

Abstract
Activity-dependent synaptic plasticity plays a critical role in the refinement of circuitry during postnatal development and may be disrupted in conditions that cause intellectual disability, such as Down syndrome (DS). To test this hypothesis, visual cortical plasticity was assessed in Ts65Dn mice that harbor a chromosomal duplication syntenic to human chromosome 21q. We find that Ts65Dn mice demonstrate a defect in ocular dominance plasticity (ODP) following monocular deprivation. This phenotype is similar to that of transgenic mice that express amyloid precursor protein (APP), which is duplicated in DS and in Ts65DN mice; however, normalizing APP gene copy number in Ts65Dn mice fails to rescue plasticity. Ts1Rhr mice harbor a duplication of the telomeric third of the Ts65Dn-duplicated sequence and demonstrate the same ODP defect, suggesting a gene or genes sufficient to drive the phenotype are located in that smaller duplication. In addition, we find that Ts65Dn mice demonstrate an abnormality in olfactory system connectivity, a defect in the refinement of connections to second-order neurons in the olfactory bulb. Ts1Rhr mice do not demonstrate a defect in glomerular refinement, suggesting that distinct genes or sets of genes underlie visual and olfactory system phenotypes. Importantly, these data suggest that developmental plasticity and connectivity are impaired in sensory systems in DS model mice, that such defects may contribute to functional impairment in DS, and that these phenotypes, present in male and female mice, provide novel means for examining the genetic and molecular bases for neurodevelopmental impairment in model mice in vivo.
SIGNIFICANCE STATEMENT Our understanding of the basis for intellectual impairment in Down syndrome is hindered by the large number of genes duplicated in Trisomy 21 and a lack of understanding of the effect of disease pathology on the function of neural circuits in vivo. This work describes early postnatal developmental abnormalities in visual and olfactory sensory systems in Down syndrome model mice, which provide insight into defects in the function of neural circuits in vivo and provide an approach for exploring the genetic and molecular basis for impairment in the disease. In addition, these findings raise the possibility that basic dysfunction in primary sensory circuitry may illustrate mechanisms important for global learning and cognitive impairment in Down syndrome patients.
Keywords: APP, Down syndrome, ocular dominance plasticity, olfactory glomerular refinement, Ts1Rhr, Ts65Dn
Introduction
Down syndrome (DS) patients experience profound delay in the attainment of developmental milestones as well as impairment in multiple cognitive domains, including speech, language production, and short-term memory (for review, see Chapman and Hesketh, 2000). To better understand the physiologic and molecular bases for these phenotypes, mouse models of DS have been generated. DS model mice that harbor duplications of mouse sequence syntenic to human chromosome 21 (Reeves et al., 1995; Sago et al., 1998; Olson et al., 2007; Pereira et al., 2009; Yu et al., 2010) or that carry an independently segregating copy of human chromosome 21 (O'Doherty et al., 2005) demonstrate learning and behavioral abnormalities, suggesting that some aspects of human impairment may be modeled in mice. The genetic bases for these phenotypes, the effects of gene dosage dysregulation on the function of intact circuits in vivo, and whether postnatal functional deficits are the result of in utero developmental abnormalities or are exacerbated by ongoing gene dysregulation throughout life are all issues critical to the design of therapeutic strategies to prevent or reverse cognitive impairment in DS.
Little is known about the function of the DS model cortex during early postnatal development, when neural activity sculpts cortical connectivity. We hypothesize that the dysregulation of gene expression caused by trisomy 21 may impair early cortical plasticity and contribute to functional impairment of the cerebral cortex in DS. To assess circuit-level plasticity in DS model mice in vivo, we have taken advantage of two well-characterized forms of plasticity in the visual and olfactory systems. We find that Ts65Dn DS model mice demonstrate a defect in visual cortical plasticity, ocular dominance plasticity (ODP). Our previous work suggested that overexpression of amyloid precursor protein (APP) and amyloid β (Aβ) are sufficient to cause impairment of critical period ODP in mouse models of Alzheimer's disease (William et al., 2012), leading to the hypothesis that overexpression of APP in the trisomy 21 model might contribute to plasticity defects. Surprisingly, however, we find that rescuing APP trisomy in Ts65Dn mice by breeding a null allele of APP into the line, restoring the number of copies of APP to 2N, does not prevent ODP impairment in those mice, suggesting that other duplicated genes are likely sufficient to drive the phenotype in Ts65Dn mice. We have also found that the Ts1Rhr DS model strain, harboring a duplication containing ∼33 genes (excluding APP) in what has been termed the DS critical region, demonstrates the same plasticity defect, reinforcing the conclusion that the plasticity defect is not dependent on triplication of the APP locus.
In addition, we have found that Ts65Dn mice demonstrate impairment in the refinement of synaptic connections by olfactory neurons in the olfactory bulb (OB). Further genetic dissection suggests that APP gene dosage does not drive the phenotype, nor is the Ts1Rhr duplication sufficient to reproduce the defect. These data reveal the possibility of multiple chromosome 21-linked, activity-dependent abnormalities in sensory-system plasticity and connectivity that may contribute to the extent of cognitive impairment in DS patients.
Materials and Methods
Mouse strains.
Mice with segmental trisomy for chromosome 16 (Ts65Dn; 171665Dn/J; strain 005252, Jackson Laboratory), mice with a duplication of a smaller portion of the sequence duplicated in the Ts65Dn line [Ts1Rhr mice; B6.129S6-Dp(16Cbr1-Fam3b)1Rhr/J; gift from William Mobley; strain 005383, Jackson Laboratory; maintained on a C57BL/6 and C3H hybrid background for comparison with Ts65Dn mice], and their nontrisomic littermates were studied in these experiments (Reeves et al., 1995; Olson et al., 2007). In addition, mice heterozygous for APP (APP+/− mice; B6.129S7-Apptm1Dbo/J; strain 004133, Jackson Laboratory) were obtained from Jackson Laboratory and bred with Ts65Dn mice to produce Ts65Dn;APP+/− mice (Ts65Dn;APP2N mice; Zheng et al., 1995). Animal experiments were performed in accordance with United States National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital.
Surgery.
Monocular deprivation (MD), via unilateral lid suture, was performed on postnatal day P27–P29 transgenic and nontransgenic mice, with lid reopening and Arc induction, as described by William et al. (2012), 4–6 d after MD. For optical imaging studies, P23–P25 mice underwent reinforced thin-skull preparation with headpost implantation. Reinforced thin-skull preparation was performed as described by Drew et al. (2010). Following recovery from surgery and restraint training, mice underwent pre-MD optical imaging and contralateral lid suture (MD) at P27–P29. After 6 d, the sutured lid was reopened and post-MD imaging performed. All surgeries and subsequent analyses were performed blind to genotype.
