

Abstract
Fragile X mental retardation protein (FMRP) loss causes Fragile X syndrome (FXS), a major disorder characterized by autism, intellectual disability, hyperactivity, and seizures. FMRP is both an RNA- and channel-binding regulator, with critical roles in neural circuit formation and function. However, it remains unclear how these FMRP activities relate to each other and how dysfunction in their absence underlies FXS neurological symptoms. In testing circuit level defects in the Drosophila FXS model, we discovered a completely unexpected and highly robust neuronal dye iontophoresis phenotype in the well mapped giant fiber (GF) circuit. Controlled dye injection into the GF interneuron results in a dramatic increase in dye uptake in neurons lacking FMRP. Transgenic wild-type FMRP reintroduction rescues the mutant defect, demonstrating a specific FMRP requirement. This phenotype affects only small dyes, but is independent of dye charge polarity. Surprisingly, the elevated dye iontophoresis persists in shaking B mutants that eliminate gap junctions and dye coupling among GF circuit neurons. We therefore used a wide range of manipulations to investigate the dye uptake defect, including timed injection series, pharmacology and ion replacement, and optogenetic activity studies. The results show that FMRP strongly limits the rate of dye entry via a cytosolic mechanism. This study reveals an unexpected new phenotype in a physical property of central neurons lacking FMRP that could underlie aspects of FXS disruption of neural function.
SIGNIFICANCE STATEMENT FXS is a leading heritable cause of intellectual disability and autism spectrum disorders. Although researchers established the causal link with FMRP loss >;25 years ago, studies continue to reveal diverse FMRP functions. The Drosophila FXS model is key to discovering new FMRP roles, because of its genetic malleability and individually identified neuron maps. Taking advantage of a well characterized Drosophila neural circuit, we discovered that neurons lacking FMRP take up dramatically more current-injected small dye. After examining many neuronal properties, we determined that this dye defect is cytoplasmic and occurs due to a highly elevated dye iontophoresis rate. We also report several new factors affecting neuron dye uptake. Understanding how FMRP regulates iontophoresis should reveal new molecular factors underpinning FXS dysfunction.
Keywords: Drosophila, dye coupling, gap junction, giant fiber circuit
Introduction
Fragile X syndrome (FXS), resulting from fragile X mental retardation protein (FMRP) loss, is the leading heritable cause of autism and intellectual disability, and displays comorbidity with numerous other neurological symptoms including attention deficit, hyperactivity and childhood seizures (Cowley et al., 2016). FMRP is a complex protein with multiple neuronal functions. The canonical FMRP role is direct RNA-binding translation control, regulating activity-dependent expression of a large but indeterminate number of neuronal proteins (Darnell et al., 2011). Without FMRP, numerous proteins become overexpressed, significantly altering the neuronal proteome (Zhang et al., 2001; Cvetkovska et al., 2013; Ifrim et al., 2015). The major non-canonical FMRP role is direct channel-binding, which regulates functional channel gating properties (Brown et al., 2010; Strumbos et al., 2010). Without FMRP, K+ and Ca2+ channels are dysregulated, altering both action potentials and synaptic transmission (Deng et al., 2013; Contractor et al., 2015). A number of other diverse functions have also been ascribed to FMRP, such as miRNA regulation and chromatin stability control (Jin et al., 2004; Alpatov et al., 2014), expanding the already complex involvement of FMRP in neuronal development and function. Targeted and effective therapies for patients will require an understanding of how these diverse FMRP roles relate to FXS symptoms.
Drosophila disease models provide a potent toolkit to discover novel neuronal mechanisms underlying neurological symptoms. The well established Drosophila FXS disease model exhibits phenotypes analogous to human symptoms, including synaptic overgrowth, hyperactivity, and learning/memory deficits (Zhang et al., 2001; Dockendorff et al., 2002; Bolduc et al., 2008), and continues to provide key new insights into FXS (Doll and Broadie, 2014, 2015, 2016; Golovin and Broadie, 2016). Elegant Drosophila brain neural circuit maps reveal individual neurons with single-cell resolution, enabling links between neuronal molecular changes and FXS circuitry defects. An excellent set of neurons for such work is the giant fiber (GF) circuit, a well characterized escape circuit comprised of five readily identifiable neurons (King and Wyman, 1980; Allen et al., 1998), which require FMRP for proper circuit function (Martinez et al., 2007). This circuit collects sensory information and relays it to motoneurons via the large GF interneuron (GFI), enabling rapid escape from aversive stimuli (Tanouye and Wyman, 1980). The toolkit for this circuit includes highly specific genetic reporters (Sun and Wyman, 1996) and an array of transgenic drivers that provide exquisite labeling and manipulation of individual GF neurons (Godenschwege et al., 2002; Lee and Godenschwege, 2015). The large size and easy accessibility of the GFI allows iontophoretic dye injection via sharp electrodes, a critical tool for circuit study (Boerner and Godenschwege, 2011).
Dye iontophoresis represents a classic strategy for neural circuit mapping and uncovering genes required for electrical synapse formation, with many studies in the extensively dye-coupled GF circuit (Phelan et al., 1996; Kudumala et al., 2013; Lee and Godenschwege, 2015). Small ionic dyes, such as neurobiotin and lucifer yellow, pass through gap junctions, labeling the electrically-coupled circuit (Lapper and Bolam, 1991; Hanani, 2012). Most experiments assay simple dye transfer between coupled neurons; however, a recent study tested quantitative relative dye transfer levels via gap junctions (Orr et al., 2014). Expanding on this quantitative dye iontophoresis approach, we began exploring GF circuit dye coupling in the FXS model to test hypothesized changes in circuit connectivity. Instead, we stumbled upon an unexpected and robust dye injection phenotype: Drosophila FMR1 (dfmr1)-null mutant neurons take up dramatically more small, charged dyes injected via iontophoretic current. This defect is not related to a multitude of neuronal properties, including size, porosity, and circuit connectivity. Indeed, we found a trend toward fewer gap junction electrical synapses in dfmr1-null neurons, and a persistent dye iontophoresis defect in neurons completely lacking all gap junction connections. We conclude FMRP regulates a cytosolic mechanism determining ionic uptake rates. This study provides new insights into quantitative iontophoresis, broadens understanding of multiple factors modulating neuronal dye injection, and identifies a new neurological disruption in this FXS disease model.
Materials and Methods
Drosophila genetics.
All animals were maintained on standard cornmeal/agar/molasses Drosophila food in a 12 h light/dark cycling incubator at 25°C. For channelrhodopsin experiments, food was made with 100 μm all-trans retinal (ATR) or EtOH vehicle as a control (Ataman et al., 2008), and animals were reared in constant darkness. The following Drosophila lines were used for genetic crosses, with female offspring used for all experiments: w1118 (RRID:BDSC_3605) | w1118; dfmr150M/TM6B, GFP, Hu (Zhang et al., 2001; RRID:BDSC_6930) | w1118; dfmr12/TM6, Hu (Dockendorff et al., 2002) | w1118; UAS-dfmr1, dfmr150M/TM6B, GFP, Hu (9557.3; Zhang et al., 2001) | w1118; elav-gal4/CyO (RRID:BDSC_8765) | w1118; elav-gal4/CyO; dfmr150M/TM6, Hu | shakB2 (Blagburn et al., 1999) | shakB2; dfmr150M/TM6, GFP | w1118; P{GMR91H05-Gal4}attP2 (Jenett et al., 2012; RRID:BDSC_40594) | w1118; UAS-ChR2-XXL (Dawydow et al., 2014) |w1118; 10XUAS-IVS-mCD8::RFP (RRID:BDSC_32219) | w1118; P{UAS-shaw.RA.FLAG}12B (Hodge et al., 2005; RRID:BDSC_55719) | w1118; UAS-irk2A/CyO, GFP (Dahal et al., 2012). All genotypes were verified with visible genotype markers when possible, and PCR when necessary. All crosses were allowed to lay eggs for 2–3 d, with offspring rearing densities carefully matched between compared genotypes.
Dye iontophoresis.
Experimental animals were selected from age-matched rearing tubes 10–13 d after egg laying, yielding staged adults 0–4 d in age. Genotyped animals were pinned submerged in physiological saline (Jan and Jan, 1976), and then cut along the dorsal midline to access the cervical connective (CC), as previously reported (Boerner and Godenschwege, 2011). Sharp glass electrodes (Kwik-Fil Borosilicate glass 1B100F-4, World Precision Instruments) were pulled on a laser electrode puller (Model P-2000, Sutter Instrument) to 10 MΩ resistance (3 m KCl). Electrodes were filled by capillary action with one of the following dye solutions: 0.25% tetramethylrhodamine (TRITC)-dextran (10 kDa, Life Technologies) and 7% Neurobiotin (Vector Laboratories, RRID:AB_2313575), or 1% lucifer yellow (LY) potassium salt (ThermoFisher Scientific). Dyes were dissolved in ddH2O or 2 m potassium acetate (KAc), as indicated. Filled electrodes were placed on a silver-chloride wire mounted on a PCS-5000 micromanipulator (Burleigh). The electrode tip was inserted into the GFI at the CC, while a ground electrode was placed in the bath saline. Voltage was manually controlled with a Grass square-pulse stimulator (S48, Astro-Med), providing 7.5 100 ms pulses/s for 2 min, unless otherwise noted. Delivery of 20 nA current was monitored by tracking the voltage drop across a 1 MΩ resistor in the electrode circuit with an AxoClamp2B amplifier, connected to a Digidata 1320A data acquisition system (Molecular Devices), controlled by Clampex software v9.2. Reversed polarity was used for LY experiments.
Dye injection manipulations.
For channelrhodopsin experiments, preparations were illuminated with filtered blue light (440–470 nm) from an EBQ100 isolated mercury lamp (Leistungselektronik Jena GmbH) for the duration of dye injection. Channelrhodopsin function was tested by behavioral response to blue light. For K+ channel blocking, 4-aminopyridine (4-AP; Sigma-Aldrich; 10 mm) was added in physiological saline before imaging (Singh and Singh, 1999). For mixture experiments, tetraethylammonium chloride (TEA; Sigma-Aldrich; 10 mm) was combined with 10 mm 4-AP in the bath, with cesium chloride (140 mm) dissolved in KAc-free neurobiotin (NB) dye injection solution. Drug was added at the beginning of the dissection, with exposure for ≥5 min before dye injection. All drug trial preparations were rinsed three times in physiological saline with a 3 min rest before fixation to prevent interaction between drug and fixative. Forced leak experiments were performed following the standard dye injection, followed by a second electrode containing 0.25% TRITC in 2 m KAc that was placed into the dye-injected neuron. The order of genotypes assayed in all trials was changed for experiments each day to avoid any possible ordering experimental artifacts.
Confocal imaging.
