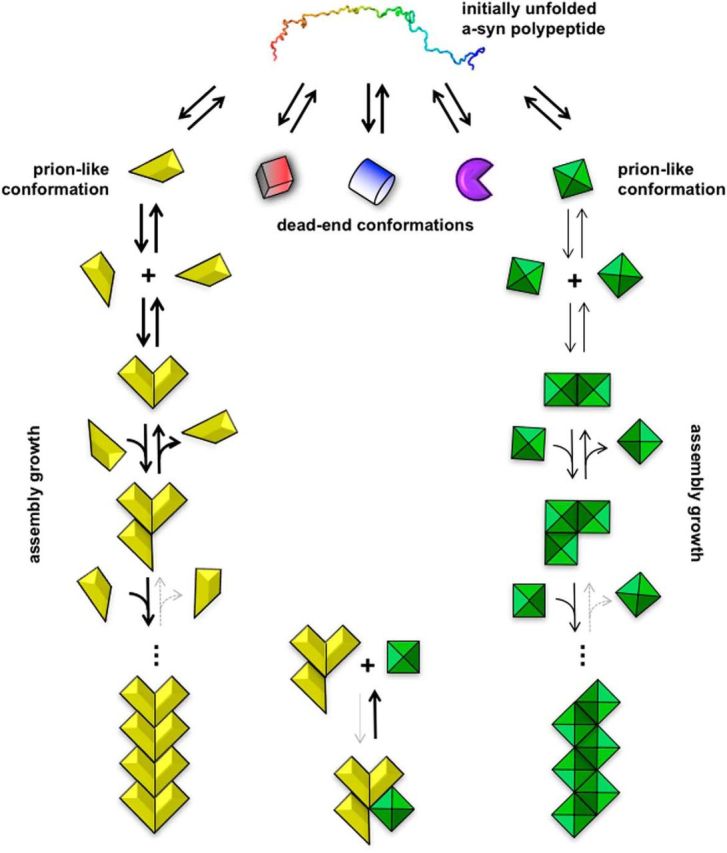
Mechanism of pathologic protein assembly and the notion of strains. Natively unfolded monomeric α-SYN multicolor molecule (top) populates different folding intermediates that expose specific amino acid stretches that determine their ability to establish defined sets of intermolecular interactions. This is schematized by the different shapes (folding intermediates) and colors (exposed amino acid stretches) of the folding intermediates (second row). The molecule in the conformation in yellow is capable of establishing longitudinal and lateral interactions with molecules in the same conformation. As long as only longitudinal or lateral interactions are established, the assemblies are transient. This is illustrated schematically by arrows, of the same size, in two directions. When both longitudinal and lateral interactions are established, a thermodynamically stable assembly is generated. This assembly can grow indefinitely by incorporation of molecules in the same conformation. This is illustrated schematically by a solid arrow pointing toward growth. Molecules constituting such an assembly can dissociate only from its ends; this is represented by a dashed arrow pointing toward disassembly. The molecule in the green conformation behaves in a similar manner, yielding fibrils made of green diamonds. The other conformers (red, blue, and purple) cannot establish thermodynamically stable intermolecular interactions. They do not yield assemblies. These folds can be considered as dead-end conformations. The intermolecular interactions the conformation in green is capable of establishing upon docking to an assembly made of yellow conformers (bottom), and vice versa, are unstable as they do not outweigh the entropic cost of binding. Thus, no mixed assemblies made of yellow and green conformers can form. The yellow and green assemblies expose distinct amino acid stretches that define among other things their interactomes, resistance to cellular clearance, tropism for different cell types in the nervous system, and the pathology they cause. The intermolecular interactions within the yellow and green assemblies and their surfaces that define their interactomes are at the origin of the notion of strains.