Arc induction.
In a procedure adapted from Tagawa et al. (2005), the eye opposite the one to be stimulated was removed the day before Arc induction. Mice were then placed in a ventilated, dark chamber for 15 h and exposed to light 30 min for Arc mRNA induction or 2 h for Arc protein induction.
Arc in situ hybridization and quantification.
In situ hybridization was performed as described by William et al. (2012). Briefly, in situ hybridization with an Arc antisense probe was performed on 10 μm, coronal, primary visual cortical sections. The visual cortex ipsilateral to the stimulated eye is photographed and a rectangle is drawn across layer 2/3 of the Arc-positive domain for quantitation of Arc domain width. A plot profile is generated along the long axis of the rectangle, parallel to the cortical surface, averaging staining intensity across the short axis and the width of the Arc-positive domain is calculated as the full width at half maximum (FWHM). Averages of staining intensity medial to the Arc-positive domain and within the area of greatest staining intensity are used to determine the value for half-maximum above background. Five or more sections were used in the quantitation of responses per mouse. Image analysis is performed in FIJI (National Institutes of Health).
Immunohistochemistry, Wisteria floribunda agglutinin staining, and cell counting.
Immunohistochemical and Wisteria floribunda agglutinin (WFA; see below) stains were performed on coronal sections of the brains of six Ts65Dn trisomic and six nontrisomic littermate mice that underwent 2 h Arc induction, to facilitate subsequent labeling of lateral visual cortex using Arc immunohistochemistry, followed by PBS cardiac perfusion, immersion fixation in 4% paraformaldehyde for 24 h, and embedding in optimal cutting temperature compound (23-730-571, Thermo Fisher Scientific). Ten micrometer coronal sections through the posterior brain were serially collected on 10 slides with 200 μm of distance between each series of sections every 100 μm and serially distributed across 10 slides. Sections were stained by overnight incubation with guinea pig anti-Arc (catalog #156 005, Synaptic Systems; 1:1000), mouse anti-NeuN (MAB377, Millipore; 1:1000), mouse anti-parvalbumin (P3088, Sigma-Aldrich; 1:4000), rabbit anti-somatostatin (ab64053, Abcam; 1:1000), and mouse anti-Gad67 (MAB5406, Millipore; 1:1000) antibodies and/or WFA (FL-1351, Vector Laboratories; 1:1000) after 10 min postfixation with 4% PFA, permeabilization with 0.3% Tween 20, and blocking with 10% goat serum. WFA-stained slides were coverslipped using Slowfade mounting medium without glycerol (S-2828, Thermo Fisher Scientific). All other slides were mounted using Vectashield with DAPI (H-1200, Vector Laboratories).
The portion of each coronal section that contained the visual cortex was determined by costaining for Arc or by comparison to a serial section stained for Arc in which the outline of the Arc-positive and medial monocular visual cortex were outlined using Computer Assisted Stereological Toolbox (CAST) software (Olympus) to then allow measuring of the area counted as well as the number of cells within each area. For NeuN counts, 30% of the analyzed area was sampled in an unbiased, stereologic approach using the CAST system.
Olfactory glomerular refinement assay.
As described by Cao et al. (2012), we examined the precision of olfactory sensory neuron (OSN) axon targeting in Ts65Dn, Ts65Dn;APP2N, Ts1Rhr, and nontrisomic mice with ages ranging from P34 to P45. To visualize the OSN axonal projection patterns, we immunostained the OBs from these mice with antibodies against the M71 and MOR28 olfactory receptors (ORs). The numbers of glomeruli receiving either M71 or MOR28 receptor neuron projections per half bulb were quantified for each mouse.
Immunohistochemistry of OB glomerular refinement assay.
As described by Cao et al. (2012), the OBs of Ts65Dn and Ts1Rhr mice were immunostained using antibodies against the M71 and MOR28 ORs (Barnea et al., 2004). The OBs were dissected, embedded in M1 (1310TS; Thermo Fisher Scientific) and frozen in a dry-ice/ethanol bath. Twenty micrometer slices of the OB were cryosectioned and collected on SuperFrost Plus slides (12-550-15, Thermo Fisher Scientific). Sections were postfixed with 4% paraformaldehyde in PBS for 8 min at room temperature, washed three times in PBS for 10 min, permeabilized in 0.1% Triton X-100 in PBS (PT) for 30 min at room temperature, blocked in 5% heat-inactivated donkey serum in PT (PTS) for 1 h at room temperature, and incubated with primary antibody (rabbit anti-MOR28, 1:3000; guinea pig anti-M71, 1:1000; Barnea et al., 2004) in PTS under a Hybrislip (H18202, Thermo Fisher Scientific) overnight in a humidified chamber at 4°C. The following day, sections were washed three times with PT for 10 min, blocked with PTS for 30 min at room temperature, incubated with a fluorescent-conjugated secondary antibody (Cy3-conjugated donkey anti-guinea pig IgG; Jackson Immunoresearch; 1:500) in PTS for 2 h at room temperature, washed briefly in PBS, and coverslipped with Fluoromount G with DAPI (0100-20, Southern Biotech). Slides were imaged using a Zeiss inverted fluorescent microscope and analyzed using FIJI (National Institutes of Health).
Optical imaging of intrinsic signals.
Mice were presented with an upward drifting grating (0.03 cycles/°, 2 Hz) occupying a 16° area of the visual field to the right of the vertical midline, 20° above the horizontal meridian. Stimulus was presented to left or right eye for 5 s, followed by 20 s of blank screen at mean luminance (for a detailed description of imaging parameters and analysis, see William et al., 2012).
Experimental design and statistical analysis.
Pde6b genotyping was performed to ensure that experimental mice did not harbor rd1 mutations due to the C3H component of the mixed background in which all mice were maintained. Littermate control mice were used in all experiments for comparison with trisomic Ts65Dn or Ts1Rhr mice. Male (n = 101) and female (n = 65) mice were used in the experiments described in this study and the gender composition of study arms was similar in each experiment. Two-tailed Student's t tests and two-way ANOVAs were calculated as described in the text using StatView (Version 5.0.1, SAS Institute).
Results
Critical period visual cortical plasticity defects in Ts65Dn mice
To test whether DS model mice demonstrate defects in cortical plasticity during early postnatal development, ODP was assessed in one of the best-characterized mouse models of DS, the Ts65Dn line, which harbors a large duplication of mouse chromosome 16 that is syntenic to the majority of human chromosome 21q (Reeves et al., 1995). ODP can be triggered in the primary visual cortex by a brief loss of vision in one eye via lid suture (MD) that results in several changes in the subsequent responsiveness of the visual cortex to stimulation of each eye: expansion of the area of the visual cortex that responds to the nondeprived eye (ipsilateral to the nondeprived eye), weakening of responses to stimulation of the deprived eye, and strengthening of responses to stimulation of the nondeprived eye (Gordon and Stryker, 1996; Tagawa et al., 2005). The cortical response to monocular challenge is most robust during a critical period at 1 month of age (Gordon and Stryker, 1996), during which all of the following studies were performed. Two complementary approaches were used to measure cortical responses following MD: induction of the immediate early gene Arc to label the visual cortex responsive to the nondeprived eye and optical imaging of intrinsic signals to measure the strength of cortical responses to stimulation of each eye.