After dye injections, the entire CNS (brain and thoracic ganglion) was dissected out and fixed for 30 min in 4% paraformaldehyde (Electron Microscopy Services)/4% sucrose (ThermoFisher Scientific) in PBS, pH 7.2 (Life Technologies). Fixed preparations were rinsed three times with PBS, and blocked for 1 h with 1% bovine serum albumin (BSA; Sigma-Aldrich) in PBST (1× PBS, 0.2% Triton X-100; ThermoFisher Scientific). Labels were diluted in PBST with 0.2% BSA. The following labels were used: streptavidin::Cy5 (1:20, Life Technologies), rabbit anti-ShakB (1:250; Phelan et al., 1996) and 633-conjugated goat anti-rabbit (1:250; Life Technologies, RRID:AB_141419). Neurobiotin was labeled with streptavidin::Cy5. Incubations were conducted either overnight at 4°C or 2 h at room temperature. Following antibody incubations, preparations were rinsed three times for 30 min in PBST before a final 30 min rinse in PBS. Preparations were mounted on glass slides (Probe On Plus 25 × 75 × 1.0 mm, ThermoFisher Scientific) in 2, 2′-thiodiethanol (Staudt et al., 2007). To prevent crushing, double-sided poster tape (Scotch) was placed on each side of the brains. Coverslips (No. 1.5H, Zeiss) were sealed with nail polish (Hard as Nails, Sally Hansen). Fluorescent images were collected using a Zeiss LSM 510 META laser-scanning confocal microscope with a 40× oil-immersion objective, focusing on the prothoracic and mesothoracic ganglia regions (Mu et al., 2014). Imaging settings were maintained across all samples, with the exception of GFI dendrites, which required increased brightness for the TRITC injection of w1118 and dfmr150M, and the NB injection of shakB2 and shakB2; dfmr150M. Images show maximum Z-stack projections.
Protein quantification.
Brains were dissected in PBS from adult females (0–4 d at 25°C). Four brains were collected on ice in lysis buffer (50 mm Tris, 100 mm NaCl, 1 mm EDTA, 1 mm PMSF, Protease Inhibitor Cocktail; Sigma-Aldrich), sonicated (Branson Model 102C, Sonifier 250 microtip) for 20 s, returned to ice for 20 min, then centrifuged at 14,000 rpm for 10 min. Protein samples were prepared using a BCA kit (ThermoFisher) and analyzed with a Nanodrop 2000c spectrophotometer (ThermoFisher).
Data analyses.
FIJI software was used for data quantification and processing (Schindelin et al., 2012; RRID:SCR_002285). For dye injection intensity quantification, the number of pixels at each intensity (0–255) was calculated for each 8-bit image stack. A pixel cutoff of 59 was empirically determined to remove background with minimal signal loss. The total number of pixels >;59 in the full stack is reported as “dye injected”. GFI dendrites were quantified in the same manner after masking cell bodies, axons, and dye-coupled neurons. A threshold of 90 was used for the TRITC dendrite quantification to offset changes in imaging brightness. Intensity quantification was automated using an ImageJ Macro script to remove any experimenter analytical bias. The dendritic structure was calculated by measuring the GFI primary branches using the FIJI Simple Neurite Trace plugin (Longair et al., 2011). For w1118 and dfmr150M, the TRITC signal was used, whereas for shakB2 and shakB2; dfmr150M NB signal was used for the structural comparisons. For anti-ShakB fluorescence quantification, the TRITC dye injection signal was used to create the region-of-interest (ROI) encompassing the GFI bend. This ROI was then overlaid onto the ShakB channel, and intensities of each pixel above the background threshold (40) were summed for all optical slices containing the GFI bend. All statistical analyses were performed using Prism software v7 (GraphPad, RRID:SCR_002798). All single pairwise comparisons were performed by two-tailed Student's t test for Gaussian distributions, and Mann–Whitney for non-Gaussian distributions. All multiple comparisons were performed using unpaired one-way ANOVA, with Tukey–Kramer pairwise post hoc tests. Slope comparisons were performed using the ANCOVA test. In all figures, graphs show mean ± SEM with the statistical comparisons displayed as not significant (NS) p >; 0.05, *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
FMRP selectivity limits small dye iontophoresis into neurons
While studying Drosophila neural connectivity changes in dfmr1-null mutants, we discovered a surprising dye iontophoresis phenomenon (Fig. 1): neurons lacking FMRP accept far more NB (287 Da), but no more dextran-TRITC (10 kDa), compared with matched genetic background controls. We pursued this intriguing finding in the GF circuit by delivering controlled iontophoretic current to coinject both NB and TRITC dyes into the GFI (Boerner and Godenschwege, 2011). TRITC provides a real-time confirmation of neuron identity, but cannot leave the neuron via gap junctions due to its large size (Phelan et al., 1996). In contrast, the much smaller NB readily crosses gap junctions and labels all of the electrically-coupled neurons within the circuit (Boerner and Godenschwege, 2010). Each brain hemisphere contains one GFI cell body and dendritic arbor (not pictured), with a large descending axon into the thoracic ganglion terminating in the mesothoracic ganglion with a characteristic bend visible by TRITC labeling (Fig. 1A, top, arrow). The GFI axon forms electrical synapses with peripherally synapsing interneurons (PSIs), visible by NB dye-coupled labeling (Fig. 1A, bottom, arrows), and the tergotrochanteral motor neuron (TTMn), which projects to leg muscles (King and Wyman, 1980; Allen et al., 1998). We compared dye labeling in dfmr1-null mutants (w1118; dfmr150M) with genetic background controls (w1118). Representative images and data summaries are shown in Figure 1.
Figure 1.
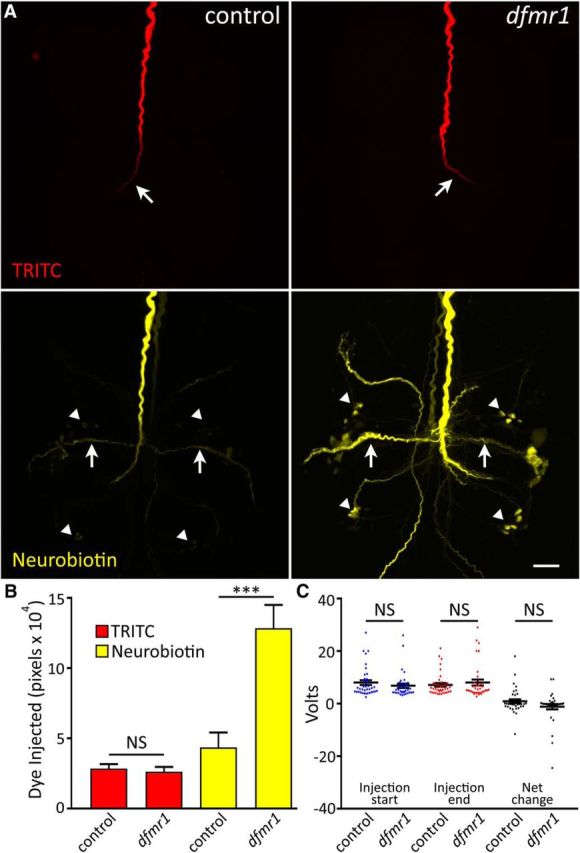
Selective small dye iontophoresis is increased in dfmr1-null neurons. A, Representative images of the GFI coinjected with 10 kDa dextran-conjugated TRITC (red; top) and 287 Da NB (yellow; bottom) dissolved in 2 m KAc. Images show the thoracic ganglion for w1118 genetic background (control; left) and dfmr150M-null mutant (right). TRITC-dextran is too large to escape the injected GFI neuron, whereas NB passes through gap junctions to dye-coupled neurons in the GF neural circuit. Arrows indicates GFI bend (top) and PSI (bottom). Arrowheads indicate coupled cell bodies. Scale bar, 20 μm. B, Quantification of TRITC (red) and NB (yellow) dye injection displayed as mean ± SEM. TRITC: control, n = 36; dfmr1, n = 33; NB: control, n = 37; dfmr1, n = 36. C, Voltage required to deliver 20 nA of current to the GFI neuron in control and dfmr1-null mutants. Voltage is shown at the start (left) and end (middle) of each 2 min injection, and also as the net voltage change after a 2 min injection (right). Control, n = 38; dfmr1, n = 35. Significance determined from two-tailed unpaired t tests (B) or two-tailed Mann–Whitney tests (C): ***p < 0.001.
Upon injection, TRITC distributes uniformly along the GFI axon to the distal terminating bend in both controls (Fig. 1A, top left) and dfmr1-nulls (Fig. 1A, top right). Dimmer signal toward the bend results from the axon moving progressively deeper into the semi-opaque tissue, which restricts light transmission. There was no quantifiable difference in TRITC dye loading between the two genotypes (Fig. 1B). Mean TRITC signal was 2.8 × 104 ± 0.3 × 104 pixels in controls (n = 36) versus 2.6 × 104 ± 0.3 × 104 pixels in dfmr1-nulls (n = 33), an indistinguishable difference in loading (p = 0.64, two-tailed unpaired t test). In sharp contrast, control GFIs contain visibly less NB dye in the axon, especially at the terminal bend (Fig. 1A, bottom left), compared with dfmr1-null mutants (Fig. 1A, bottom right). Moreover, the mutants display much more NB signal in the coupled PSIs (arrows) and other labeled neurons, including their cell bodies (arrowheads). Null dfmr1 GFIs are not visibly coupled to any neurons not labeled in controls and maintain a normal circuit pattern. Upon quantification, we found dfmr1-null neurons take up nearly threefold more NB dye than controls (Fig. 1B). Mean NB dye intensity in controls was 4.4 × 104 ± 1.1 × 104 (n = 37) compared with 12.8 × 104 ± 1.7 × 104 in dfmr150M mutants (n = 36), a very highly significant increase (p = 6.2 × 10−5, two-tailed unpaired t test). This selective dye iontophoresis defect is highly replicable and extremely robust, providing an excellent new measure for the effects of FMRP loss. Furthermore, such a genotypic difference in iontophoretic dye loading has never been reported for any mutant condition, to our knowledge.
To test whether current delivery differences might cause the differential dye loading between genotypes, we next recorded the voltage required to deliver a constant current (Fig. 1C). Any voltage change must be coupled with a resistance change to maintain a constant current. Differing voltage requirements between genotypes would reveal altered conductivity throughout the dye delivery circuit, such as altered plasma membrane permeability. Comparing genetic background controls to dfmr1-null mutants, we recorded the voltage required to deliver 20 nA currents over a 2 min interval, but found no significant differences between the two genotypes (Fig. 1C). The voltage did not differ at the onset of current injection (control: 8.0 ± 0.9 V, n = 38; dfmr150M: 6.8 ± 0.8 V, n = 35, p = 0.21, two-tailed Mann–Whitney test; Fig. 1C) or at the end of current injection (control: 7.1 ± 0.7 V; dfmr150M: 8.0 ± 1.1 V, p = 0.66, two-tailed Mann–Whitney test; Fig. 1C). The net voltage change also did not differ between the genotypes (control: 0.9 ± 0.7 V; dfmr150M: −1.2 ± 1.0 V, p = 0.44, two-tailed Mann–Whitney test; Fig. 1C). Detailed analyses of instant current readings (1 kHz) throughout the injection period also revealed no differential fluctuations between genotypes that could account for loading differences (data not shown). These findings show the electrical circuit resistance does not differ between dfmr1-nulls and background controls, and indicate that the elevated NB loading in mutants must result from altered fundamental neuron properties in the absence of FMRP.
