

ABSTRACT
Anaerobic phenylalanine (Phe) degradation in the betaproteobacterium Aromatoleum aromaticum involves transamination and decarboxylation to phenylacetaldehyde, followed by oxidation to phenylacetate. The latter reaction is catalyzed simultaneously by two enzymes, a highly specific phenylacetaldehyde dehydrogenase (PDH) and a rather unspecific tungsten-dependent aldehyde oxidoreductase (AOR). Attempting to establish increased synthesis of AOR, we constructed a mutant lacking the gene for PDH. This mutant still grew on phenylalanine, exhibiting increased AOR activities on medium containing tungstate. In the absence of tungstate, the mutant showed initially severe growth deficiency, but it resumed growth on Phe after longer incubation times. Moreover, the growth rates of the mutant increased during several reinoculation cycles on either tungstate-proficient or -deficient media, reaching the same values as recorded in wild-type strains. We confirmed AOR as the major alternative enzyme serving Phe degradation under tungstate-supplied conditions and identified and characterized the alternative NAD-dependent aldehyde dehydrogenase AldB taking over the function under tungstate-deficient conditions. Sequence analysis of the respective genes from adapted cultures under either growth condition revealed a mutation in the upstream region of the aor operon and a mutation within the coding region of aldB, which are likely involved in the observed adaptation of the deletion mutant to regain fast growth on Phe.
IMPORTANCE The betaproteobacterium Aromatoleum aromaticum degrades many aromatic compounds under denitrifying conditions. One of the steps of phenylalanine degradation is catalyzed by two simultaneously induced enzymes, a NAD(P)-dependent phenylacetaldehyde dehydrogenase and a W-containing aldehyde oxidoreductase. We report here that the latter fully complements a constructed deletion mutant lacking the gene for phenylacetaldehyde dehydrogenase and is overproduced after several reinoculations. Moreover, an alternative NAD-dependent dehydrogenase is recruited to resume growth in tungstate-free medium, which does not allow the production of aldehyde oxidoreductase. This alternative enzyme is overproduced and seems to have acquired a point mutation in the active center. Our research illustrates the flexibility of environmentally important bacteria in adapting their metabolic pathways to new challenges within only a few generations.
KEYWORDS: anaerobic metabolism, phenylacetaldehyde, tungsten enzyme, aldehyde:ferredoxin oxidoreductase, aldehyde dehydrogenase, adaptation, mutation
INTRODUCTION
The denitrifying betaproteobacterium Aromatoleum aromaticum is known to degrade many toxic aromatic compounds such as the common environmental pollutants toluene, ethylbenzene or phenol, and others like the aromatic amino acid phenylalanine (Phe) (1–3). The anaerobic Phe metabolism has been well studied in bacteria of the Azoarcus/Thauera cluster (4–6), following a similar pathway in the hyperthermophilic archaeon Ferroglobus placidus (7), whereas a different pathway has been reported for the sulfate-reducing bacterium Desulfobacula toluolica (8). Anaerobic Phe degradation in A. aromaticum or the related species Thauera aromatica is initiated by transamination to phenylpyruvate, which is then decarboxylated to phenylacetaldehyde (PAld) (4, 5, 9). The latter is oxidized to phenylacetate (PA), which is activated to the coenzyme A (CoA) thioester and further degraded via phenylglyoxylate and benzoyl-CoA along a well-characterized pathway (9–11).
PAld oxidation to PA in A. aromaticum appears to be mainly catalyzed by a specifically induced phenylacetaldehyde dehydrogenase (PDH) coupled to either NAD or NADP but partially also by a simultaneously induced aldehyde:ferredoxin oxidoreductase (AOR) (12). Under these growth conditions, A. aromaticum synthesizes the W-dependent AOR and the Mo-containing enzymes phenylacetyl-CoA:acceptor oxidoreductase (13) and nitrate reductase (NAR) (2, 14) at the same time and needs to be able to discriminate the respective metals needed for cofactor synthesis and incorporation (12).