A brief period of visual stimulation induces Arc expression in many responding excitatory neurons in the visual cortex, highlighting the binocular visual cortex ipsilateral to the stimulated eye (Fig. 1A–D; Tagawa et al., 2005). In normally-reared mice, the pattern and width of the responsive domains of the visual cortex are similar in Ts65Dn and control mice (width of Arc-positive domain in nontrisomic cortex, 871 ± 56 μm, n = 6; width in Ts65Dn cortex, 879 ± 32 μm, n = 6 mice; p = 0.14, t test; Fig. 1A,B). Following 4–6 d of MD, stimulation of the nondeprived eye revealed an expansion in the area of the visual cortex responsive to that eye, indicating a normal plastic response in control mice (width post-MD, 1108 ± 69 μm, n = 14 mice; p = 7.75 × 10−7, t test; Fig. 1C,E). This expansion was not seen in Ts65Dn mice (874 ± 80 μm, n = 7; p = 0.95, t test; Fig. 1D,E). A significant interaction exists between genotype and MD (p < 0.0001; two-way ANOVA), confirming that Ts65Dn mice do not demonstrate the normal plastic response to MD. To assess whether the abnormality that we observe in critical period plasticity in Ts65Dn mice is caused by a delay in the timing of the critical period, we have tested plasticity following 6 d of MD in 2–3-month-old Ts65Dn mice and found a similar defect (nontrisomic width of Arc-positive domain post-MD, 1041 ± 7 μm, n = 2; Ts65Dn width, 801 ± 51 μm, n = 4 mice; p = 0.003, t test).
Figure 1.

Defect in MD-induced expansion of nondeprived eye response territory in Ts65Dn mice. A–D, Coronal sections of the visual cortex demonstrating the pattern of Arc mRNA induction following stimulation of the ipsilateral eye in nontrisomic (A, C) and Ts65Dn (B, D) mice. In normally-reared mice, nontrisomic (A) and Ts65Dn (B) responses are similar. Following 4–6 d (from P27–P29 to P31–P35) of MD, nontrisomic mice demonstrate the expected expansion in width of the domain responsive to the nondeprived eye (C). Ts65Dn mice do not demonstrate this expansion (D). E, Quantitation of results. Arrowheads denote medial and lateral edges of responsive domains. Cortical layers are labeled in B. Error bars denote SD. Scale bar, 500 μm; ***p < 1.24 × 10−6; n.s., not significant; t test. Two-way ANOVA for interaction between genotype and MD, p < 0.0001.
Intrinsic imaging is an alternative assay to measure visual cortical responses in vivo. We first assessed whether a primary defect in vision might underlie the observed difference in critical period plasticity. Ts65Dn mice demonstrate responses of comparable magnitude to those of control mice (Fig. 2B) and the width of the peak of response at half of the maximum value is similar in nontrisomic and Ts65Dn mice (nontrisomic width at half-maximum, 934 ± 279 μm, n = 6; Ts65Dn width at half-maximum, 866 ± 130 μm, n = 7; p = 0.29, t test), suggesting similar responsiveness of the Ts65Dn cortex to visual stimulation. Following 6 d of MD, nontrisomic mice demonstrated weakening of responses to the deprived eye, contralateral to the imaged cortex, and strengthening of responses to the ipsilateral, nondeprived eye (Fig. 2B, black bars, Pre-MD and Post-MD). Following MD, Ts65Dn mice demonstrated weakening of responses to the deprived eye. However, they failed to demonstrate strengthening of responses to the nondeprived eye (Fig. 2B, red bars, Pre-MD and Post-MD). As a result, the shift in ocular dominance observed in nontrisomic mice is not as robust in Ts65Dn mice (Fig. 2C,D). A significant interaction is present between genotype and shift in the ocular dominance index (ODI) post-MD (nontrisomic ODI pre-MD, 0.186 ± 0.030, n = 8; post-MD, −0.312 ± 0.080, n = 8; Ts65Dn ODI pre-MD, 0.117 ± 0.047, n = 10; post-MD, −0.038 ± 0.057, n = 6; p = 0.0058, two-way ANOVA), consistent with a defect in ODP in Ts65Dn mice.
Figure 2.

Defect in ocular dominance response shift following MD in Ts65Dn mice. A, Optical imaging schematic. B, Quantitation of responses. Following MD, nontrisomic (black) and Ts65Dn (red) mice demonstrate weakening of responses to the deprived, contralateral (contra) eye. However, only nontrisomic mice demonstrate strengthening of responses to the nondeprived, ipsilateral (ipsi) eye. #p = 0.072, *p = 0.013, ***p = 0.00034. C, D, Shifts in the ODI (contra − ipsi/contra + ipsi), for individual mice (C) and the averages for each group (D) demonstrate that the expected shift that occurs in nontrisomic mice is less robust in Ts65Dn mice. Error bars denote SE. #p = 0.06, *p = 0.024, ***p = 4.44 × 10−5, t test.