FMRP-dependent and charge-independent dye iontophoresis defect
We next tested whether the altered neuronal properties were specifically due to loss of FMRP. We first compared the w1118 genetic background control (Fig. 2A, top left) with a second, independent dfmr1-null allele, dfmr12 (Fig. 2A, top right). To rule out recessive background effects, we tested the heteroallelic combination with dfmr150M. This mutant replicated the specific NB dye-loading defect (control: 3.9 × 104 ± 1.3 × 104, n = 6; dfmr12/50M: 12.1 × 104 ± 1.1 × 104, n = 6, p = 0.0008, two-tailed unpaired t test; Fig. 2B), supporting a causative FMRP role. We next tested whether transgenic expression of wild-type dFMRP in dfmr1-null neurons rescues the phenotype using neuron-specific elav-Gal4 as the driver (Brand and Perrimon, 1993). Dye loading in the rescue condition was comparable to the paired control, and greatly reduced compared with the dfmr1-null (Fig. 2A,B). Quantification supports the conclusion that wild-type dFMRP reintroduction rescues the dye-loading phenotype (elav-Gal4/+: 6.1 × 104 ± 1.3 × 104, n = 17; elav-Gal4/+; dfmr150M, UAS-dfmr1/dfmr150M: 7.8 × 104 ± 1.5 × 104, n = 18; elav-Gal4/+; dfmr150M/dfmr150M: 13.4 × 104 ± 1.9 × 104, n = 13), with no significant difference between control and rescue conditions (p = 0.68), but a significant difference existing between the rescue and mutant (p = 0.03) and persisting between the control and mutant (p = 0.005, unpaired ANOVA, Tukey–Kramer pairwise post hoc test; Fig. 2B). Together, these findings confirm that FMRP specifically regulates NB dye iontophoresis, ruling out genetic background and nonspecific effects.
Figure 2.
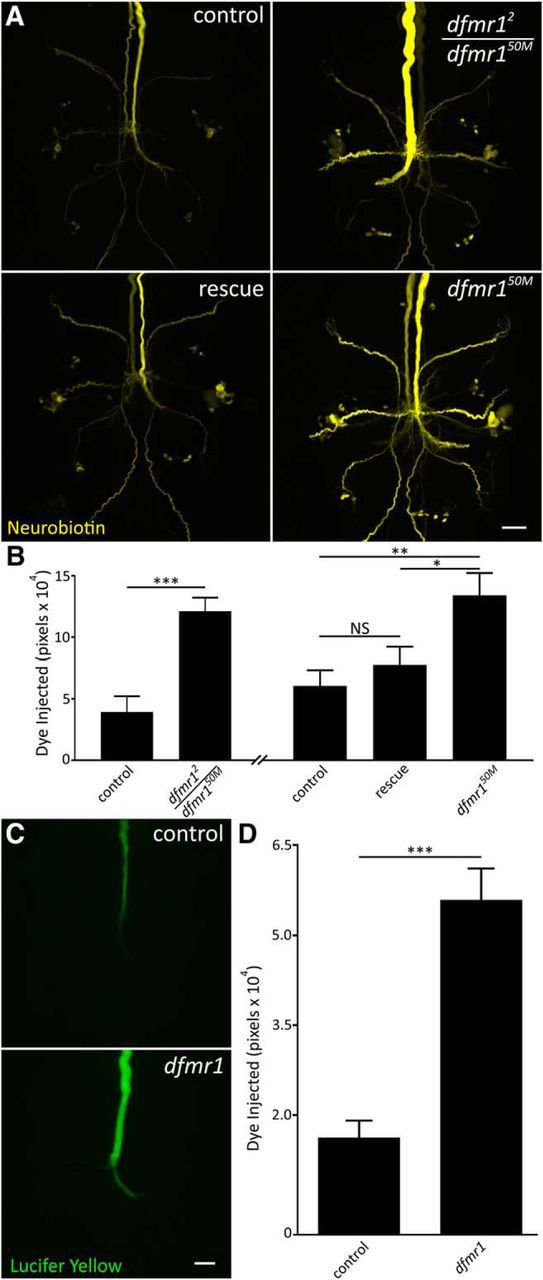
Dye iontophoresis defect is FMRP dependent and charge independent. A, Representative NB images of GFI injections (2 m KAc) for w1118 genetic background (control; top left), heteroallelic dfmr1-null (dfmr12/dfmr150M; top right), wild-type UAS-dFMRP driven with neural elav-Gal4 (rescue; bottom left) in dfmr150M-null background and the dfmr150M-null alone (bottom right). Scale bar, 20 μm. B, Quantification of dye injection in the above four genotypes. The heteroallelic combination and transgenic rescue experiment occurred independently and are displayed with their separate genetic controls. All graphs show data as mean ± SEM. Heteroallelic: control, n = 6; dfmr12/50M, n = 6; rescue control, n = 17; rescue, n = 18; dfmr150M, n = 13. C, Representative images of LY dye injections (in ddH2O) into the GFI neuron in the w1118 genetic background (control; top) and dfmr150M-null mutant (bottom). Scale bar, 20 μm. D, Quantification of LY injection in both genotypes. Control, n = 13; dfmr1 n = 12. Significance determined from two-tailed unpaired t test (B, left; D) and unpaired ANOVA (B, right): *p < 0.05, **p < 0.01, ***p < 0.001.
We next tested whether the dye-loading phenotype is NB-specific or occurs with other small dyes. We repeated the dye injection studies with the commonly used negatively charged small dye, LY (430Da), which had the added benefit of testing for a role of charge polarity (Hanani, 2012). LY is fluorescent and visible during injection, requiring no amplification before imaging. Using the same iontophoresis paradigm, but with reversed polarity, we again found a dramatic increase in dye uptake in the dfmr1 mutants (Fig. 2C). Compared with w1118 genetic controls (top), dfmr1-nulls load far more LY dye (bottom), with increased signal continuously along the descending axon and terminating bend (Fig. 2C). LY passage to electrically-coupled neurons is much slower than NB, thereby largely limiting analysis to the injected GFI (Boerner and Godenschwege, 2011). Similar to NB dye, quantification shows controls (1.6 × 104 ± 0.3 × 104, n = 13) loaded threefold less LY dye than dfmr1-null mutants (5.6 × 104 ± 0.5 × 104, n = 12), a very highly significant difference (p = 3.1 × 10−6, two-tailed unpaired t test; Fig. 2D). These results show the dfmr1 mutant defect is generalizable for small dyes, regardless of dye charge polarity. The selective elevation in small dye iontophoresis, with large TRITC dye loading unchanged, strongly suggested the phenotype arises from a gap junction mechanism.
Increased dye iontophoresis persists in the absence of gap junctions
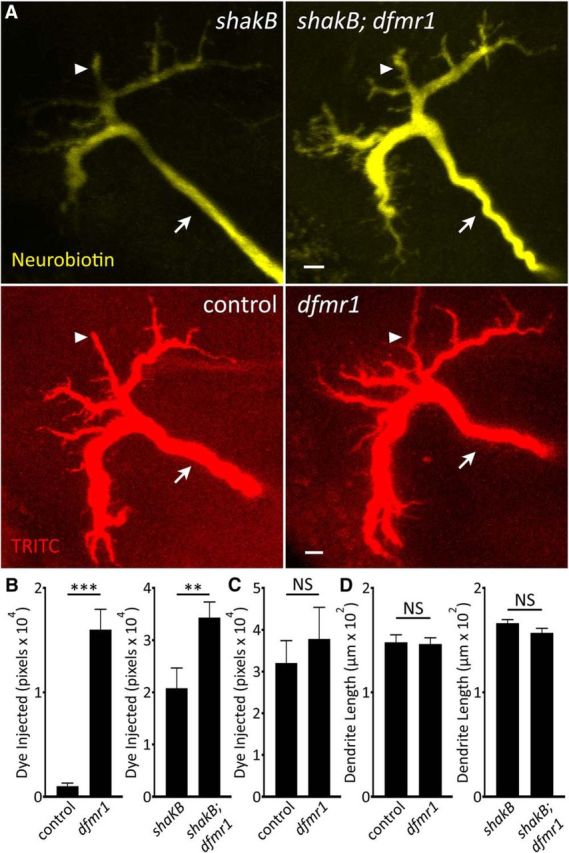
The obvious factor that could determine dye loading based on dye size is gap junction electrical synapses. FXS patients and models exhibit synaptic overgrowth (Comery et al., 1997; Irwin et al., 2000; Zhang et al., 2001; Doll and Broadie, 2015), but these analyses have focused on chemical, not electrical, synapses. We hypothesized that increased electrical connectivity mirrors increased chemical synapse connectivity in the FXS disease state, promoting dye movement into electrically-coupled neurons and thereby bringing more total dye into the GF circuit. All GFI electrical synapses require the Shaking B (ShakB) N+16 isoform (Phelan et al., 2008), allowing tests of gap junction abundance between controls and dfmr1-nulls. Using a ShakB N+16 antibody, TRITC-injected neurons were assayed for fluorescence intensity at TTMn synapses along the GFI bend (Fig. 3A). Contrary to our hypothesis, we found ShakB levels in controls (top) trend higher than in dfmr1-nulls (bottom). ShakB punctae along the GFI bend (arrows) connect to the TTMn (Phelan et al., 2008). Punctae were reduced in dfmr1 mutants, potentially indicating weaker connections (Fig. 3A). Quantification of ShakB intensity shows weak mutant signal (control: 2.5 × 105 ± 0.5 × 105, n = 27; dfmr150M: 1.6 × 105 ± 0.2 × 105, n = 26), although the decrease is not significant with a large sample size (p = 0.13, two-tailed unpaired t test; Fig. 3B). This surprising finding runs counter to our hypothesis that the dye iontophoresis defect results from increased gap junction connectivity. However, it remained possible that gap junctions in dfmr1 mutants are more conductive or spend more time in an open conformation than controls (Niculescu and Lohmann, 2014), thus promoting a loading increase.
Figure 3.

Null dfmr1 neurons manifest dye injection defect without gap junctions. A, Representative images of anti-ShakB labeling in the GFI for the w1118 genetic background (control; top) and dfmr150M-null mutant (bottom). White outline indicates GFI bend as labeled by injected TRITC signal. ShakB signal intensity is represented as a heat map. Arrows indicate ShakB punctae. Scale bar, 5 μm. B, Quantification of ShakB signal intensity in both genotypes, displayed as mean ± SEM. Control, n = 27; dfmr1, n = 26. C, Representative NB injections into the GFI (2 m KAc) for the shakB2 single-mutant (left) and shakB2; dfmr150M double-mutant (right). Scale bar, 10 μm. D, Quantification of the injected dye levels in both genotypes, displayed as mean ± SEM. Control, n = 30; dfmr1, n = 31. Significance was determined from two-tailed unpaired t tests: **p < 0.01.