Animal models of α-SYN pathology propagation
Experiments in several types of animal models have also demonstrated that α-SYN can transfer between neurons, and also from neurons to glial cells, in the adult brain (Rey et al., 2016a). The animal models used first were based on experimental cell transplantation paradigms that mimicked the clinical transplants, which originally had triggered interest in the prion hypothesis for PD (Desplats et al., 2009; Hansen et al., 2011; Kordower et al., 2011; Angot et al., 2012). While these experiments were an important proof of principle, they did not allow for a demonstration of long-distance propagation of pathology between distant brain regions, which is a key element of the prion hypothesis for PD. Instead, this was shown in subsequent experiments in rodents and nonhuman primates using intracerebral injections of one of three sources of misfolded α-SYN. These injections contained the following: (1) brain homogenates from patients with PD or other synucleinopathies, (2) brain tissue from transgenic animal models that had developed α-SYN pathology, or (3) preparations of fibrils generated from recombinant α-SYN (Luk et al., 2012a, b; Mougenot et al., 2012; Rey et al., 2013, 2016b; Sacino et al., 2013, 2014; Recasens et al., 2014; Peelaerts et al., 2015; Shimozawa et al., 2017). For example, injections of preformed α-SYN fibrils into the striatum lead to the generation of α-SYN aggregates, and subsequent death of dopamine neurons akin to what is seen in PD, in the substantia nigra that directly innervates the striatum. Notably, in the initial reports, α-SYN pathology was not observed when injections are made into α-SYN-null mutant mice, suggesting that endogenous α-SYN is required for the amplification and gradual propagation of pathology. Even injections of α-SYN fibrils into muscle or bloodstream can lead to α-SYN aggregates in the brain (Sacino et al., 2014; Peelaerts et al., 2015). Importantly, injections of α-SYN fibrils into either the gut wall, which is innervated by the vagal nerve, or the olfactory bulb leads to pathology in interconnected regions of the nervous system (Rey et al., 2013, 2016a; Holmqvist et al., 2014). This is particularly important in light of the fact that the Braak model posits that the enteric nervous system and the olfactory bulb might be the two sites where α-SYN pathology appears first in PD (Hawkes et al., 2007). When α-SYN is, instead, overexpressed in the medulla oblongata following injection of a viral vector into the vagal nerve leading to retrograde transport of the virus in the nerve, the α-SYN pathology propagates rostrally from the brainstem (Ulusoy et al., 2013). If the integrity of the neuronal circuitry is destroyed by neurodegeneration, the propagation of pathology is interrupted (Helwig et al., 2016). In the model involving injections into the mouse olfactory bulb, α-SYN aggregates slowly spread throughout interconnected brain regions during the year that follows the injection, and the aggregates express multiple markers that are characteristic of Lewy bodies and neurites (Rey et al., 2016b). They gradually involve additional brain regions that are located one or more synapses away from the injection site, and olfactory deficits develop in the mice in parallel (Rey et al., 2016b). Eventually, aggregated α-SYN appears in brainstem nuclei, including the substantia nigra. These observations are thought-provoking because they mimic some of the α-SYN pathology that has been suggested to underlie “prodromal” PD (Rey et al., 2016c). This condition is suggested to last 5–10 years and involves, for example, hyposmia long before the first signs of motor deficits develop (Mahlknecht et al., 2015). Together, the evidence is compelling that exogenously delivered α-SYN (either via injection of fibrils or via overexpression using a viral vector) can trigger aggregation of endogenous α-SYN. The propagation is relatively slow (several months in rodents, which equates to decades in humans), requires the presence of the endogenous α-SYN protein, and appears to follow known anatomical pathways that must remain intact for the process to continue effectively.
Together, we consider that the bulk of evidence from animal studies supports the idea that α-SYN aggregates are transported from one brain region to another along defined neural pathways, and that they then can be propagated to neighboring neurons through trans-synaptic transmission. Additional support for this model is described in the next section, namely, that different specific “strains” of α-SYN fibrils exist and that each type of strain seeds aggregates with defined and shared properties.
Evidence for different strains of α-SYN aggregates
Monomeric α-SYN is considered natively unfolded as it populates a very large ensemble of conformational states that are affected by physical-chemical conditions (pH, viscosity, ionic strength, and nature of ions, etc.) (Uversky, 2003). Each assembly-competent conformational state of α-SYN monomers exposes specific amino acid stretches that determine its ability to establish defined sets of intermolecular interactions. These interactions lead to assemblies that exhibit different intrinsic structures and have distinct amino acid stretches exposed at their surfaces. The intermolecular interactions, lateral and longitudinal, that maintain α-SYN molecules within the assemblies govern the ability of a given α-SYN assembly to grow by incorporating additional monomeric α-SYN molecules in well-defined conformations and in a thermodynamically stable manner. Importantly, the exposed amino acid stretches influence with which partner proteins, receptors, and lipids a given α-SYN assembly will interact. Thus, it is evident that this can result in highly specific (patho)biological properties of a given α-SYN assembly. In essence, the exposed amino acid in a specific type of α-SYN assembly dictates its seeding propensity, resistance to cellular clearance machineries, cytotoxicity, tropism for different cell types in the nervous system, etc. Therefore, the concept of different α-SYN fibril “strains” is very important for our understanding of the molecular pathogenesis of PD and other synucleinopathies.
For the prion protein PrP, aggregation in a given organism yields distinct diseases with characteristic incubation time, brain lesions, and proteolytic cleavage patterns (Poggiolini et al., 2013). Key questions for the prion hypothesis for PD are whether different strains of α-SYN aggregates exist (Melki, 2015) and whether they, at least in part, explain why different synucleinopathies exhibit α-SYN aggregates with dissimilar cellular and anatomical predilections (Halliday et al., 2011; McCann et al., 2014). Indeed, recent important findings show that intracerebral or systemic injections into experimental animals of fractionated brain homogenates from clinical cases with MSA or PD induce distinct neuropathologies (Watts et al., 2013; Prusiner et al., 2015). The resulting pathologies each share some characteristics with the synucleinopathy (MSA vs PD) from which the injected brain homogenate was derived. These findings suggest that specific α-SYN strains with different pathogenic effects exist. The underlying mechanisms can now be addressed by systematic experiments using well-defined α-SYN assemblies. Thus, by varying the experimental conditions, it is possible to generate different types of α-SYN fibrillar assemblies, which exhibit differential toxicity in cell cultures (Bousset et al., 2013) and give rise to distinct forms of neuropathology with features that resemble PD or MSA (Peelaerts et al., 2015). If cell-autonomous factors (oxidative stress, failing protein clearance, energy failure) were the primary reasons that α-SYN aggregates develop in different parts of the brain of an experimental animal or patient, one would not expect that all cells and brain regions would have α-SYN aggregates with the same characteristics. Thus, the findings of “strain-specific” neuropathologies in experimental models add further support to the idea that α-SYN fibrils propagate in a prion-like manner.