Histologic evaluation of the visual cortex in DS model mice
Several groups have reported abnormalities in neurogenesis involving excitatory and inhibitory neurons in DS model mice (Chakrabarti et al., 2007, 2010; Pérez-Cremades et al., 2010; Haydar and Reeves, 2012). To assess whether significant defects in the generation of cortical neurons are present in the visual cortex and may contribute to the defect in plasticity that we observe in Ts65Dn mice, we performed immunohistologic analysis of P28–P32 trisomic and littermate control mice. We observed no significant difference in the thickness of the visual cortex in Ts65Dn and control mice (nontrisomic cortical thickness, 1479 ± 83 μm, n = 6; Ts65Dn cortical thickness, 1428 ± 32 μm, n = 6; p = 0.19, t test). The density of NeuN+ neurons was similar in the nontrisomic and Ts65Dn visual cortex (nontrisomic NeuN+ neuronal density, 2380 ± 324 neurons/mm2, n = 6; Ts65Dn density, 2115 ± 556 neurons/mm2; p = 0.34, t test; Fig. 3A–C). We counted comparable numbers of Gad67+ inhibitory neurons (nontrisomic Gad67+ neuronal density, 117.4 ± 32.3 neurons/mm2, n = 6; Ts65Dn density, 123.1 ± 23.5 neurons/mm2, n = 6; p = 0.73, t test; Fig. 3D–F) in Ts65Dn and nontrisomic mice and comparable numbers of parvalbumin-expressing (nontrisomic parvalbumin+ neuronal density, 102 ± 36 neurons/mm2, n = 6; Ts65Dn density, 103 ± 39 neurons/mm2, n = 6; p = 0.99, t test; Fig. 3G–I) and somatostatin-expressing inhibitory neuronal subtypes (nontrisomic somatostatin+ neuronal density, 33.3 ± 7.7 neurons/mm2, n = 6; Ts65Dn density, 32.1 ± 6.8 neurons/mm2, n = 6; p = 0.39, t test; Fig. 3J–L) in nontrisomic and Ts65Dn mice. Similarly, we found no significant difference in the number of inhibitory neurons enshrouded in perineuronal nets (PNNs; nontrisomic density of PNNs, 82.5 ± 30.8 neurons/mm2, n = 6; Ts65Dn density, 71.9 ± 11.1 neurons/mm2, n = 6; p = 0.29, t test), a chondroitin sulfate proteoglycan-rich extracellular matrix that matures during the critical period and aids in inhibition of plasticity after the critical period (Pizzorusso et al., 2002; Ye and Miao, 2013). These data suggest that the plasticity impairment we observe in Ts65Dn mice cannot be easily attributed to a significant decrease in the number of neurons, an abnormality in the number of inhibitory neurons, or by aberrant or delayed maturation of the PNN matrix in the primary visual cortex.
Figure 3.
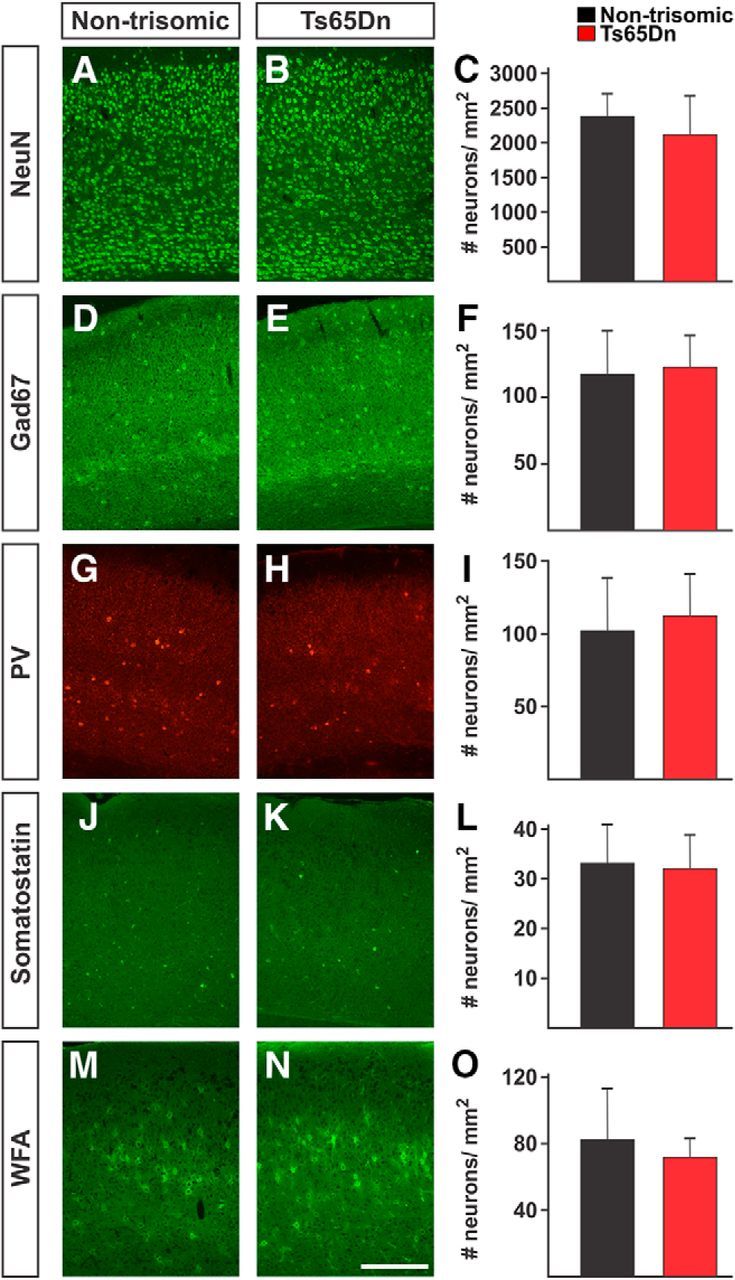
Histologic assessment of neuronal populations in the Ts65Dn visual cortex. A–O, Representative immunostains (A–L) and WFA staining (M–O) of nontrisomic littermate (A, D, G, J, M) and Ts65Dn (B, E, H, K, N) visual cortex. No significant differences were observed in the density of NeuN+ (A–C), Gad67+ (D–F), parvalbumin+ (G–I), somatostatin+ (J–L), or WFA-stained neurons in Ts65Dn and nontrisomic visual cortex. C, F, I, L, O, Quantitation of cell counts in nontrisomic (black) and Ts65Dn (red) cortex. Scale bar, 500 μm. Error bars denote SD. n = 6 per group.
APP duplication does not drive the ODP defect in Ts65Dn mice
We had previously demonstrated that expression of a mutant allele of the APP gene was sufficient to block critical period ODP in transgenic mice (William et al., 2012), and Ts65Dn mice have been demonstrated to overexpress APP (Reeves et al., 1995), raising the possibility that overexpression of APP and its proteolytic cleavage product, Aβ, may be responsible for the plasticity impairment we observe in Ts65Dn mice. To test this hypothesis, a mutant allele of APP was crossed into the Ts65Dn strain and Ts65Dn mice heterozygous for the endogenous APP locus (Ts65Dn; APP2N mice) were assessed for their ability to exhibit ODP. Following 6 d of MD, Ts65Dn;APP2N mice failed to demonstrate an expansion in the domain of the visual cortex responsive to the nondeprived eye (width of Arc-positive visual cortex ipsilateral to the nondeprived eye, 866 ± 37 μm, n = 7). Moreover, no strengthening of responses to nondeprived eye stimulation was observed following 6 d of MD (pre-MD response to nondeprived eye stimulation, 0.49 ± 0.05%; post-MD response, 0.46 ± 0.04%, n = 12; p = 0.2, t test; Fig. 4A). A significant, but modest decline in the ODI was observed in Ts65Dn;APP2N mice (pre-MD ODI, 0.22 ± 0.04; post-MD ODI, −0.03 ± 0.07; p = 0.007, t test; Fig. 4B) driven entirely by weakening of responses to the deprived eye (pre-MD response to deprived eye stimulation, 0.74 ± 0.04%; post-MD response, 0.43 ± 0.05%, n = 11; p = 6.03 × 10−5, t test; Fig. 4A). These data are a phenocopy of the results obtained in Ts65Dn mice and they suggest that duplication of the APP gene is not required to drive ODP impairment in Ts65Dn mice.