To determine whether gap junction changes cause increased dye loading in dfmr1-nulls, we examined GFI dye injection patterns in the shakB2-null mutant, an allele that does not produce the ShakB N+16 isoform, thus completely uncoupling GFI connectivity (Blagburn et al., 1999). We performed NB dye injections on both shakB2-null mutants and shakB2; dfmr150M double-null mutants. The shakB2 allele completely abolished all dye coupling in both cases, preventing injected NB dye from leaving the GFI (Fig. 3C). Again, against our expectations, the excessive NB dye-loading defect persisted in dfmr1-nulls in the absence of gap junctions. In shakB2 single-mutants (left), the NB signal is much reduced all along the descending GFI axon as well as terminal bend, compared with the relatively highly elevated signal in shakB2; dfmr150M double-mutants (Fig. 3C, right). Quantification of dye loading shows nearly twice the signal in dfmr1-nulls compared with the matched single-mutant controls (shakB2: 1.7 × 104 ± 0.3 × 104, n = 30; shakB2; dfmr150M: 3.1 × 104 ± 0.4 × 104, n = 31), a very significant increase (p = 0.0025, two-tailed unpaired t test; Fig. 3D). Note that we used only 10 s injections in these shakB2 studies, as the >;2 min injection paradigm caused complete loss of NB/TRITC signals, presumably due to GFI lysis from overloading a constrained volume via electroosmotic effects (see Discussion). Together, these findings indicate the dye iontophoresis defect in dfmr1-null neurons does not depend on gap junction electrical synapses. Having ruled out connectivity changes, we next turned to intrinsic neuron properties to trace the impacted mechanism.
GFI dendrites are structurally unaltered and display dye-loading defect
The polarized structure of neurons provides one hypothesis for the mechanism underlying increased dye loading: dye compartmentalization varies between genotypes. We therefore tested whether increased dfmr1-null axonal dye levels result from reduced dendritic dye accumulation, due either to inhibited retrograde dye movement or decreased dendrite architecture. We measured dendritic dye loading, volume, and coverage in both the w1118 and shakB2 genetic backgrounds to address dye-coupled and -uncoupled circuits. Representative images in Figure 4A show the complete GFI dendritic field, with a dorsal projection connecting the cell body (arrowhead), and the GFI axon projecting toward the bottom right in each image (arrow). Control dendrites (e.g., shakB2; top left) load significantly less NB dye than dfmr1 mutants (shakB2; dfmr150M; top right), in the absence of discernable structural differences. Quantification of NB signal in w1118 controls versus dfmr1-nulls (Fig. 4B, left), and shakB2 versus shakB2; dfmr150M (Fig. 4B, right), shows elevated dye loading in dendrites replicating axonal phenotypes. Both conditions show significant increases in dfmr1-nulls (control: 0.1 × 104 ± 0.03 × 104, n = 20; dfmr150M: 1.6 × 104 ± 0.2 × 104, n = 20, p = 2.7 × 10−7, two-tailed unpaired t test; shakB2: 2.1 × 104±0.4 × 104, n = 27; shakB2; dfmr150M: 3.4 × 104 ± 0.3 × 104, n = 30, p = 0.009, two-tailed unpaired t test; Fig. 4B). This finding indicates there is not a reduction in dye in dfmr1-null dendrites relative to controls that could offset the increase seen in axons.
Figure 4.
Specific small dye iontophoresis is increased in dfmr1 GFI dendritic arbors. A, Representative images of the GFI dendritic arbors in shakB2 single-mutant (top left) and shakB2; dfmr150M double-mutants (top right) labeled with NB injection (yellow; ddH2O) and w1118 genetic control (bottom left), and dfmr150M-null (bottom right) labeled with TRITC injection (red; ddH2O). Arrowheads indicate dendritic projections connected to the cell body (out of focus). Arrows indicate the descending axons. Scale bars, 5 μm. B, NB injection quantification of GFI dendritic arbor for w1118 control versus dfmr150M-null (left), and shakB2 versus shakB2; dfmr150M (right), displayed as the mean ± SEM. w1118, n = 20; dfmr150M, n = 20; shakB2, n = 27; shakB2; dfmr150M, n = 30. C, TRITC injection quantification of GFI dendritic arbor for w1118 control versus dfmr150M-null, displayed as the mean ± SEM. w1118, n = 17; dfmr150M, n = 17. D, Dendritic branch length quantification for w1118 control versus dfmr150M-null (left), and shakB2 versus shakB2; dfmr150M (right), displayed as the mean ± SEM. w1118, n = 17; dfmr150M, n = 18; shakB2, n = 26; shakB2; dfmr150M, n = 28. Significance was determined from two-tailed unpaired t tests: **p < 0.01, ***p < 0.001.
To determine whether the dfmr1-null GFI dendritic architecture is reduced, thereby forcing more injected NB dye into axons, we measured dendritic length, volume, and coverage. TRITC labeling in the GFI dendrites shows no detectable changes in the GFI dendritic field comparing w1118 controls (left) and dfmr1-nulls (Fig. 4A, right bottom). Quantification of the injected TRITC dye shows dendrite volume in the controls (3.2 × 104 ± 0.5 × 104, n = 17) trends smaller than dfmr1-nulls (3.8 × 104 ± 0.8 × 104, n = 17), but the difference is not significant (p = 0.54, two-tailed unpaired t test; Fig. 4C). Measurement of the primary dendritic branches also shows similar dendritic branch length between w1118 controls (148.2 ± 7.1 μm, n = 17) and dfmr1-null mutants (dfmr150M: 146.4 ± 6.0 μm, n = 18, p = 0.85, two-tailed unpaired t test; Fig. 4D, left). Likewise, dendritic branching was similar in the dye-uncoupled circuit comparing shakB2 versus shakB2; dfmr150M double-mutants (shakB2: 166.6 ± 3.3 μm, n = 26; shakB2; dfmr150M: 157.3 ± 4.2 μm, n = 28, p = 0.09, two-tailed unpaired t test; Fig. 4D, right). Overall, dye loading is increased in dfmr1-null dendrites, similar to our findings in axons, indicating the dye loading increase occurs throughout the neuron. Moreover, gross dendritic arbor volume and branching is unchanged in dfmr1 mutants, ruling out the possibility that smaller dfmr1 dendrites drive more dye entry into axons. It is worth noting that there is similarly elevated dye loading in the dfmr1-null cell bodies relative to controls (not pictured). Given this global increase in dye iontophoresis, we next tested multiple other neuronal properties that could impact dye uptake.
Intrinsic neuronal properties unrelated to dye injection defect
We first tested whether an increase in axon size could elevate NB dye loading in dfmr1-nulls, despite unaltered TRITC uptake (Fig. 1). We hypothesized larger volume could accommodate more injected dye, causing the noticeable increase in signal along the GFI axon. We used the TRITC signal, an internal control unchanged between genotypes, to measure GFI axon diameter in control (left) versus dfmr1-null (right) neurons (Fig. 5A). We found no difference in diameter between genotypes over a GFI axonal length of 40 μm (Fig. 5B), with controls averaging 4.9 ± 0.2 μm (n = 15) and dfmr1-nulls 5.0 ± 0.2 μm (n = 16) in diameter, a nonsignificant difference (p = 0.79, two-tailed unpaired t test; Fig. 5B). We confirmed this finding by labeling the GFI with membrane RFP (UAS-mCD8::RFP) driven with a GFI-specific Gal4 driver (91H05-Gal4; Jenett et al., 2012). With this independent transgenic label (data not shown), we again found similar axonal diameters comparing dfmr1-nulls (5.9 ± 0.4 μm, n = 12) versus controls (5.2 ± 0.3 μm, n = 12; p = 0.16, two-tailed unpaired t test), showing FMRP loss does not impact size. We also tested dfmr1 mutants for elevated protein levels as a consequence of increased translation by extracting protein from control versus dfmr1-null brains, and comparing protein levels using the BCA assay. Quantified comparisons of total brain protein levels confirm a highly significant increase in dfmr1-nulls relative to controls by 14.5% (w1118 control: 1.40 ± 0.04 μg/brain, n = 11; dfmr150M: 1.61 ± 0.03 μg/brain, n = 11, p = 0.0003, two-tailed unpaired t test). Thus, neuron size is unchanged in dfmr1 mutants but protein concentration is elevated, consistent with expectations.
Figure 5.
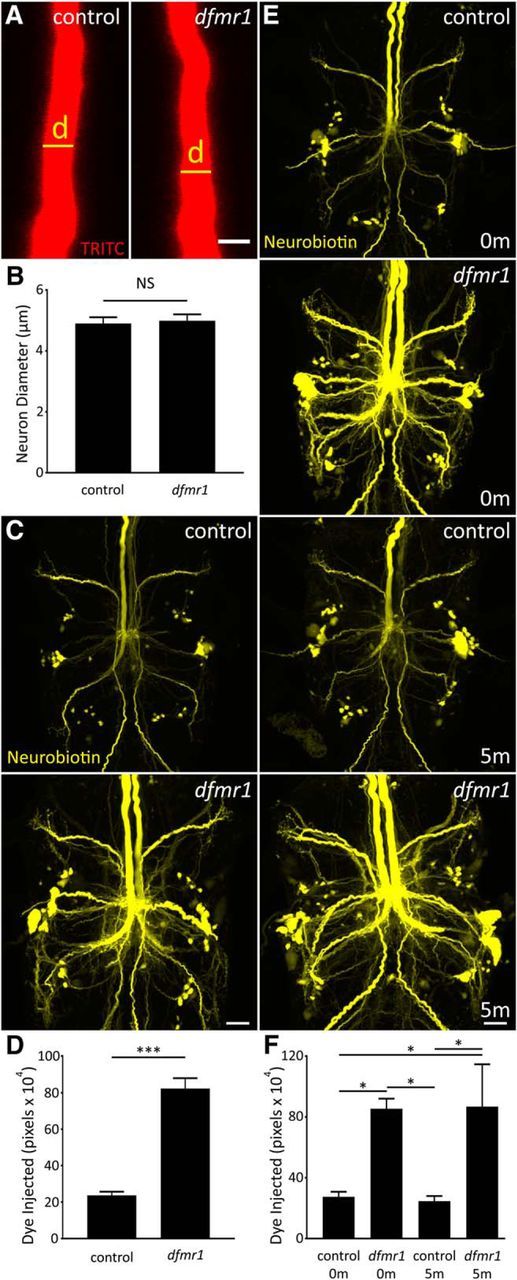
Dye injection defect is not related to multiple GFI neuron properties. A, Representative TRITC injections (ddH2O) showing the GFI descending axon for w1118 genetic background (control; left) and dfmr150M-null mutant (right). Scale bar, 5 μm. B, Quantification of the axon diameter in both genotypes, displayed as the mean ± SEM. Control, n = 15; dfmr1, n = 16. C, Representative NB dye injections into GFI for the w1118 genetic background (control; top) and the dfmr150M-null mutant (bottom) using only KAc-free dye solution (ddH2O). Scale bar, 20 μm. D, Quantification of the injected dye levels, displayed as mean ± SEM. Control, n = 25; dfmr1,n = 26. E, Representative NB injections (ddH2O) into GFI for w1118 genetic background (control; top two panels) and dfmr150M-null mutant (bottom two panels). After dye injection, samples were either immediately dissected (0m) or injected with TRITC for 5 min (5m) with positive current before dissection. Scale bar, 20 μm. F, Quantification of injected dye levels, displayed as the mean ± SEM. 0 m: control, n = 6; dfmr1, n = 6; 5 m: control, n = 6; dfmr1, n = 5. Significance determined from two-tailed unpaired t test (D) and unpaired ANOVA (F): *p < 0.05, ***p < 0.001.