Critics of the prion hypothesis argue that neural connections are not the key paths of pathology spread
It has been argued that neural connections are unlikely to be the preferred routes by which pathogenic α-SYN assemblies spread. Critics who instead consider that “cell-autonomous” mechanisms govern which cells develop α-SYN aggregates have pointed out that in postmortem brain examinations the proposed progressive increase in α-SYN pathology in PD does not seem to have followed neural connections (Burke et al., 2008; Surmeier et al., 2017a). Thus, even in advanced PD, there is no pathology in some “nearest neighbors” to nuclei in the brainstem and diencephalon, which do exhibit LP (Surmeier et al., 2017a). While we still argue that neural pathways are major highways for the spreading of aggregation-prone α-SYN assemblies between different brain areas, we acknowledge that several additional factors can be in play. For example, it is possible that certain neuronal connections are more likely to display release of pathogenic α-SYN assemblies depending on their rates of synaptic activity. Neuron-to-neuron spread of tau aggregates has been shown to depend upon neuronal activity; therefore, the propagation of pathology is not necessarily equal between all interconnected brain regions (Wu et al., 2016). Interestingly, postmortem studies show that α-SYN aggregates do not accumulate and form extracellular plaques upon neuronal death. Therefore, upon release from a dying cell into the extracellular space, aggregated α-SYN might be degraded or cleared by one or more of several mechanisms. For example, extracellular metalloproteases can efficiently degrade α-SYN assemblies (E. M. Kim and Hwang, 2011; Pampalakis et al., 2017). Alternatively, extracellular α-SYN might be removed via glymphatics (i.e., a perivascular clearance pathway, which transports, e.g., soluble amyloid-β from brain interstitium) (Iliff et al., 2012). Recent work has shown that α-SYN assemblies exist in CSF of PD patients, opening up for the possibility that aggregation-prone α-SYN spreads between brain regions via the CSF circulation (Shahnawaz et al., 2017). Cerebral vasculature could potentially be an additional path for removal of α-SYN assemblies from the extracellular space and a route for long-distance spread of pathology between brain regions. Animal experiments have shown that injections of α-SYN assemblies into peripheral blood vessels seed α-SYN aggregation in the brain (Peelaerts et al., 2015). Finally, glial cells, which can take up α-SYN from the extracellular space (Lee et al., 2008b, 2010), could constitute another vector for dissemination of α-SYN aggregates. Astroglia and microglia express little or no endogenous α-SYN but might migrate to nearby brain regions, release some of the aggregated protein, which then is taken up by neurons and seeds further aggregation. For tauopathies, it has been proposed that microglia harbor pathological tau, migrate to neighboring brain regions, and release it via exosomes (Asai et al., 2015), but this has not yet been demonstrated for α-SYN. A spreading neuroinflammatory process has also been considered to contribute to the propagation of α-SYN aggregates. Intracerebral injections of an inflammagen can cause post-translational modifications of α-SYN and promote aggregation locally (Gao et al., 2008). However, there is no experimental evidence that spreading neuroinflammation actually can promote progressive dissemination of α-SYN aggregates in the brain. As an alternate explanation for why the progression of neuropathology is similar between patients with the same disease, regional differences in brain levels of proteins that either promote or counteract the accumulation of aggregation-prone proteins have been proposed to explain differential susceptibility of brain regions in Alzheimer's disease (Freer et al., 2016). This concept of “region-autonomous” mechanisms (Surmeier et al., 2017a) is not supported, however, by animal experiments that instead indicate that the pattern of slowly spreading of α-SYN pathology is directly related to the site of α-SYN fibril inoculation and follows a temporal course consistent with propagation along neural tracts, involving trans-synaptic transmission.
What are the initial triggers of α-SYN aggregation and where does it start?
Assuming that cell-to-cell transfer of α-SYN assemblies plays an important role in PD pathogenesis, two important follow-up questions are how and why does this process start? In this section, we discuss these questions.
One can view misfolding and aggregation of α-SYN as a stochastic event that occurs throughout life. Under some cell stress conditions (e.g., toxic insults, local inflammation, oxidative stress), which we discuss further below, α-SYN misfolding is promoted. Normally, neurons clear this “garbage” (i.e., the misfolded α-SYN species), but we suggest that on rare occasions the proteostasis mechanisms fail and then the pathogenic process starts (Xilouri et al., 2013).