Figure 4.

Restoration of APP copy number in Ts65Dn mice fails to rescue ODP defect. A, Following MD, Ts65Dn;APP2N mice demonstrate weakening of responses to the deprived eye (contra), but fail to demonstrate strengthening of responses to the nondeprived eye (ipsi). B, Plots of the ODIs of individual Ts65Dn;APP2N mice (n = 11). Error bars denote SE. ***p = 6.03 × 10−5, t test.
The DS critical region is sufficient to drive ODP impairment
To begin to define a minimal region within the Ts65Dn duplication sufficient to cause visual system plasticity impairment, ODP was assessed in the Ts1Rhr mouse line that harbors a duplication approximately one-third the size of that of the Ts65Dn line (Olson et al., 2007). The sequence included in this duplication corresponds to a region of human chromosome 21 that has been termed the DS critical region due to its ability to confer a number of DS-associated phenotypes when duplicated in patients with partial duplications of chromosome 21 (Rahmani et al., 1989). In the absence of a challenge, Ts1Rhr mice and nontrisomic littermates demonstrate similar patterns of Arc expression upon stimulation of the ipsilateral eye (nontrisomic Arc+ domain width, 870 ± 25 μm, n = 6; Ts1Rhr domain width, 898 ± 42 μm, n = 5; p = 0.21, t test; Fig. 5A,B,E). Following 6 d of MD, however, Ts1Rhr mice demonstrated a defect in the plastic expansion in the cortex responsive to the nondeprived eye as assessed by Arc expression (post-MD nontrisomic Arc+ domain width, 1156 ± 72 μm, n = 9; compared with normally reared, p = 4.4 × 10−7, t test; post-MD Ts1Rhr domain width, 899 ± 52 μm, n = 9; compared with normally reared, p = 0.97, t test; Fig. 5C–E). A significant interaction was present between genotype and deprivation (p < 0.0001, two-way ANOVA). Ts1Rhr mice demonstrated robust responses comparable to those of nontrisomic mice (Fig. 5A), with similar response widths (nontrisomic FWHM of response to contralateral eye stimulation, 665 ± 162 μm, n = 6; Ts1Rhr FWHM, 691 ± 202 μm, n = 6; p = 0.8, t test). Following a challenge of 6 d of MD, nontrisomic mice demonstrated weakening of responses to the deprived eye (contra) and strengthening of responses to the nondeprived eye (ipsi; Fig. 5F, black bars, Pre-MD and Post-MD). Ts1Rhr mice demonstrated weakening of responses to the deprived eye following deprivation, but also demonstrated weakening of responses to the nondeprived eye (pre-MD nondeprived eye dF/F, 0.74 ± 0.04%; post-MD dF/F, 0.58 ± 0.04%; p = 0.02, t test; Fig. 5F, red bars, Pre-MD and Post-MD). The shifts in the ODI that occured in nontrisomic mice were less robust in Ts1Rhr mice and a significant interaction was observed between genotype and challenge (p = 0.024, two-way ANOVA; Fig. 5G,H). These data indicate that Ts1Rhr mice demonstrate a defect in ODP similar to that of Ts65Dn mice, an inability of the visual cortex to strengthen responses to stimulation of the nondeprived eye following the loss of input from the contralateral eye.
Figure 5.

ODP defect in Ts1Rhr mice. A–H, Similar to Ts65Dn mice, Ts1Rhr mice demonstrate a defect in ODP by Arc induction (A–E) and optical imaging (F–H) assays. The expansion in the width of the responsive cortex ipsilateral to the nondeprived eye that occurs in nontrisomic mice (C) does not occur in Ts1Rhr mice (D). Scale bar, 500 μm. E, Quantitation of Arc data. ***p = 1.86 × 10−7, t test. F, Following MD, Ts1Rhr mice demonstrate weakening of responses to the nondeprived (ipsi) eye, instead of strengthening. *p < 0.048, t test. G, H, ODI shifts in Ts1Rhr mice are much less robust and driven entirely by weakening of responses to the deprived eye. *p = 0.0237, two-way ANOVA.
Olfactory glomerular target refinement is disrupted in DS model mice
The plasticity impairments observed in the visual systems of Ts65Dn and Ts1Rhr mice raise the possibility that plasticity in other sensory systems may be perturbed in DS model mice. During early postnatal development, the synapses between OSNs in the olfactory epithelium and tufted and mitral cells in the OB undergo activity-dependent refinement (Zou et al., 2004). The synapses between OSNs and second-order neurons in the bulb are organized into discrete spheres of neuropil, termed glomeruli, such that OSNs that express the same odorant receptor all synapse within the same glomerulus (Vassar et al., 1994; Mombaerts et al., 1996). In the mature bulb, the axon projections of OSNs expressing a given odorant receptor converge on a single glomerulus in each half of the OB. Through a process that requires OSN activity, refinement of connections to a single glomerulus per hemibulb occurs (Yu et al., 2004). A critical window exists during which OSN activity is required for refinement to occur (Zou et al., 2004; Ma et al., 2014; Tsai and Barnea, 2014). For example, OSNs expressing the odorant receptor M71 demonstrate an activity-dependent window that closes by P25 (Zou et al., 2004), suggesting that by P34, much of this refinement has already occurred. To assess glomerular refinement in the olfactory system, the distal projections of M71-expressing neurons were labeled in the OBs of Ts65Dn mice and nontrisomic littermates using an antibody to M71 (Barnea et al., 2004). Nontrisomic mice demonstrated 1–2 glomeruli targeted by M71 neurons (average number of glomeruli per hemibulb, 1.36 ± 0.13, n = 14 foci; Fig. 6A,C); this is expected as M71 glomerular refinement continues until P60–P90 in wild-type mice (Zou et al., 2004). Ts65Dn mice, however, exhibit a larger number of M71-targeted glomeruli per hemibulb (average number of glomeruli per hemibulb, 1.81 ± 0.12, n = 26 foci; p = 0.027, t test; Fig. 6B,C). Refinement of projections of neurons expressing the odorant receptor MOR28 to single glomeruli occurs normally in Ts65Dn mice (number of glomeruli per nontrisomic hemibulb, 1.0 ± 0.0, n = 10 hemibulbs from 4 mice; number of glomeruli per Ts65Dn hemibulb, 1.0 ± 0.0, n = 10 hemibulbs from 5 mice), suggesting that this population of OSNs is less sensitive to perturbation than the population expressing M71. Consistent with our visual system plasticity data, Ts65Dn;APP2N mice demonstrate a defect in M71 receptor glomerular refinement (average glomeruli per hemibulb, 1.77 ± 0.17, n = 13 hemibulbs from 4 mice; p = 0.86 compared with Ts65Dn mice, t test; Fig. 6C), suggesting the connectivity abnormality observed in the olfactory systems of Ts65Dn mice is not driven by duplication of APP alone. Surprisingly, the smaller duplication present in Ts1Rhr mice does not appear to be sufficient to cause impairment of refinement of glomerular connectivity (average number of glomeruli per hemibulb in nontrisomic bulbs, 1.29 ± 0.16, n = 14 hemibulbs; average number of glomeruli per Ts1Rhr hemibulb, 1.39 ± 0.16, n = 18 hemibulbs; p = 0.66, t test; Fig. 6D–F), suggesting either that the neural system sensitivity to a plasticity-disrupting genetic landscape differs between olfactory and visual cortices, or that duplicated genes present outside of the Ts1Rhr duplication, but included in the Ts65Dn duplication, are required to cause this defect in refinement of OSN connections.