We next explored the possibility that the standard 2 m KAc in the dye-injection electrode (Phelan et al., 1996; Kudumala et al., 2013; Lee and Godenschwege, 2015) is related to the iontophoretic differences between genotypes. We hypothesized that dfmr1 mutants preferentially accept NB+ ions over K+ ions from the electrode due to intrinsic differences in membrane K+ conductivity, characteristic of FXS disease models (Strumbos et al., 2010; Lee et al., 2011). We repeated dye injections with NB/TRITC dissolved in ddH2O and found far more NB loaded in both controls and mutants (Fig. 5C). Incredibly, KAc-free injection controls contained as much dye signal as 2 m KAc injected dfmr1-null mutants typically display (compare Figs. 5C, 1A). KAc-free injection in dfmr1-nulls revealed far more dye-coupled neurons, showing the extraordinary degree of GF circuit connectivity. In both genotypes, signal increased in GFIs and coupled neurons; however, the relative difference between dfmr1-null and control remained unchanged (Fig. 5C,D). Mutants show a 3.5-fold increase in NB signal over controls (control: 23.6 × 104 ± 2.1 × 104, n = 25; dfmr150M: 82.3 × 104 ± 5.7 × 104, n = 26), a very significant elevation (p = 6.2 × 10−11, two-tailed unpaired t test; Fig. 5D). Although this does not rule out a role for neuronal [K+] or K+ channel function in the dye-loading phenotype, we found K+ in the dye solution has no demonstrable impact on relative dye loading between controls and dfmr1-nulls. We further explore the role for K+ currents later (see next section), but first we tested a simpler explanation: does loss of FMRP reduce NB dye leakage from neurons?
In addition to gap junctions, many neuronal membrane pores permit escape of small ionic molecules (like NB) to the extracellular space, including innexin hemi-channels, transient receptor potential channels and purinergic receptors (Bennett et al., 2003; Meyers et al., 2003). The broad FMRP role in mRNA translational regulation makes disruption of these protein levels a possibility in the FXS disease state, although only tenuous links are currently known (Darnell et al., 2011; Naviaux et al., 2013; Kong et al., 2014). We used multiple experiments to test the hypothesis that membrane pores cause controls to leak more dye than dfmr1-null neurons. First, we modified our protocol to rapidly fix brains after 2 m KAc dye injection, dramatically shortening the potential NB dye leak period. When comparing the new rapid-fix w1118 control and dfmr1-null neurons, we found no correction of the NB dye-loading defect (control: 5.3 × 104 ± 1.7 × 104, n = 9; dfmr150M: 16.4 × 104 ± 2.1 × 104, n = 10), with a highly significant increase in mutants (p = 0.0007, two-tailed unpaired t test; data not shown). Second, we performed KAc-free injections and then allowed preparations to sit for 5 min (5m) after injection, to intensify the effects of putative dye leakage. Quantification of dye loading shows no difference in signal with increased time for leakage [no delay (0m) w1118: 30.1 × 104 ± 4.8 × 104, n = 7; delay (5m) w1118: 38.8 × 104 ± 6.6 × 104, n = 8; p = 0.32, two-tailed unpaired t test; data not shown]. Together, these two studies do not support a passive dye leakage effect causing the differential dye loading.
In a final trial, we tested the hypothesis that NB dye leakage occurs only during the actual current injection. The hypothesis was that controls produce more membrane pores than dfmr1-nulls, but only current can drive NB through these pores, permitting leakage solely during iontophoresis. We tested this idea by performing KAc-free NB/TRITC injections for 2 min, and then injecting TRITC dissolved in 2 m KAc for another 5 min. We found no difference between standard injections (Fig. 5E, 0m) and additional current injections (Fig. 5E, 5m) for either controls or dfmr1 mutants, clearly ruling out the possibility of an electrically induced dye leak from the GFI. Moreover, the dye iontophoresis defect was not modulated, with far more NB signal in dfmr1 mutants compared with controls (Fig. 5E). Quantification shows that injected dye remains following prolonged postinjection current for 5 min [0m control (c0): 27.6 × 104 ± 3.2 × 104, n = 6; 0m dfmr150M (d0): 85.5 × 104 ± 6.7 × 104, n = 6; 5m control (c5): 24.7 × 104 ± 3.4 × 104, n = 6; 5m dfmr150M (d5): 86.8 × 104 ± 27.9 × 104, n = 5], with significant differences persisting in all comparisons between controls and mutants (c0 vs d0: p = 0.018; c0 vs c5: p = 0.998; c0 vs d5: p = 0.022; c5 vs d0: p = 0.013; c5 vs d5: p = 0.015; d0 vs d5: p = 1.00; unpaired ANOVA, Tukey-Kramer pairwise post hoc test; Fig. 5F). After ruling out the possibility that dye leaks from the circuit, we turned our attention to cation movement to test whether changes in endogenous K+ conductance alters NB injection.
Ionic manipulations alter dye loading but do not resolve dfmr1 defect
A recent large body of work links FMRP to K+ channel expression and function (Brown et al., 2010; Strumbos et al., 2010; Gross et al., 2011; Lee et al., 2011; Deng et al., 2013). We therefore hypothesized that dfmr1 neurons more efficiently expel K+ ions, reducing charge buildup to permit accumulation of more NB+ ions. The work in the previous section supports this hypothesis by showing that removing KAc from the injection solution increases dye signal (Fig. 5C,D). We first used the K+ channel blocker 4-AP (10 mm; Singh and Singh, 1999) to test whether reduced K+ conductance impairs NB dye loading (Fig. 6A). Comparing control (top) and dfmr1-nulls (bottom) without (left) or with 4-AP (right), we found blocking K+ channels greatly reduced dye loading in both genotypes. Importantly, however, a significant difference between drug-treated controls and dfmr1-null mutants remains despite the major reduction in dye uptake (Fig. 6A). Quantification shows that 4-AP treatment strongly reduces NB loading, but also that the dfmr1 defect persists [control 0 mm 4-AP (c−): 8.4 × 104 ± 1.2 × 104, n = 15; dfmr150M 0 mm 4-AP (d−): 18.2 × 104 ± 1.7 × 104, n = 15; control 10 mm 4-AP (c+): 2.2 × 104 ± 0.7 × 104, n = 15; dfmr150M 10 mm 4-AP (d+): 10.1 × 104 ± 1.2 × 104, n = 15], with highly significant differences remaining (c− vs d−: p < 0.0001; c− vs c+: p = 0.004; c− vs d+: p = 0.79; c+ vs d−: p = 0.0001; c+ vs d+: p = 0.0002, d− vs d+: p = 0.0001; unpaired ANOVA, Tukey–Kramer pairwise post hoc test; Fig. 6B). Thus, K+ channel function appears not to explain the dfmr1 defect.
Figure 6.

K+ channel block reduces dye loading without correcting dfmr1 defect. A, Representative NB dye injections (2 m KAc) into GFI in w1118 genetic background (control; top) and dfmr150M-null mutant (bottom). Samples were bathed in standard saline (left) or standard saline + 10 mm 4-AP (right). Scale bar, 20 μm. B, Quantification of injected dye levels, displayed as mean ± SEM. 0 mm 4-AP: control, n = 15; dfmr1, n = 15; 10 mm 4-AP: control, n = 15; dfmr1, n = 15. C, Representative NB injections into GFI (ddH2O) for elav-Gal4-driven UAS-ChR2-XXL animals raised on EtOH vehicle (top) or ATR cofactor (bottom). Scale bar, 20 μm. D, Quantification of injected NB dye levels for both elav-GAL4 and 91H05-Gal4 (images not shown) driven UAS-ChR2-XXL, displayed as mean ± SEM; elav: EtOH, n = 14; ATR, n = 14; 91H05: EtOH, n = 10; ATR, n = 12. E, Representative NB dye injections into GFI (ddH2O) using an internal ground also within the GFI for the w1118 genetic background (top) and dfmr150M-null mutant (bottom). Scale bar, 20 μm. F, Quantification of injected dye levels for both genotypes, displayed as mean ± SEM. Control, n = 9; dfmr1, n = 10. Significance determined with two-tailed unpaired t test (F) and unpaired ANOVA (B, D): **p < 0.01, ***p < 0.001.
To rule out the possibility that 4-AP was not fully blocking all K+ channels involved in the phenotype, we assayed a mixture consisting of both 4-AP (10 mm) and TEA (10 mm) in the bath, and cesium chloride (140 mm) in the injection pipette. We again found reduced NB dye loading, but the expanded mixture still does not correct the relative dfmr1 mutant phenotype [control untreated (c−): 24.3 × 104 ± 3.4 × 104, n = 16; dfmr150M untreated (d−): 49.4 × 104 ± 5.4 × 104, n = 14; control K+ mixture (c+): 6.3 × 104 ± 1.2 × 104, n = 17; dfmr150M K+ mixture (d+): 19.4 × 104±2.7 × 104, n = 16; c− vs d−: p < 0.0001; c− vs c+: p = 0.0007; c− vs d+: p = 0.29; c+ vs d−: p < 0.0001; c+ vs d+: p = 0.0124; d− vs d+: p < 0.0001; unpaired ANOVA, Tukey–Kramer pairwise post hoc test). We also repeated assays with the LY− dye to test whether the reduced dye loading caused by K+ channel blockade is specific to cationic dyes. We found that 10 mm 4-AP during LY injection also reduces dye uptake in both control and dfmr1-null neurons and maintains the pattern of increased loading in the absence of FMRP [LY control 0 mm 4-AP (c−): 1.0 × 104 ± 0.3 × 104, n = 35; LY dfmr150M 0 mm 4-AP (d−): 2.3 × 104 ± 0.4 × 104, n = 34; LY control 10 mm 4-AP (c+): 0.5 × 104 ± 0.2 × 104, n = 36; LY dfmr150M 10 mm 4-AP (d+): 0.7 × 104 ± 0.2 × 104, n = 36; c− vs d−: p = 0.017; c− vs c+: p = 0.57; c− vs d+: p = 0.85; c+ vs d−: p = 0.0002; c+ vs d+: p = 0.96; d− vs d+: p = 0.0012; unpaired ANOVA, Tukey–Kramer pairwise post hoc test]. Note that the lowered dye injection levels following 4-AP application makes fluorescence readings more variable and complicates determination of the significance of the LY effects. We conclude that 4-AP reduces NB/LY dye loading in both genotypes, but blocking K+ channels does not alleviate differential dye loading between dfmr1 and controls. To further test the role of ionic conductance, we next assayed increased conductivity.