How can α-SYN misfold and aggregate under normal conditions? Even if we assume that each of the 140 amino acid residues within the natively unfolded α-SYN can adopt a limited number of conformations, the number of possible conformations α-SYN could adopt would still be immense. For example, if we assume, as a Gedankenexperiment, that the number of conformations for each amino acid is 3 (1 trans and 2 gauche) with 2 torsions each, the number of possible conformations for the 140 amino acids be 3139 × 2, although in equilibrium, the concentration and lifespan of each conformation will be specific to each conformer and dependent on its interaction with the solvent, ions, and partner molecules (e.g., lipids and proteins). Thus, at any time, there is a significant probability that a newly synthesized α-SYN molecule can populate conformers that are capable of establishing well-defined intermolecular interactions with molecules that are in a compatible conformation. As a consequence of this simplified view, α-SYN aggregates form at a relatively low rate in a stochastic manner throughout life (Jarrett and Lansbury, 1993).
As stated above, the thermodynamic stability of such intermolecular interactions depends on the concentration of the assembly competent conformers (Oosawa and Asakura, 1975). Point mutations within the α-SYN encoding gene, SNCA, increase or decrease the number of possible conformations α-SYN adopts, and affect the lifespan and cellular concentration of these conformations. Duplication and triplication of SNCA also affect the lifespan and concentration of assembly competent conformers. This is why certain point mutations and gene duplication/triplication are associated with increased aggregation propensity and early onset of PD-like conditions (Devine et al., 2011). Furthermore, single nucleotide polymorphisms in a distal SNCA enhancer are associated with altered PD risk (Nalls et al., 2014), and experiments in neurons differentiated from induced pluripotent stem cells suggest that very modest changes in α-SYN expression significantly impact life-time PD (i.e., α-SYN aggregation) (Soldner et al., 2016).
Several exogenous factors and genes beyond SNCA can also increase the risk for α-SYN aggregation. For example, certain viral infections have been reported to upregulate α-SYN levels in the brain and could therefore elevate the risk of aggregate formation (Massey and Beckham, 2016). Exposure to Escherichia coli, which generate the extracellular amyloid protein curli, has been suggested to trigger α-SYN accumulation in the gut and brain (Chen et al., 2016). Certain forms of cell stress (e.g., those associated with exposure to pesticides and environmental toxins) also increase α-SYN levels (Manning-Bog et al., 2002; Cicchetti et al., 2009; Kumar et al., 2016). Reduced calcium buffering capacity and raised free calcium have been associated with an induction of α-SYN assemblies (Rcom-H'cheo-Gauthier et al., 2014). Oxidative stress, and other stimuli, can trigger post-translational modifications in α-SYN that affect the propensity for aggregation (Duda et al., 2000; Giasson et al., 2000; Paxinou et al., 2001; Nonaka et al., 2005; Levin et al., 2011; Barrett and Timothy Greenamyre, 2015; Oueslati, 2016). Several studies, including seminal work by Surmeier et al. (2017b), have implicated calcium dyshomeostasis, mitochondrial failure, and oxidative stress in PD, and these observations tie in well with the idea that α-SYN aggregation in a few ailing neurons can trigger a widespread synucleinopathy (Guzman et al., 2010; Goldberg et al., 2012; Surmeier et al., 2017b). Mitochondrial failure and oxidative stress can also be results of rare PD mutations (e.g., parkin, PINK1, DJ-1) that are increasingly linked to mitochondrial function and quality control (Guzman et al., 2010; Truban et al., 2017). Inflammation in the nervous system is considered a potential cause of increased levels of α-SYN and inflammatory mediators also promote undesirable post-translational modifications of α-SYN, both of which are changes that could trigger a cascade of α-SYN pathology propagation (Gao et al., 2008; Hirsch and Hunot, 2009; Lema Tomé et al., 2013; Lim et al., 2016; Ransohoff, 2016). Together, the observations described above are consistent with longstanding ideas from a large body of epidemiological and experimental research literature that has implicated environmental toxins, mitochondrial failure, oxidative stress, and neuroinflammation in the etiology and pathogenesis of PD.
When do pathogenic α-SYN assemblies escape cellular clearance mechanisms?