Figure 6.

Ts65Dn mice demonstrate a defect in olfactory glomerular refinement. A–F, Refinement of glomerular targeting was assessed in Ts65Dn (A–C) and Ts1Rhr (D–F) mouse models at P34–P36. Ts65Dn mice (B) demonstrate impairment in refinement of connections to single glomeruli when compared with littermate controls (A). C, Quantitation of results in nontrisomic mice, Ts65Dn mice, and Ts65Dn mice with two copies of APP (Ts65Dn;APP2N). Fourteen hemibulbs from six nontrisomic mice, 26 hemibulbs from seven Ts65Dn mice, and 13 hemibulbs from four Ts65Dn; APP2N mice were analyzed. Ts1Rhr mice (E) and littermate controls (D) demonstrate similar degrees of target refinement at P34–P36 (p = 0.86; quantitation in F). Fourteen hemibulbs from four nontrisomic mice and 18 hemibulbs from five Ts1Rhr mice were analyzed. Scale bar, 200 μm; *p = 0.027, t test.
Discussion
To explore the possibility that defects in synaptic plasticity during early postnatal development may contribute to functional impairment in DS, we assessed critical period visual cortical plasticity in two mouse models of DS, the Ts65Dn and Ts1Rhr strains. Both demonstrate impairment in ODP. The specific component of the plastic response that is impaired in these mice is the ability of the cortex to strengthen responses to the nondeprived eye following 4–6 d of contralateral lid closure, evidenced also as a lack of expansion in the proportion of the visual cortex that responds to the nondeprived eye.
Ts65Dn and Ts1Rhr mice demonstrate robust responses to visual stimulation in the absence of challenge and appropriate weakening of responses to the deprived eye following challenge, suggesting that many aspects of visual circuit function are intact in these mice. Blank et al. (2011) have described a retinogeniculate refinement abnormality in Ts65Dn and Ts1Rhr mice in which, before eye opening, trisomic mice demonstrate excessive refinement of contralateral and ipsilateral projections from each eye to target dorsal lateral geniculate nuclei. Ts65Dn mice demonstrate normal retinal wave activity, suggesting that the phenotype is not secondary to altered retinal physiology (Blank et al., 2011). The impact of a retinogeniculate refinement phenotype on the cortical plastic response is not clear, though these data suggest that abnormalities are present at multiple levels along the visual circuit.
Another possible explanation for an impairment in the cortical plastic response lies in abnormalities in cortical development. Using markers of differentiated neurons, inhibitory neurons, subtypes of inhibitory neurons, and a marker of inhibitory circuit maturation, we find no significant differences when comparing the Ts65Dn and the nontrisomic visual cortex. Abnormalities in neurogenesis and in inhibitory neuron numbers have been reported in the adult somatosensory cortex of Ts65Dn mice (Chakrabarti et al., 2010; Pérez-Cremades et al., 2010). Those differences were not identified in our analysis of the visual cortex of P28–P32 Ts65Dn mice; whether defects in neurogenesis are more apparent at different ages in different cortical areas may be borne out in future studies that more specifically address this question.
Transgenic overexpression of mutant alleles of APP can impair critical period ODP in mouse models of Alzheimer's disease (William et al., 2012; Maya-Vetencourt et al., 2014), raising the possibility that overexpression of wild-type APP in Ts65Dn mice may contribute to the impairment observed in developmental plasticity. APP overexpression causes degeneration of basal forebrain cholinergic neurons in adult Ts65Dn mice (Salehi et al., 2006), but whether APP overexpression contributes to impairment at an earlier age is less clear. Two lines of evidence argue against a required role for trisomy of APP in the ODP phenotype present in Ts65Dn mice. Removing one functional allele of APP in Ts65Dn mice or studying mice with a smaller duplication of sequence that excludes the APP locus, we find that APP triplication is not necessary to drive either visual or olfactory system phenotypes that we observe in Ts65Dn mice. Overexpression of wild-type murine APP via duplication of the endogenous locus would be expected to exert a milder effect on the organism than exogenous expression of the mutant human gene, though twofold increases in the concentration of wild-type, murine Aβ have been demonstrated to impair hippocampal synaptic plasticity in vitro (Abramov et al., 2009). Another possibility is that the effect of duplication of murine APP on ODP may be masked by the effects of duplication of other genes. Trisomy of murine APP may contribute to the defects that we observe. However, overexpression of other genes included in the Ts65Dn duplication appears to be sufficient to drive impairment in ODP and olfactory glomerular refinement.
The finding that the Ts1Rhr sequence duplication is sufficient to cause a defect in cortical plasticity narrows down the list of possible genes responsible for the phenotype to a group of ∼33 candidates. Overexpression of one gene duplicated in the Ts1Rhr sequence, dual-specificity tyrosine-phosphorylated and tyrosine-regulated kinase 1A, DYRK1A, has been implicated in neurogenesis (Guedj et al., 2012), hippocampal plasticity impairment in vitro, and in learning and memory impairment in transgenic mice in vivo (García-Cerro et al., 2014). Overexpression of DYRK1A can produce cognitive impairment in mice (Branchi et al., 2004). Detailed dissection of the sequence duplicated in Ts1Rhr mice implicates the DYRK1A gene as contributing to neural system impairment in model mice, though DYRK1A duplication does not account for the entire phenotype in either Ts65Dn or Ts1Rhr mice (García-Cerro et al., 2014; Jiang et al., 2015). Overexpression of DSCAM (Down syndrome cell adhesion molecule) can phenocopy the excessive refinement of retinogeniculate connections observed in Ts1Rhr mice, though restoring the number of DSCAM genes to 2N in Ts1Rhr mice fails to rescue the phenotype (Blank et al., 2011). Therefore, DYRK1A, APP, and DSCAM overexpression each causes defects in plasticity, but reducing them individually to 2N in the context of a larger trisomy does not rescue impairment, suggesting complex gene–gene interactions. Thus, the visual plasticity and olfactory refinement phenotypes explored here, and the cognitive phenotypes previously described in Ts1Rhr mice (Belichenko et al., 2009), suggest a complicated picture in which multiple genes may be involved. A more detailed understanding of the effects of gene dosage on visual-system plasticity will provide insight into the basis for dysfunction across circuits required for higher cognitive functions.