We increased K+ conductance via K+ channel overexpression, including Irk2 (inwardly rectifying K+ channel 2; mammalian Kir2.1) and Shaw (Shaker cognate w; mammalian Kv3.2). Neural elav-Gal4 driven Irk2A causes a trending but nonsignificant increase in NB dye loading (elav/+ control: 22.2 × 104 ± 2.5 × 104, n = 25; elav/UAS-Irk2: 30.9 × 104 ± 4.1 × 104, n = 22, p = 0.08, two-tailed unpaired t test). Neural elav-Gal4 Shaw overexpression proved to be lethal and therefore could not be examined, but targeted GFI-specific 91H05-Gal4-driven Shaw expression has no discernable effect on dye loading (91H05/+: 57.5 × 104 ± 4.7 × 104, n = 14; 91H05/UAS-Shaw: 58.4 × 104 ± 7.0 × 104, n = 14, p = 0.91, two-tailed unpaired t test). We also used blue light-gated channelrhodopsin with the ATR cofactor (Nagel et al., 2003) to provide temporal and pharmacological control over elevated cation conductivity. To maximize current, we used high-conductance ChR2-XXL channels (Dawydow et al., 2014) both pan-neuronally (elav-Gal4) and targeted to the GFI (91H05-Gal4). Both drivers show no difference in dye loading between controls fed the EtOH vehicle (Fig. 6C, top) and experimentals fed the essential ATR cofactor (bottom). Quantification shows ChR2-XXL activation causes no increase in dye loading for either driver (elav-Gal4 EtOH: 52.4 × 104 ± 5.5 × 104, n = 14; elav ATR: 55.4 × 104 ± 8.9 × 104, n = 14, p = 0.78, two-tailed unpaired t test; 91H05-Gal4 EtOH: 63.5 × 104 ± 8.4 × 104, n = 10; 91H05 ATR: 55.7 × 104 ± 7.9 × 104, n = 12, p = 0.5, two-tailed unpaired t test; Fig. 6D). These results show increased K+ permeability is unlikely to be the cause of increased dfmr1 dye loading.
To more broadly test whether conductance across the membrane alters dye loading we inserted both dye iontophoresis and ground electrodes into the GFI neuron, short-circuiting current flow across the membrane. We hypothesized that this “closed circuit” should prevent the dye-loading phenotype if the dfmr1 dye defect arises from membrane conductivity differences. With this dye injection paradigm (KAc-free), the dfmr1 dye defect persisted (Fig. 6E). Controls showed the normal basal labeling of the injected GFI and dye-coupled neurons (top), whereas dfmr1-null mutants showed an elevated, intense dye signal (bottom). Quantification of injected dye shows an internal ground maintains the iontophoresis defect (control: 15.0 × 104 ± 3.0 × 104, n = 9; dfmr150M: 56.0 × 104 ± 6.3 × 104, n = 10), with a highly significant effect (p = 5.5 × 10−5, two-tailed unpaired t test; Fig. 6F). Thus, results from removing the plasma membrane from the injection circuit suggest the defect does not arise from membrane ionic flux differences between the genotypes. Together, blocking K+ channels with 4-AP reduces total dye loading dramatically, but does not equalize signal in controls and dfmr1 mutants, dye loading is not impacted by introducing Drosophila K+ or exogenous cation channels and removing membrane roles does not correct differential dye loading. This suggests that cytoplasmic changes in the absence of FMRP must contribute to the rate at which small charged dyes accumulate in neurons during iontophoresis.
Elevated intracellular dye iontophoresis rate in dfmr1-null neurons
To test how more NB dye enters the GF circuit over time in dfmr1-null mutants, we performed KAc-free dye injections for a series of durations: 10 s, 30 s, 1 min, and 2 min (Fig. 7). We hypothesized the dye signal would increase linearly as a function of time in both genotypes, with a reduced slope in controls compared with mutants. To our surprise, timed injection results showed a much more striking difference in dye loading (Fig. 7A): control neurons display relatively limited dye uptake increases after the first 10 s of injection (left) compared with dfmr1-null's steady, strong increase at each time point (right). In controls, the GFI axon and PSI cell bodies did not load significantly more dye over time, and the coupled neurons remained relatively weakly labeled after they reached an apparent equilibrium point (Fig. 7A). In sharp contrast, the dfmr1 mutants began with similar NB dye levels compared with controls in the first 10 s, but then continuously accumulated more dye in the injected GFI as well as dye-coupled neurons, as evidenced by the dramatically broadening circuit dye incorporation over time. In dfmr1-nulls, dye-coupled neurons that were not visible after 10 s of iontophoresis began to reach the quantification threshold after 30 s, and greatly contributed to the overall signal by 2 min (Fig. 7A). Note that dye was present in the same circuit neurons for both genotypes, and the broader pattern in dfmr1-nulls results from more neurons crossing the quantification threshold, rather than new circuit partners.
Figure 7.
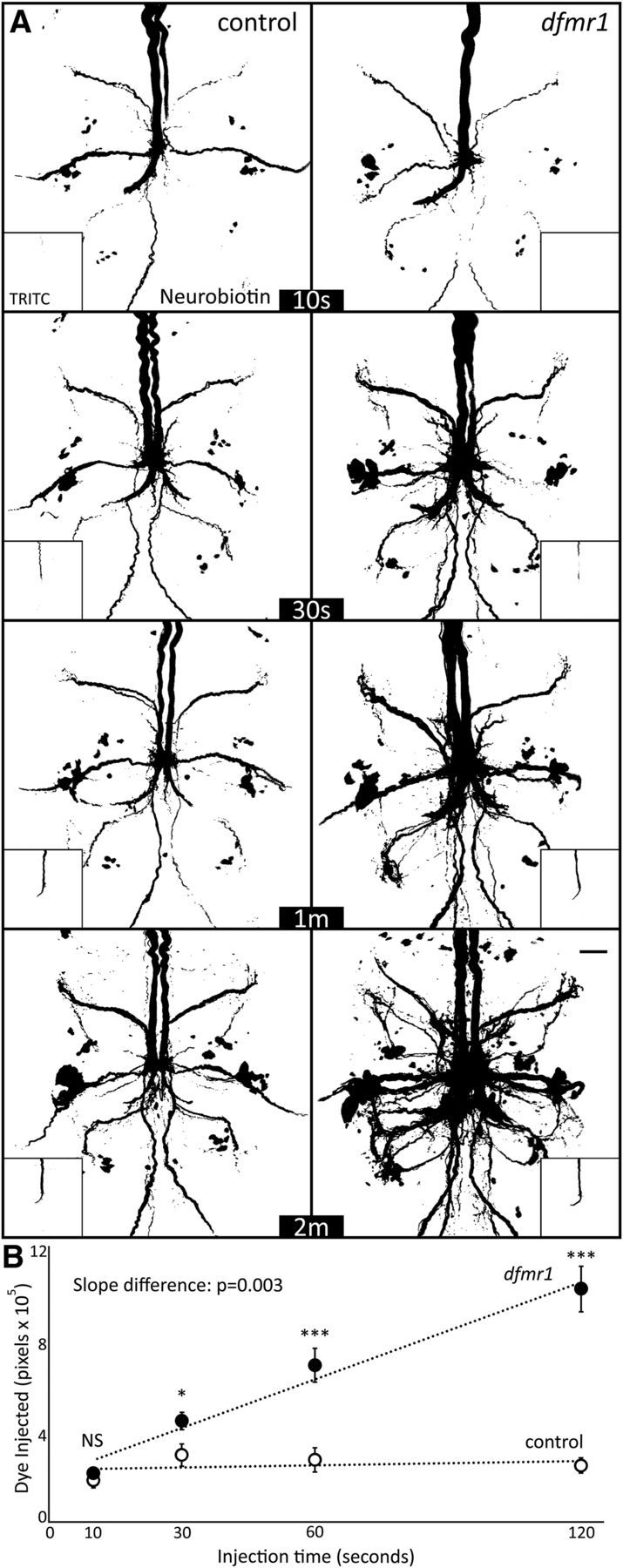
Dye iontophoresis rate is highly elevated in dfmr1-null mutant neurons. A, Representative NB dye injection (ddH2O) images of GFI for the w1118 genetic background (control, left) and dfmr150M-null mutant (right) with progressively increasing periods of iontophoresis (10, 30, 60, and 120 s). The main images show NB dye loading, and insets show coinjected TRITC dye loading within the same neurons. All images display pixels above the threshold of 59. Scale bar, 20 μm. B, Quantification of injected dye levels at each of the four time points for both genotypes, displayed as mean ± SEM. Control 10 s, n = 10; control 30 s, n = 10; control 1 m, n = 10; control 2 m, n = 10; dfmr1 10 s, n = 10; dfmr1 30 s, n = 10; dfmr1 1 m, n = 9; dfmr1 2 m, n = 10. Significance determined from two-tailed unpaired t test (points) and unpaired ANCOVA (slope): *p < 0.05, **p < 0.01, ***p < 0.001.
Quantified comparisons show a significantly elevated rate of dye loading in dfmr1 mutants (Fig. 7B). After 10 s of injection, there was no significant difference (control: 18.0 × 104 ± 3.4 × 104, n = 10; dfmr150M: 20.9 × 104 ± 2.3 × 104, n = 10; p = 0.48), but by 30 s a small but significant elevation was apparent in the mutants (control: 29.1 × 104 ± 5.0 × 104, n = 10; dfmr150M: 44.0 × 104 ± 3.9 × 104, n = 10; p = 0.03). After a full minute, a 2.5-fold increase was present in dfmr1-nulls (control: 27.0 × 104 ± 5.4 × 104, n = 10; dfmr150M: 68.3 × 104±7.5 × 104, n = 9; p = 4.7 × 10−4), which grew to more than a fourfold increase by 2 min of injection (control: 24.6 × 104 ± 3.5 × 104, n = 10; dfmr150M: 101.4 × 104 ± 10.1 × 104, n = 10; p = 1.6 × 10−5, two-tailed unpaired t test; Fig. 7B). The result is a significant difference in the dye loading slope (p = 0.003, unpaired ANCOVA), with a nearly 25-fold increase in the dfmr1-nulls compared with the matched controls (Fig. 7B). In contrast to NB dye, TRITC levels increased linearly at a comparable rate between the two genotypes (Fig. 7A, insets), with no significant difference between the slopes (p = 0.61, unpaired ANCOVA), supporting our findings that FMRP loss does not affect TRITC dye loading (Fig. 1). Together, these results show a highly selective FMRP-dependent dye iontophoresis rate defect, with dfmr1-null mutants losing a temporal restriction on iontophoresis. We conclude the FXS model lacks a barrier to small charged dye uptake, increasing dye loading in injected neurons and, consequently, signal throughout the electrically-coupled circuit.