In the previous section, we discussed conditions that promote the accumulation pathogenic α-SYN assemblies. In most situations, these assemblies will be cleared from the cells through normal proteostatic mechanisms (Chen et al., 2011; Y. E. Kim et al., 2013). Under certain conditions, however, the cellular clearance mechanisms fail and are simply not able to keep up with a high production of pathogenic assemblies (Ciechanover and Kwon, 2015). The roles of molecular chaperones and the Ubiquitin Proteasome System in sensing and maintaining normal cellular proteostasis are widely acknowledged (Gidalevitz et al., 2011; Morimoto, 2011; Brehme et al., 2014). The efficacy of these quality control cellular machineries decreases with increasing age (Kaushik and Cuervo, 2015); thus, aggregates that could have been efficiently dismantled or degraded actually end up escaping in aged cells (Auluck et al., 2002). They further imbalance proteostasis and their clearance. Misfolded α-SYN is largely cleared via the lysosomal autophagy pathway, and with increasing age the efficacy of this clearance system also gradually declines (Gan-Or et al., 2015; D. K. Kim et al., 2016), potentially in part driven by age-induced changes in epigenetic control of lysosomal enzymes (Jin et al., 2016). This is a particularly important observation considering that increasing age is the greatest risk factor for idiopathic PD. Heterozygous mutations in the lysosomal enzyme glucocerebrosidase, encoded by GBA, are associated with a marked increase in PD risk (Sidransky et al., 2009), and single nucleotide polymorphisms near the GBA locus affect PD risk (Nalls et al., 2014; Gan-Or et al., 2015). Collectively, all these findings lend further support for the idea that lysosomes play a pivotal role as protectors against synucleinopathy. A reduction in function of the lysosomal autophagy system could specifically drive the prion behavior of misfolded α-SYN (Chu et al., 2009; D. K. Kim et al., 2016), both by extending the time that the aberrant α-SYN assembly can act as a permissive template in the cytoplasm and by increasing the rate of α-SYN excretion into the extracellular space making it accessible to neighboring neurons. Indeed, experimental inhibition of lysosomes in cultured cells leads to increased excretion of α-SYN to the extracellular medium (Alvarez-Erviti et al., 2011), providing support to the idea that poor lysosomal function promotes cell-to-cell transfer of α-SYN assemblies. Loss-of-function mutations in the membrane protein ATP13A2 are associated with a neurological syndrome that includes juvenile-onset parkinsonism and α-SYN accumulation. Experimental studies suggest that ATP13A2 levels influence the excretion of α-SYN in exosomes (Kong et al., 2014; Tsunemi et al., 2014), which, in turn, could change the dynamics of cell-to-cell transfer of α-SYN assemblies.
The bottom line is that genetic factors and aging can both favor the prion behavior of α-SYN by impairing the clearance of misfolded assemblies and increasing excretion of the protein. It is reasonable to suggest that multiple factors (even within the different domains of, e.g., lysosomal dysfunction, calcium handling, oxidative stress, inflammation) have additive or synergistic effects on the risk for α-SYN aggregation.
The seminal postmortem studies by Braak and colleagues suggest that, in PD, α-SYN aggregates appear in circumscript parts of the nervous system and then gradually spread along neural tracts to other areas as the disease progresses (Braak et al., 2003). Although some PD patients do not appear to follow precisely the six Braak stages of neuropathology (Jellinger, 2009), his work is a cornerstone of the prion hypothesis for PD. Notably, Braak and his team, and other investigators who have followed up his pioneering work, suggests that two potential starting points for the α-SYN pathology are the enteric nerves innervating the gut and the olfactory bulb (Hawkes et al., 2007; Angot et al., 2010). These two systems exhibit abundant LP in PD, and this pathology is potentially also present in “prodromal” PD. Notably, both the gut and the olfactory bulb are particularly exposed to the exogenous insults (e.g., viruses, bacteria, fungi, pollutants, toxins) because they are exposed to the surrounding environment by virtue of anatomy (Rey et al., 2016c; Sampson et al., 2016). Together, a model where the α-SYN aggregation is triggered in enteric nerves and/or olfactory bulb in predisposed individuals (as discussed above), and then spreads in a prion-like fashion, is attractive. Naturally, cross-sectional neuropathology studies on postmortem brain tissue have shortcomings in that they do not provide insight into the precise temporal development of α-SYN pathology. Therefore, it is imperative that imaging ligands specific to pathogenic α-SYN assemblies are developed, which will allow us to define in detail how LP progresses in a given individual.
Why do not all synucleinopathies look and behave the same?
The aggregation of nonmutant α-SYN can cause distinct synucleinopathies in humans. The fact that α-SYN can form fibrillar assemblies with distinct structural characteristics, as discussed above, has led to the hypothesis that α-SYN strains may account for the different clinicopathological traits that characterize DLB, MSA, and PD. Indeed, α-SYN molecules adopt different conformations leading to distinct molecular stacking within distinct fibrillar assemblies and surfaces dissimilarities. As explained above, in addition to having different biophysical properties that reflect into different seeding, persistence, and macroscopic appearance, those assemblies have surface characteristics that likely account for distinct interactomes. Thus, by interacting with different partners, including membranous surface proteins, diverse α-SYN strains perturb cellular proteostasis in different ways leading to strain-specific clinicopathophysiological traits. This is a potential explanation for why α-SYN aggregates mostly appear in oligodendrocytes in MSA and in neurons in PD/DLB. Patients with DLB, PD, and PD with dementia also exhibit different initial clinical symptoms and diverse rates of progression. While this potentially is also influenced by the predominant strain of α-SYN, another explanation might be that the primary initial trigger site of LP is different (e.g., gut vs olfactory system).
A unifying hypothesis
Prion propagation of α-SYN is an important molecular mechanism that contributes significantly to disease progression. There exist several potential triggers for cellular α-SYN misfolding that have either genetic or environmental backgrounds (Fig. 2). Whether the initial misfolding actually leads to a cascade of LP spreading depends on several factors, including the age and genetic background of the individual. Thus, we recognize that mitochondrial dysfunction, oxidative stress, failure of the lysosomal autophagy and ubiquitin proteasome systems, and neuroinflammation all probably play crucial roles in enabling the initial misfolding of α-SYN or in facilitating the cell-to-cell transfer of pathogenic α-SYN assemblies. Future research should invest in improving our understanding of how these mechanisms interact with each other. Furthermore, we need better insight into how different α-SYN fibril strains and anatomical trigger sites influence the type of synucleinopathy that develops. Ultimately, new insights into these mechanisms can aid the development of novel therapies that slow disease progression by interfering with fundamental steps in the pathogenesis and progressive worsening of synucleinopathies.