Ts65Dn mice demonstrate a defect in refinement of OSN projections onto single OB glomeruli. This process has been demonstrated to require OSN activity as it can be impaired by nares closure (Zou et al., 2004). The refinement defect present in Ts65Dn mice is less severe than that described in mice subjected to nares closure, suggesting that trisomy in this model does not completely impair activity-dependent target refinement. Whether the refinement defect we observe persists into adulthood is an important question that will need to be pursued with additional time points in older mice. The underlying genetic cause of this phenotype may overlap with the activity-dependent impairment we observe in the visual cortex, though the abatement of this phenotype in Ts1Rhr mice suggests that additional or different genes contribute to the glomerular refinement defect in Ts65Dn mice (Table 1). Reduction of APP copy number to 2N in Ts65Dn mice failed to rescue plasticity in the visual system or to rescue glomerular refinement in the OB. Wild-type human APP does not have the same ability to disrupt glomerular refinement as human APPswe when expressed in OSNs (Cao et al., 2012), which is consistent with our data suggesting that other genes drive the olfactory system phenotype in Ts65Dn mice. Thus, the current data revealing marked defects in developmental plasticity in both olfactory and visual systems suggest that refinement of connectivity and plasticity is impaired across circuits throughout the nervous system in the developing DS brain.
Table 1.
Comparison of visual cortical plasticity and olfactory glomerular refinement phenotypes in mouse models of Alzheimer's disease and DS
| Genotype | ODP | Olfactory glomerular refinement |
|---|---|---|
| APPswe | Impaired (William et al., 2012) | Impaired (Cao et al., 2012) |
| Wild-type human APP overexpression | Not tested | Normal (Cao et al., 2012) |
| Ts65Dn | Impaired | Impaired |
| Ts65Dn;APP2N | Impaired | Impaired |
| Ts1Rhr | Impaired | Normal |
This table summarizes the results of assessments of ODP and of olfactory glomerular refinement in mouse strains expressing mutant (Swedish mutation K670N, M671L;APPswe) or wild-type alleles of human APP and in DS model mice trisomic (Ts65Dn) or disomic (Ts65Dn;APP2N and Ts1Rhr) for APP. References to published data are cited.
DS patients demonstrate abnormalities in visual function secondary to refractive errors, strabismus, lens opacities, and other defects (van Splunder et al., 2003); visual function may be further impaired by defects in activity-dependent plasticity in the visual cortex. Reduced spine density and dendritic arborization abnormalities have been described in the visual cortex of DS infants (Takashima et al., 1981), structural findings that may also be secondary to defects in the functional wiring of the visual system. A recent study revealed significant olfactory deficits in DS patients, even in a group of younger adults (ages 18–29 years; Cecchini et al., 2016). Patients with visual and hearing deficits have higher rates of intellectual disability (Määttä et al., 2006), suggesting that abnormalities in sensory circuits could contribute to impairment in learning and in cognitive development. Our data provide a model system to test the molecular underpinnings of these sensory defects, and to potentially provide a testbed for future behavioral or therapeutic interventions.
Footnotes
This work was supported by the National Institutes of Health (Grant K08 NS069811 and Pilot Project Grant from 5P50AG005134 to C.M.W.; Grant 5P50AG005134 to B.T.H.; and Grants R21NS085711 and R21NS094861 to M.W.A.). We thank Natalia Garcia de Marco for advice on somatostatin immunohistochemistry. We thank members of the Hyman and Albers laboratories for advice and discussion.
The authors declare no competing financial interests.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I (2009) Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12:1567–1576. 10.1038/nn.2433 [DOI] [PubMed] [Google Scholar]
- Barnea G, O'Donnell S, Mancia F, Sun X, Nemes A, Mendelsohn M, Axel R (2004) Odorant receptors on axon termini in the brain. Science 304:1468. 10.1126/science.1096146 [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC (2009) The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci 29:5938–5948. 10.1523/JNEUROSCI.1547-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M, Fuerst PG, Stevens B, Nouri N, Kirkby L, Warrier D, Barres BA, Feller MB, Huberman AD, Burgess RW, Garner CC (2011) The Down syndrome critical region regulates retinogeniculate refinement. J Neurosci 31:5764–5776. 10.1523/JNEUROSCI.6015-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchi I, Bichler Z, Minghetti L, Delabar JM, Malchiodi-Albedi F, Gonzalez MC, Chettouh Z, Nicolini A, Chabert C, Smith DJ, Rubin EM, Migliore-Samour D, Alleva E (2004) Transgenic mouse in vivo library of human Down syndrome critical region 1: association between DYRK1A overexpression, brain development abnormalities, and cell cycle protein alteration. J Neuropathol Exp Neurol 63:429–440. 10.1093/jnen/63.5.429 [DOI] [PubMed] [Google Scholar]
- Cao L, Schrank BR, Rodriguez S, Benz EG, Moulia TW, Rickenbacher GT, Gomez AC, Levites Y, Edwards SR, Golde TE, Hyman BT, Barnea G, Albers MW (2012) Abeta alters the connectivity of olfactory neurons in the absence of amyloid plaques in vivo. Nat Commun 3:1009. 10.1038/ncomms2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini MP, Viviani D, Sandri M, Hähner A, Hummel T, Zancanaro C (2016) Olfaction in people with Down syndrome: a comprehensive assessment across four decades of age. PLoS One 11:e0146486. 10.1371/journal.pone.0146486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF (2007) Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci 27:11483–11495. 10.1523/JNEUROSCI.3406-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Best TK, Cramer NP, Carney RS, Isaac JT, Galdzicki Z, Haydar TF (2010) Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat Neurosci 13:927–934. 10.1038/nn.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RS, Hesketh LJ (2000) Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev 6:84–95. 10.1002/1098-2779(2000)6:2%3C84::AID-MRDD2%3E3.3.CO%3B2-G [DOI] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, Akassoglou K, Tsai PS, Kleinfeld D (2010) Chronic optical access through a polished and reinforced thinned skull. Nat Methods 7:981–984. 10.1038/nmeth.1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cerro S, Martínez P, Vidal V, Corrales A, Flórez J, Vidal R, Rueda N, Arbonés ML, Martínez-Cué C (2014) Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS One 9:e106572. 