Discussion
We have discovered an unexpected new effect of FMRP loss in the Drosophila FXS model: dye iontophoresis rates dramatically increase in neurons lacking FMRP, with the effect dependent on dye size but not charge. The defect is gap junction independent, but also increases dye transfer to downstream electrically-coupled neurons within the circuit. Evidence that genotype differences can affect neural dye iontophoresis is totally unprecedented, to our knowledge, despite extensive dye injection studies over many decades (Schofield, 2008; Lanciego and Wouterlood, 2011; Hanani, 2012). Both NB and LY dye-loading defects are striking, but the absence of loading differences for the larger TRITC dye complicates the findings. Transdermal studies show electromigration primarily drives rapid electrophoretic ion movement for small molecules, whereas the slower movement of larger molecules depends more on electroosmosis (Marro et al., 2001; Kalia et al., 2004). Electroosmosis is an electric field-dependent solvent flow caused by bulk movement of ions that accumulate along solid–liquid interfaces (Pikal, 2001). Here, this would result in ionic flux along the electrode sides, carrying a slow flow of solvent and larger dye molecules into the neuron. This effect may explain why larger TRITC responds differently than smaller NB and LY dyes: the slow electroosmotic solvent flow remains unchanged in dfmr1 mutants, whereas the rapid electromigration rate becomes significantly disrupted.
Several tests show FMRP loss specifically causes dye iontophoresis elevation. First, we replicated the defect with a second dfmr1-null allele, ruling out nonspecific recessive effects. Second, we rescued the phenotype with wild-type FMRP targeted to neurons, showing neuronal FMRP restricts dye uptake. Third, we showed the defect in shakB2 mutants in the Oregon-R background, not only ruling out gap junction connectivity defects, but also showing persistence across genetic backgrounds. We found the defect is not NB-specific by repeating with negatively charged LY, eliminating NB-specific explanations, such as biotinidase degradation (Mishra et al., 2010), and positive-charge-specific theories, such as anionic precipitation. Gap junction coupling only allows passage of small molecules (<1 kDa), regardless of charge, and seemed the obvious explanation here (Weber et al., 2004). Increased electrical synapses appeared a predicted consequence of well reported FXS synaptic overgrowth and hyperconnectivity (Comery et al., 1997; Irwin et al., 2001; Zhang et al., 2001; Doll and Broadie, 2015). Indeed, recent work hints that FXS models have elevated gap junctions (Kong et al., 2014). However, we found dfmr1-nulls show a trending gap junction reduction (ShakB levels), and complete gap junction loss (shakB2-null) does not correct the dfmr1 dye loading phenotype. Likewise, LY loading was primarily limited to the GFI under our injection conditions, similar to shakB2 results, further confirming that dye movement to coupled neurons is not required for differential dye loading. Note the dye loading differences between dfmr1 and control were reduced in the shakB2 background, suggesting partial rescue. The assay's shorter injection times may explain the difference, but it is possible that elevated interneuron connectivity is a separable neuronal property causing dfmr1-nulls to increase dye uptake.
In our experiments, the only manipulation that increased dye loading was removing K+ from the injection electrode, although the dfmr1 phenotype persisted. The likely explanation is that dye no longer competes with smaller, more mobile, K+ ions and thus constitutes more of the injection current; a shift well known in transdermal iontophoresis (Pikal, 2001). K+-free injection allows lower currents to deliver more dye over shorter periods, an advantage used in subsequent experiments. The dfmr1 dye-loading defect holds for dyes of both polarities, so it remained possible, although not parsimonious, that FMRP loss disrupts multiple ion channel classes, altering anionic and cationic flow. We first tested K+ channel function, known to be disrupted in FXS models (Brager and Johnston, 2014; Contractor et al., 2015). Importantly, application of K+ channel blocker 4-AP (Singh and Singh, 1999) reduces dye loading, although the relative dfmr1 phenotype persisted. A relationship between K+ currents and iontophoretic loading has not been previously reported, to our knowledge, but suggests that increased dfmr1 dye loading could result from elevated K+ currents (Contractor et al., 2015). However, we could not significantly increase dye loading by elevating conductance with Irk2 or Shaw K+ channels, or transgenic channelrhodopsin. Of course, changes to endogenous K+ conductance in dfmr1-nulls may not be sufficiently replicated with channel manipulations. However, persistence of the phenotype after removal of the plasma membrane from the injection circuit supports the conclusion that ion channels are not the primary cause of altered dfmr1-null dye loading.
Another possibility is that dfmr1-null neurons have a shifted iontophoretic current composition that does not affect net current flow. The current composition is the percentage of charge carried by each ion group in the system. Each ionic contribution is determined by its charge, concentration, size, and surrounding environment, represented by the transference number (Sackin and Boulpaep, 1981). FMRP loss could shift current composition, making it more difficult for neuronal ions to enter the electrode, favoring instead dye ions entering the neuron. This could be accomplished either by changing the ratio of ion groups in the cytoplasm or the cytoplasmic viscosity ions experience. The importance of ion ratios was demonstrated when we removed the small K+ ion from the dye injection electrode and caused more dye to enter neurons without changing the iontophoretic current. A related change could occur in dfmr1-null neurons, with the mutant cytoplasm containing a higher ratio of large ions than controls, thus reducing average cytoplasmic ion speed and favoring dye transfer into the neuron. An increase in large ion concentration may be a consequence of the known disruptions to multiple ion channel classes in the FXS model, altering neuronal resting ionic concentrations (Contractor et al., 2015). Testing such a hypothesis may require more sophisticated channel manipulations. Alternatively, the increased protein concentrations in dfmr1-nulls, confirmed in this work, could shift current composition to favor dye uptake if their charged residues alter the ionic composition of the cytosol (Qin et al., 2005; Tessier and Broadie, 2008). A similar shift could come from byproducts of disrupted enzymatic metabolism in the FXS condition (Lima-Cabello et al., 2016). Importantly, correcting protein levels via slowing the elevated translation rate ameliorates many FXS symptoms (Bolduc et al., 2008; Osterweil et al., 2013), and could prove equally efficacious for the increased dfmr1-null dye loading.
In timed iontophoresis experiments, control neurons approach dye equilibrium after just 10 s of injection, whereas dfmr1-null neurons exhibit constant dye accumulation over minutes. The limitation on dye loading is not caused by leakage, as fluorescence intensity remains constant regardless of time between dye injection and fixation. Dye loading was increased in all regions of dfmr1-null neurons, including dendrites and axons, showing that differential dye compartmentalization or preferential subcellular filling does not explain the defect. We also rule out neuronal dye capacity, as both dendrite and axon sizes are unaltered. Moreover, any internal capacity differences would similarly affect the larger TRITC dye, which loaded equally in both genotypes. Further, iontophoretic differences based on neuronal capacity would not permit the increased dye loading caused by removing K+ from the injection electrode. It is possible that increased cytoplasmic viscosity in dfmr1-nulls shifts the ratio of incoming to outgoing ions by altering ionic flux (Kühn et al., 2011). This defect could arise from increased cytosolic concentration of proteins resulting from the loss of FMRP translational repression (Qin et al., 2005; Tessier and Broadie, 2008), Such a disruption would have wide-ranging developmental and functional effects on neural circuits in the FXS disease state.
In conclusion, we have uncovered a robust new consequence of FMRP loss in Drosophila FXS model neurons. Quantitative dye iontophoresis reveals dfmr1-null neurons selectively lack an impediment to small charged dye entry. The FMRP mechanism limits iontophoretic rate, strictly capping the maximum dye transfer. The dfmr1 mutant defect is limited to small NB+1 and LY−1 dyes, with no effect on the larger TRITC+1 dye. Increased gap junction coupling seemed the obvious explanation for our findings, but persistence of the NB loading defect in shakB2-nulls lacking gap junctions, as well as the GFI-restricted LY loading defect in dfmr1-nulls, rules out neuronal coupling as causative. Neuron size, membrane porosity, and ion channel function all appear to play negligible roles in the phenotype. This study has also shed new light on the classic iontophoretic dye-injection technique, showing that: (1) genotype can dictate dye iontophoresis rate, (2) ionic composition can limit dye loading, and (3) K+ channel currents can enhance dye transfer. All of these findings are unprecedented, despite extensive dye studies for decades, and force us to expand our understanding of this classic technique. FMRP regulates numerous transcript targets (Darnell et al., 2011), primarily repressing translation (Cvetkovska et al., 2013; Ifrim et al., 2015), and also regulates numerous ion channel classes (Contractor et al., 2015), providing several avenues for future investigation of the FMRP intersection with dye loading. Determining the causative mechanism(s) underlying disrupted dye iontophoresis will be a difficult, albeit important goal because it may help identify neuronal causes of FXS symptoms.
Footnotes
This work was supported by National Institutes of Health Grants MH084989 (K.B.), and NS092250 and HD007502 (T.K.). We thank the Bloomington Drosophila Stock Center (Indiana University) for numerous lines, Dr. Tanja Godenschwege (Florida Atlantic University) and Dr. Pauline Phelan (University of Kent, UK) for ShakB transgenic lines, Dr. Jonathan Bacon (University of Sussex, UK) for the kind gift of ShakB antibody, and Mary Lynn Dear and Dr. Caleb Doll for comments on the paper.
The authors declare no competing financial interests.