Figure 2.

Schematic diagram depicting a possible central cascade leading to cell-to-cell transfer of α-SYN. The central process is likely affected by several other disease mechanisms that have already been implicated in PD (shown inside the circle) and that form an interdependent network of molecular events, which in combination or each on their own can promote the central cascade. Weak genetic risk factors and aging are depicted as potential triggers or promoters of cell-to-cell transfer of α-SYN (for details of the different mechanisms, see text).
Footnotes
P.B. was supported by Van Andel Research Institute, Michael J. Fox Foundation, National Institutes of Health Grant R21 NS09399302 and R01 DC016519, Cure Parkinson's Trust, Peter C. and Emajean Cook Foundation, East Tennessee Foundation, and Campbell Foundation. Research performed in the R.M. laboratory was supported by the Centre National de la Recherche Scientifique, European Commission Joint Programme on Neurodegenerative Diseases JPND-NeuTARGETs-ANR-14-JPCD-0002-02 and ANR-15-JPWG-0012-03, France Parkinson, Fondation de France, Fondation Simone et Cino Del Duca of the Institut de France, Fondation Bettencourt-Schueller, and Fondation Recherche Médicale.
Dr. Brundin has received commercial support as a consultant from Renovo Neural, Roche, Teva, Lundbeck, AbbVie, NeuroDerm, Axial Biotherapeutics and Cellular Dynamics International. Additionally, he has received commercial support for grants/research from Renovo, Teva and Lundbeck. Dr. Brundin has ownership interests in Acousort AB, Parkcell AB. Dr. Melki declares no competing financial interests.
References
- Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C (2016) Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J 35:2120–2138. 10.15252/embj.201593411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Reish HE, Standaert DG (2015) Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 5:1–19. 10.3233/JPD-140491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM (2011) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42:360–367. 10.1016/j.nbd.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot E, Steiner JA, Hansen C, Li JY, Brundin P (2010) Are synucleinopathies prion-like disorders? Lancet Neurol 9:1128–1138. 10.1016/S1474-4422(10)70213-1 [DOI] [PubMed] [Google Scholar]
- Angot E, Steiner JA, Lema Tomé CM, Ekström P, Mattsson B, Björklund A, Brundin P (2012) Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One 7:e39465. 10.1371/journal.pone.0039465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulić S, Le TT, Moda F, Abounit S, Corvaglia S, Casalis L, Gustincich S, Zurzolo C, Tagliavini F, Legname G (2014) Defined α-synuclein prion-like molecular assemblies spreading in cell culture. BMC Neurosci 15:69. 10.1186/1471-2202-15-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295:865–868. 10.1126/science.1067389 [DOI] [PubMed] [Google Scholar]
- Barrett PJ, Timothy Greenamyre J (2015) Post-translational modification of α-synuclein in Parkinson's disease. Brain Res 1628:247–253. 10.1016/j.brainres.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. 10.1007/s00401-009-0538-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson's disease. J Neurochem 139 [Suppl 1]:216–231. [DOI] [PubMed] [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. 10.1038/ncomms3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. 10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Brahic M, Bousset L, Bieri G, Melki R, Gitler AD (2016) Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol 131:539–548. 10.1007/s00401-016-1538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9:1135–1150. 10.1016/j.celrep.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Li JY, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci 9:741–745. 10.1038/nrn2477 [DOI] [PubMed] [Google Scholar]
- Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11:301–307. 10.1038/nrm2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 64:485–491. 10.1002/ana.21541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Odin P (2010) The challenge of non-motor symptoms in Parkinson's disease. Prog Brain Res 184:325–341. 10.1016/S0079-6123(10)84017-8 [DOI] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J (2011) Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 3:a004374. 10.1101/cshperspect.a004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, Jagadapillai R, Liu R, Choe K, Shivakumar B, Son F, Jin S, Kerber R, Adame A, Masliah E, Friedland RP (2016) Exposure to the functional bacterial amyloid protein Curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 6:34477. 10.1038/srep34477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398. 10.1016/j.nbd.2009.05.023 [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Drouin-Ouellet J, Gross RE (2009) Environmental toxins and Parkinson's disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci 30:475–483. 10.1016/j.tips.2009.06.005 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147–216. 10.1038/emm.2014.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009) Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 111:192–203. 10.1111/j.1471-4159.2009.06324.x [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. 10.1186/1750-1326-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–13015. 10.1073/pnas.0903691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Gwinn K, Singleton A, Hardy J (2011) Parkinson's disease and α-synuclein expression. Mov Disord 26:2160–2168. 10.1002/mds.23948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson's disease. J Parkinsons Dis 3:461–491. 10.3233/JPD-130230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM, Trojanowski JQ (2000) Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol 157:1439–1445. 10.1016/S0002-9440(10)64781-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30:6838–6851. 10.1523/JNEUROSCI.5699-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, Bogetofte H, Lang C, Ryan BJ, Sardi SP, Badger J, Vowles J, Evetts S, Tofaris GK, Vekrellis K, Talbot K, Hu MT, James W, Cowley SA, Wade-Martins R, et al. (2016) ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson's iPSC-derived dopamine neurons. Stem Cell Rep 6:342–356. 10.1016/j.stemcr.2016.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, Wiethoff CM, Campbell EM (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. 