10.1371/journal.pone.0106572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP (1996) Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci 16:3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj F, Pereira PL, Najas S, Barallobre MJ, Chabert C, Souchet B, Sebrie C, Verney C, Herault Y, Arbones M, Delabar JM (2012) DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis 46:190–203. 10.1016/j.nbd.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Haydar TF, Reeves RH (2012) Trisomy 21 and early brain development. Trends Neurosci 35:81–91. 10.1016/j.tins.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Liu C, Yu T, Zhang L, Meng K, Xing Z, Belichenko PV, Kleschevnikov AM, Pao A, Peresie J, Wie S, Mobley WC, Yu YE (2015) Genetic dissection of the Down syndrome critical region. Hum Mol Genet 24:6540–6551. 10.1093/hmg/ddv364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wu Y, Qiu Q, Scheerer H, Moran A, Yu CR (2014) A developmental switch of axon targeting in the continuously regenerating mouse olfactory system. Science 344:194–197. 10.1126/science.1248805 [DOI] [PubMed] [Google Scholar]
- Määttä T, Kaski M, Taanila A, Keinänen-Kiukaanniemi S, Iivanainen M (2006) Sensory impairments and health concerns related to the degree of intellectual disability in people with Down syndrome. Downs Syndr Res Pract 11:78–83. 10.3104/reports.317 [DOI] [PubMed] [Google Scholar]
- Maya-Vetencourt JF, Carucci NM, Capsoni S, Cattaneo A (2014) Amyloid plaque-independent deficit of early postnatal visual cortical plasticity in the 5XFAD transgenic model of Alzheimer's disease. J Alzheimers Dis 42:103–107. 10.3233/JAD-140453 [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Wang F, Dulac C, Chao SK, Nemes A, Mendelsohn M, Edmondson J, Axel R (1996) Visualizing an olfactory sensory map. Cell 87:675–686. 10.1016/S0092-8674(00)81387-2 [DOI] [PubMed] [Google Scholar]
- O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TV, Henderson DJ, Nizetic D, Tybulewicz VL, Fisher EM (2005) An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309:2033–2037. 10.1126/science.1114535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Sengstaken CL, Peterson EA, Aquino V, Galdzicki Z, Siarey R, Pletnikov M, Moran TH, Reeves RH (2007) Trisomy for the Down syndrome ‘critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Hum Mol Genet 16:774–782. 10.1093/hmg/ddm022 [DOI] [PubMed] [Google Scholar]
- Pereira PL, Magnol L, Sahun I, Brault V, Duchon A, Prandini P, Gruart A, Bizot JC, Chadefaux-Vekemans B, Deutsch S, Trovero F, Delgado-García JM, Antonarakis SE, Dierssen M, Herault Y (2009) A new mouse model for the trisomy of the Abcg1–U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet 18:4756–4769. 10.1093/hmg/ddp438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cremades D, Hernández S, Blasco-Ibáñez JM, Crespo C, Nacher J, Varea E (2010) Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down's syndrome. J Neural Transm 117:445–455. 10.1007/s00702-010-0376-9 [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L (2002) Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298:1248–1251. 10.1126/science.1072699 [DOI] [PubMed] [Google Scholar]
- Rahmani Z, Blouin JL, Creau-Goldberg N, Watkins PC, Mattei JF, Poissonnier M, Prieur M, Chettouh Z, Nicole A, Aurias A (1989) Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc Natl Acad Sci U S A 86:5958–5962. 10.1073/pnas.86.15.5958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11:177–184. 10.1038/ng1095-177 [DOI] [PubMed] [Google Scholar]
- Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, Epstein CJ, Huang TT (1998) Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci U S A 95:6256–6261. 10.1073/pnas.95.11.6256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC (2006) Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51:29–42. 10.1016/j.neuron.2006.05.022 [DOI] [PubMed] [Google Scholar]
- Tagawa Y, Kanold PO, Majdan M, Shatz CJ (2005) Multiple periods of functional ocular dominance plasticity in mouse visual cortex. Nat Neurosci 8:380–388. 10.1038/nn1410 [DOI] [PubMed] [Google Scholar]
- Takashima S, Becker LE, Armstrong DL, Chan F (1981) Abnormal neuronal development in the visual cortex of the human fetus and infant with Down's syndrome. A quantitative and qualitative Golgi study. Brain Res 225:1–21. 10.1016/0006-8993(81)90314-0 [DOI] [PubMed] [Google Scholar]
- Tsai L, Barnea G (2014) A critical period defined by axon-targeting mechanisms in the murine olfactory bulb. Science 344:197–200. 10.1126/science.1248806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Splunder J, Stilma JS, Bernsen RM, Arentz TG, Evenhuis HM (2003) Refractive errors and visual impairment in 900 adults with intellectual disabilities in The Netherlands. Acta Ophthalmol Scand 81:123–129. 10.1034/j.1600-0420.2003.00035.x [DOI] [PubMed] [Google Scholar]
- Vassar R, Chao SK, Sitcheran R, Nuñez JM, Vosshall LB, Axel R (1994) Topographic organization of sensory projections to the olfactory bulb. Cell 79:981–991. 10.1016/0092-8674(94)90029-9 [DOI] [PubMed] [Google Scholar]
- William CM, Andermann ML, Goldey GJ, Roumis DK, Reid RC, Shatz CJ, Albers MW, Frosch MP, Hyman BT (2012) Synaptic plasticity defect following visual deprivation in Alzheimer's disease model transgenic mice. J Neurosci 32:8004–8011. 10.1523/JNEUROSCI.5369-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Miao QL (2013) Experience-dependent development of perineuronal nets and chondroitin sulfate proteoglycan receptors in mouse visual cortex. Matrix Biol 32:352–363. 10.1016/j.matbio.2013.04.001 [DOI] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O'Donnell S, Brown HE, Osborne J, Axel R, Gogos JA (2004) Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron 42:553–566. 10.1016/S0896-6273(04)00224-7 [DOI] [PubMed] [Google Scholar]
- Yu T, Liu C, Belichenko P, Clapcote SJ, Li S, Pao A, Kleschevnikov A, Bechard AR, Asrar S, Chen R, Fan N, Zhou Z, Jia Z, Chen C, Roder JC, Liu B, Baldini A, Mobley WC, Yu YE (2010) Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res 1366:162–171. 10.1016/j.brainres.2010.09.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81:525–531. 10.1016/0092-8674(95)90073-X [DOI] [PubMed] [Google Scholar]
- Zou DJ, Feinstein P, Rivers AL, Mathews GA, Kim A, Greer CA, Mombaerts P, Firestein S (2004) Postnatal refinement of peripheral olfactory projections. Science 304:1976–1979. 10.1126/science.1093468 [DOI] [PubMed] [Google Scholar]