References
- Allen MJ, Drummond JA, Moffat KG (1998) Development of the giant fiber neuron of Drosophila melanogaster. J Comp Neurol 397:519–531. 10.1002/(SICI)1096-9861(19980810)397:4%3C519::AID-CNE5%3E3.0.CO%3B2-4 [DOI] [PubMed] [Google Scholar]
- Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stützer A, Armache KJ, Simon MD, Xu C, Ali M, Murn J, Prisic S, Kutateladze TG, Vakoc CR, Min J, Kingston RE, Fischle W, Warren ST, Page DC, Shi Y (2014) A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 157:869–881. 10.1016/j.cell.2014.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V (2008) Rapid activity-dependent modifications in synaptic structure and function require bidirectional wnt signaling. Neuron 57:705–718. 10.1016/j.neuron.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MV, Contreras JE, Bukauskas FF, Sáez JC (2003) New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci 26:610–617. 10.1016/j.tins.2003.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagburn JM, Alexopoulos H, Davies JA, Bacon JP (1999) Null mutation in shaking-B eliminates electrical, but not chemical, synapses in the Drosophila giant fiber system: a structural study. J Comp Neurol 404:449–458. 10.1002/(SICI)1096-9861(19990222)404:4%3C449::AID-CNE3%3E3.3.CO%3B2-4 [DOI] [PubMed] [Google Scholar]
- Boerner J, Godenschwege TA (2010) Application for the Drosophila ventral nerve cord standard in neuronal circuit reconstruction and in-depth analysis of mutant morphology. J Neurogenet 24:158–167. 10.3109/01677063.2010.489624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerner J, Godenschwege TA (2011) Whole mount preparation of the adult Drosophila ventral nerve cord for giant fiber dye injection. J Vis Exp 52:e3080. 10.3791/3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T (2008) Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci 11:1143–1145. 10.1038/nn.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Johnston D (2014) Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull 103:11–17. 10.1016/j.brainresbull.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK (2010) Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat Neurosci 13:819–821. 10.1038/nn.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94:5401–5404. 10.1073/pnas.94.10.5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C (2015) Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87:699–715. 10.1016/j.neuron.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley B, Kirjanen S, Partanen J, Castrén M (2016) Epileptic electroencephalography profile associates with attention problems in children with fragile X syndrome: review and case series. Front Hum Neurosci 10:353. 10.3389/fnhum.2016.00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovska V, Hibbert AD, Emran F, Chen BE (2013) Overexpression of down syndrome cell adhesion molecule impairs precise synaptic targeting. Nat Neurosci 16:677–682. 10.1038/nn.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal GR, Rawson J, Gassaway B, Kwok B, Tong Y, Ptácek LJ, Bates E (2012) An inwardly rectifying K+ channel is required for patterning. Development 139:3653–3664. 10.1242/dev.078592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261. 10.1016/j.cell.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawydow A, Gueta R, Ljaschenko D, Ullrich S, Hermann M, Ehmann N, Gao S, Fiala A, Langenhan T, Nagel G, Kittel RJ (2014) Channelrhodopsin-2-XXL, a powerful optogenetic tool for low-light applications. Proc Natl Acad Sci U S A 111:13972–13977. 10.1073/pnas.1408269111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA (2013) FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77:696–711. 10.1016/j.neuron.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA (2002) Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34:973–984. 10.1016/S0896-6273(02)00724-9 [DOI] [PubMed] [Google Scholar]
- Doll CA, Broadie K (2014) Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front Cell Neurosci 8:30. 10.3389/fncel.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll CA, Broadie K (2015) Activity-dependent FMRP requirements in development of the neural circuitry of learning and memory. Development 142:1346–1356. 10.1242/dev.117127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll CA, Broadie K (2016) Neuron class-specific requirements for fragile X mental retardation protein in critical period development of calcium signaling in learning and memory circuitry. Neurobiol Dis 89:76–87. 10.1016/j.nbd.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godenschwege TA, Hu H, Shan-Crofts X, Goodman CS, Murphey RK (2002) Bi-directional signaling by semaphorin 1a during central synapse formation in Drosophila. Nat Neurosci 5:1294–1301. 10.1038/nn976 [DOI] [PubMed] [Google Scholar]
- Golovin RM, Broadie K (2016) Developmental experience-dependent plasticity in the first synapse of the Drosophila olfactory circuit. J Neurophysiol 116:2730–2738. 10.1152/jn.00616.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ (2011) Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31:5693–5698. 10.1523/JNEUROSCI.6661-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanani M. (2012) Lucifer yellow: an angel rather than the devil. J Cell Mol Med 16:22–31. 10.1111/j.1582-4934.2011.01378.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge JJ, Choi JC, O'Kane CJ, Griffith LC (2005) Shaw potassium channel genes in Drosophila. J Neurobiol 63:235–254. 10.1002/neu.20126 [DOI] [PubMed] [Google Scholar]
- Ifrim MF, Williams KR, Bassell GJ (2015) Single-molecule imaging of PSD-95 mRNA translation in dendrites and its dysregulation in a mouse model of fragile X syndrome. J Neurosci 35:7116–7130. 10.1523/JNEUROSCI.2802-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044. 10.1093/cercor/10.10.1038 [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98:161–167. 10.1002/1096-8628(20010115)98:2%3C161::AID-AJMG1025%3E3.0.CO%3B2-B [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN (1976) L-glutamate as an excitatory transmitter at the Drosophila larval neuromuscular junction. J Physiol 262:215–236. 10.1113/jphysiol.1976.sp011593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenett A, Rubin GM, Ngo TT, Shepherd D, Murphy C, Dionne H, Pfeiffer BD, Cavallaro A, Hall D, Jeter J, Iyer N, Fetter D, Hausenfluck JH, Peng H, Trautman ET, Svirskas RR, Myers EW, Iwinski ZR, Aso Y, DePasquale GM, et al. (2012) A gal4-driver line resource for Drosophila neurobiology. Cell Rep 2:991–1001. 10.1016/j.celrep.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST (2004) Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 7:113–117. 10.1038/nn1174 [DOI] [PubMed] [Google Scholar]
- Kalia YN, Naik A, Garrison J, Guy RH (2004) Iontophoretic drug delivery. Adv Drug Deliv Rev 56:619–658. 10.1016/j.addr.2003.10.026 [DOI] [PubMed] [Google Scholar]
- King DG, Wyman RJ (1980) Anatomy of the giant fibre pathway in Drosophila: I. Three thoracic components of the pathway. J Neurocytol 9:753–770. 10.1007/BF01205017 [DOI] [PubMed] [Google Scholar]
- Kong SW, Sahin M, Collins CD, Wertz MH, Campbell MG, Leech JD, Krueger D, Bear MF, Kunkel LM, Kohane IS (2014) Divergent dysregulation of gene expression in murine models of fragile X syndrome and tuberous sclerosis. Mol Autism 5:16. 10.1186/2040-2392-5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudumala S, Freund J, Hortsch M, Godenschwege TA (2013) Differential effects of human L1CAM mutations on complementing guidance and synaptic defects in Drosophila melanogaster. PLoS One 8:e76974. 10.1371/journal.pone.0076974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn T, Ihalainen TO, Hyväluoma J, Dross N, Willman SF, Langowski J, Vihinen-Ranta M, Timonen J (2011) Protein diffusion in mammalian cell cytoplasm. PLoS One 6:e22962. 10.1371/journal.pone.0022962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciego JL, Wouterlood FG (2011) A half century of experimental neuroanatomical tracing. J Chem Neuroanat 42:157–183. 10.1016/j.jchemneu.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP (1991) The anterograde and retrograde transport of neurobiotin in the central nervous system of the rat: comparison with biocytin. J Neurosci Methods 39:163–174. 10.1016/0165-0270(91)90082-B [DOI] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY (2011) Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72:630–642. 10.1016/j.neuron.2011.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LH, Godenschwege TA (2015) Structure–function analyses of tyrosine phosphatase PTP69D in giant fiber synapse formation of Drosophila. Mol Cell Neurosci 64:24–31. 10.1016/j.mcn.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Cabello E, Garcia-Guirado F, Calvo-Medina R, El Bekay R, Perez-Costillas L, Quintero-Navarro C, Sanchez-Salido L, De Diego-Otero Y (2016) An abnormal nitric oxide metabolism contributes to brain oxidative stress in the mouse model for the fragile X syndrome, a possible role in intellectual disability. Oxid Med Cell Longev 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD (2011) Simple neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27:2453–2454. 10.1093/bioinformatics/btr390 [DOI] [PubMed] [Google Scholar]
- Marro D, Kalia YN, Delgado-Charro MB, Guy RH (2001) Optimizing iontophoretic drug delivery: identification and distribution of the charge-carrying species. Pharm Res 18:1709–1713. 10.1023/A:1013370529366 [DOI] [PubMed] [Google Scholar]
- Martinez VG, Javadi CS, Ngo E, Ngo L, Lagow RD, Zhang B (2007) Age-related changes in climbing behavior and neural circuit physiology in Drosophila. Dev Neurobiol 67:778–791. 10.1002/dneu.20388 [DOI] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP (2003) Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci 23:4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Dhingra K, Schüz A, Logothetis NK, Canals S (2010) Improved neuronal tract tracing with stable biocytin-derived neuroimaging agents. ACS Chem Neurosci 1:129–138. 10.1021/cn900010d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu L, Bacon JP, Ito K, Strausfeld NJ (2014) Responses of Drosophila giant descending neurons to visual and mechanical stimuli. J Exp Biol 217:2121–2129. 10.1242/jeb.099135 [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A 100:13940–13945. 10.1073/pnas.1936192100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP, Schuchbauer MA, Rogac M, Tang Q, Dugan LL, Powell SB (2013) Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS One 8:e57380. 10.1371/journal.pone.0057380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu D, Lohmann C (2014) Gap junctions in developing thalamic and neocortical neuronal networks. Cereb Cortex 24:3097–3106. 10.1093/cercor/bht175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr BO, Borgen MA, Caruccio PM, Murphey RK (2014) Netrin and frazzled regulate presynaptic gap junctions at a Drosophila giant synapse. J Neurosci 34:5416–5430. 10.1523/JNEUROSCI.3183-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF (2013) Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77:243–250. 10.1016/j.neuron.2012.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Nakagawa M, Wilkin MB, Moffat KG, O'Kane CJ, Davies JA, Bacon JP (1996) Mutations in shaking-B prevent electrical synapse formation in the Drosophila giant fiber system. J Neurosci 16:1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Goulding LA, Tam JL, Allen MJ, Dawber RJ, Davies JA, Bacon JP (2008) Molecular mechanism of rectification at identified electrical synapses in the Drosophila giant fiber system. Curr Biol 18:1955–1960. 10.1016/j.cub.2008.10.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikal MJ. (2001) The role of electroosmotic flow in transdermal iontophoresis. Adv Drug Deliv Rev 46:281–305. 10.1016/S0169-409X(00)00138-1 [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB (2005) Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci 25:5087–5095. 10.1523/JNEUROSCI.0093-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackin H, Boulpaep EL (1981) Isolated perfused salamander proximal tubule: methods, electrophysiology, and transport. Am J Physiol 241:F39–52. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield BR. (2008) Retrograde axonal tracing with fluorescent markers. Curr Protoc Neurosci 1:Unit 1.17. 10.1002/0471142301.ns0117s43 [DOI] [PubMed] [Google Scholar]
- Singh A, Singh S (1999) Unmasking of a novel potassium current in Drosophila by a mutation and drugs. J Neurosci 19:6838–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt T, Lang MC, Medda R, Engelhardt J, Hell SW (2007) 2,2′-Thiodiethanol: a new water soluble mounting medium for high resolution optical microscopy. Microsc Res Tech 70:1–9. 10.1002/jemt.20396 [DOI] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK (2010) Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci 30:10263–10271. 10.1523/JNEUROSCI.1125-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YA, Wyman RJ (1996) Passover eliminates gap junctional communication between neurons of the giant fiber system in Drosophila. J Neurobiol 30:340–348. 10.1002/(SICI)1097-4695(199607)30:3%3C340::AID-NEU3%3E3.0.CO%3B2-4 [DOI] [PubMed] [Google Scholar]
- Tanouye MA, Wyman RJ (1980) Motor outputs of giant nerve fiber in Drosophila. J Neurophysiol 44:405–421. [DOI] [PubMed] [Google Scholar]
- Tessier CR, Broadie K (2008) Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135:1547–1557. 10.1242/dev.015867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber PA, Chang HC, Spaeth KE, Nitsche JM, Nicholson BJ (2004) The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophys J 87:958–973. 10.1529/biophysj.103.036350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K (2001) Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107:591–603. 10.1016/S0092-8674(01)00589-X [DOI] [PubMed] [Google Scholar]