10.1371/journal.pone.0062143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freer R, Sormanni P, Vecchi G, Ciryam P, Dobson CM, Vendruscolo M (2016) A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimers disease. Sci Adv 2:e1600947. 10.1126/sciadv.1600947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M (2012) Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol 72:517–524. 10.1002/ana.23747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z, Dion PA, Rouleau GA (2015) Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11:1443–1457. 10.1080/15548627.2015.1067364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM (2008) Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci 28:7687–7698. 10.1523/JNEUROSCI.0143-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290:985–989. 10.1126/science.290.5493.985 [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Prahlad V, Morimoto RI (2011) The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol 3:a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H (2013) 100 years of Lewy pathology. Nat Rev Neurol 9:13–24. 10.1038/nrneurol.2012.242 [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, Surmeier DJ (2012) Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci 15:1414–1421. 10.1038/nn.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468:696–700. 10.1038/nature09536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, Holton JL, Revesz T, Dickson DW (2011) Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 122:187–204. 10.1007/s00401-011-0852-9 [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725. 10.1172/JCI43366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599–614. 10.1111/j.1365-2990.2007.00874.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwig M, Klinkenberg M, Rusconi R, Musgrove RE, Majbour NK, El-Agnaf OM, Ulusoy A, Di Monte DA (2016) Brain propagation of transduced α-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 139:856–870. 10.1093/brain/awv376 [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8:382–397. 10.1016/S1474-4422(09)70062-6 [DOI] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110:E3138–E3147. 10.1073/pnas.1301440110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128:805–820. 10.1007/s00401-014-1343-6 [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4:147ra111. 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT Jr (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73:1055–1058. 10.1016/0092-8674(93)90635-4 [DOI] [PubMed] [Google Scholar]
- Jellinger KA. (2009) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740. 10.1016/j.bbadis.2008.07.006 [DOI] [PubMed] [Google Scholar]
- Jin SG, Zhang ZM, Dunwell TL, Harter MR, Wu X, Johnson J, Li Z, Liu J, Szabó PE, Lu Q, Xu GL, Song J, Pfeifer GP (2016) Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep 14:493–505. 10.1016/j.celrep.2015.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM (2015) Proteostasis and aging. Nat Med 21:1406–1415. 10.1038/nm.4001 [DOI] [PubMed] [Google Scholar]
- Kim DK, Lim HS, Kawasaki I, Shim YH, Vaikath NN, El-Agnaf OM, Lee HJ, Lee SJ (2016) Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy 12:1849–1863. 10.1080/15548627.2016.1207014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EM, Hwang O (2011) Role of matrix metalloproteinase-3 in neurodegeneration. J Neurochem 116:22–32. 10.1111/j.1471-4159.2010.07082.x [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82:323–355. 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA (2014) Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-synuclein externalization via exosomes. Hum Mol Genet 23:2816–2833. 10.1093/hmg/ddu099 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504–506. 10.1038/nm1747 [DOI] [PubMed] [Google Scholar]
- Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (2011) Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43:552–557. 10.1016/j.nbd.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Leinisch F, Kadiiska MB, Corbett J, Mason RP (2016) Formation and implications of alpha-synuclein radical in Maneb- and Paraquat-induced models of Parkinson's disease. Mol Neurobiol 53:2983–2994. 10.1007/s12035-015-9179-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. 10.1523/JNEUROSCI.0692-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ (2008a) Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 40:1835–1849. 10.1016/j.biocel.2008.01.017 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee SJ (2008b) Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun 372:423–428. 10.1016/j.bbrc.2008.05.045 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262–9272. 10.1074/jbc.M109.081125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ (2013) Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 45:e22. 10.1038/emm.2013.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P (2013) Inflammation and α-synuclein's prion-like behavior in Parkinson's disease: is there a link? Mol Neurobiol 47:561–574. 10.1007/s12035-012-8267-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin J, Högen T, Hillmer AS, Bader B, Schmidt F, Kamp F, Kretzschmar HA, Bötzel K, Giese A (2011) Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J Parkinsons Dis 1:205–216. 10.3233/JPD-2011-11040 [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14:501–503. [DOI] [PubMed] [Google Scholar]
- Lim S, Chun Y, Lee JS, Lee SJ (2016) Neuroinflammation in synucleinopathies. Brain Pathol 26:404–409. 10.1111/bpa.12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106:20051–20056. 10.1073/pnas.0908005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012a) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012b) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209:975–986. 10.1084/jem.20112457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht P, Seppi K, Poewe W (2015) The concept of prodromal Parkinson's disease. J Parkinsons Dis 5:681–697. 10.3233/JPD-150685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA (2002) The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 277:1641–1644. 10.1074/jbc.C100560200 [DOI] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, et al. (2016) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. 10.1126/science.aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, McMahon HT (2008) Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol 9:543–556. 10.1038/nrm2417 [DOI] [PubMed] [Google Scholar]
- Massey AR, Beckham JD (2016) Alpha-synuclein, a novel viral restriction factor hiding in plain sight. DNA Cell Biol 35:643–645. 10.1089/dna.2016.3488 [DOI] [PubMed] [Google Scholar]
- McCann H, Stevens CH, Cartwright H, Halliday GM (2014) α-Synucleinopathy phenotypes. Parkinsonism Relat Disord 20 [Suppl 1]:S62–S67. [DOI] [PubMed] [Google Scholar]
- Melki R. (2015) Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J Parkinsons Dis 5:217–227. 10.3233/JPD-150543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. (2011) The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol 76:91–99. 10.1101/sqb.2012.76.010637 [DOI] [PubMed] [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228. 10.1016/j.neurobiolaging.2011.06.022 [DOI] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46:989–993. 10.1038/ng.3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Iwatsubo T, Hasegawa M (2005) Ubiquitination of alpha-synuclein. Biochemistry 44:361–368. 10.1021/bi0485528 [DOI] [PubMed] [Google Scholar]
- Oosawa F, Asakura S (1975) Thermodynamics of the polymerization of proteins (Horecker B, Kaplan NO, Matmur J, Scheraga HA, eds) pp. 41–55. London: Academic Press, Inc. [Google Scholar]
- Oueslati A. (2016) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinsons Dis 6:39–51. 10.3233/JPD-160779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampalakis G, Sykioti VS, Ximerakis M, Stefanakou-Kalakou I, Melki R, Vekrellis K, Sotiropoulou G (2016) KLK6 proteolysis is implicated in the turnover and uptake of extracellular alpha-synuclein species. Oncotarget 8:14502–14515. 10.18632/oncotarget.13264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H (2001) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 21:8053–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- Poggiolini I, Saverioni D, Parchi P (2013) Prion protein misfolding, strains, and neurotoxicity: an update from studies on Mammalian prions. Int J Cell Biol 2013:910314–910324. 10.1155/2013/910314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112:E5308–E5317. 10.1073/pnas.1514475112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. (2016) How neuroinflammation contributes to neurodegeneration. Science 353:777–783. 10.1126/science.aag2590 [DOI] [PubMed] [Google Scholar]
- Rcom-H'cheo-Gauthier A, Goodwin J, Pountney DL (2014) Interactions between calcium and alpha-synuclein in neurodegeneration. Biomolecules 4:795–811. 10.3390/biom4030795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. 10.1002/ana.24066 [DOI] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P (2013) Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126:555–573. 10.1007/s00401-013-1160-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, George S, Brundin P (2016a) Review: Spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol 42:51–76. 10.1111/nan.12299 [DOI] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016b) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J Exp Med 213:1759–1778. 10.1084/jem.20160368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Wesson DW, Brundin P (2016c) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis. Advance online publication. Retrieved Dec. 20, 2016. 10.1016/j.nbd.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Olsson TT, Lamberts JT, Devine MJ, Kunath T, Brundin P (2015) A cell culture model for monitoring α-synuclein cell-to-cell transfer. Neurobiol Dis 77:266–275. 10.1016/j.nbd.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI (2013) Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun 1:38. 10.1186/2051-5960-1-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI (2014) Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 111:10732–10737. 10.1073/pnas.1321785111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167:1469–1480.e12. 10.1016/j.cell.2016.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C (2017) Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 74:163–172. 10.1001/jamaneurol.2016.4547 [DOI] [PubMed] [Google Scholar]
- Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M (2017) Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol Commun 5:12. 10.1186/s40478-017-0413-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Léna C, Aperia A, Melki R, Triller A (2015) α-Synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 34:2408–2423. 10.15252/embj.201591397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661. 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533:95–99. 10.1038/nature17939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM (2017a) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113. 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E (2017b) Calcium and Parkinson's disease. Biochem Biophys Res Commun 483:1013–1019. 10.1016/j.bbrc.2016.08.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM (2014) α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 7:2054–2065. 10.1016/j.celrep.2014.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM. (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol 10:583–596. 10.1038/nrm2751 [DOI] [PubMed] [Google Scholar]
- Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W (2017) PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson's disease pathobiology? J Parkinsons Dis 7:13–29. 10.3233/JPD-160989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Hamada K, Krainc D (2014) ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci 34:15281–15287. 10.1523/JNEUROSCI.1629-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Di Monte DA (2013) Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 5:1119–1127. 10.1002/emmm.201302475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. (2003) A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn 21:211–234. 10.1080/07391102.2003.10506918 [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous α-synuclein fibrils induce Lewy Body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 110:19555–19560. 10.1073/pnas.1318268110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19:1085–1092. 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Stefanis L (2013) α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 47:537–551. 10.1007/s12035-012-8341-2 [DOI] [PubMed] [Google Scholar]