Tungsten-containing enzymes are abundant among many Archaea, especially members of the anaerobic and hyperthermophilic genera Thermococcus and Pyrococcus. They belong to a class of enzymes containing at least five families of aldehyde-oxidizing enzymes, which are represented by the isoenzymes present in P. furiosus. These include the “prototype” AOR isoenzymes (15, 16), glyceraldehyde-3-phosphate oxidoreductases (17) involved in a modified glycolytic pathway, formaldehyde oxidoreductases (18), and two further W-containing oxidoreductases of unknown function (WOR4 and WOR5) (19–21). A W-containing AOR-type enzyme was also reported from the thermophilic fermentative bacterium Moorella thermoacetica (22). Very recently, another family of W-enzymes affiliated with the same class was discovered in strictly anaerobic aromatic-degrading bacteria, which were identified as catalytic subunits of a very large enzyme complex reducing the aromatic ring of benzoyl-CoA (type 2 benzoyl-CoA reductases), key enzymes of anaerobic aromatic metabolism (23).
Archaeal AOR-type enzymes are present in various activities under all growth conditions and catalyze the reversible ferredoxin-dependent oxidation of various aldehydes to the respective acids (19, 21, 24). Their main function is assumed to detoxify aldehyde side products that are produced during the fermentative degradation of amino acids (25, 26). Archaeal AORs are dimeric enzymes that contain a W-bis-molybdopterin cofactor, one Fe4S4 cluster per subunit, and another mononuclear Fe2+ ion bridging the two subunits (15, 27). In addition to the previously known AOR of Moorella thermoacetica (22), a W-containing bacterial AOR was also discovered recently in denitrifying Phe-degrading cells of A. aromaticum strain EbN1 (5, 12). However, a clearly defined metabolic role in Phe metabolism could not be assigned because the same cells contained a highly substrate-specific PDH that appears to play the major role in metabolic conversion of PAld. AOR is present simultaneously with PDH but appears not to be essential for Phe metabolism and normal growth on Phe. Instead, AOR exhibits a very broad substrate spectrum, as examined in EbN1 cell extracts, oxidizing various aromatic and aliphatic aldehydes to the corresponding carbonic acids (12). Therefore, it has been suggested that AOR is primarily involved in aldehyde detoxification, as previously proposed for archaeal AORs (12, 25, 26).
This study aims to find out more about the presence of AOR under various growth conditions and its function for cellular metabolism. We also investigate whether AOR or other enzymes can replace PDH for anaerobic growth on phenylalanine. To achieve this, a loss-of-function strain lacking PDH was constructed by deleting the pdh gene and investigated for growth on phenylalanine and the presence of relevant enzyme activities in tungsten-supplemented and -depleted media.
RESULTS
PDH and AOR activities in cells grown under different conditions.
To find out more about the function of AOR of A. aromaticum and the conditions under which it is produced, denitrifying cultures were grown anaerobically on various aromatic substrates, either intermediates of the Phe metabolic pathway or unrelated compounds like benzoate and ethylbenzene. The respective cell extracts were analyzed for the activities of the aldehyde oxidizing enzymes AOR and PDH as well as phenylglyoxylate oxidoreductase (PGOR) (28, 29) as indicator enzyme of the induced phenylacetate (PA)-metabolic pathway (Table 1). Cells of A. aromaticum grown on aromatic substrates that are not metabolized via the Phe or PA pathways (e.g., benzoate, benzaldehyde, benzyl alcohol, or ethylbenzene) showed no or very low levels of PGOR activity, whereas cells grown on Phe, PA, or other intermediates feeding into the PA-metabolic pathway (e.g., phenylpyruvate or phenylglyoxylate) exhibited specific PGOR activities larger than 80 nmol min−1 mg−1 in cell extracts. These measured activities are consistent with the minimum requirement to explain the observed growth rates on the respective substrates (30 nmol min−1 mg−1 protein equals a growth rate of 0.08 h−1). Typical activities and the observed growth rates on different substrates are listed in Table 1.
TABLE 1.
Activities of AOR, phenylglyoxylate oxidoreductase (PGOR) and PDH (NAD and NADP) in cell extracts of A. aromaticum grown anaerobically on various aromatic compoundsa
| Compoundb | AOR (nmol min−1 mg−1) | PGOR (nmol min−1 mg−1) | PDH |
μ (h−1)c | ||
|---|---|---|---|---|---|---|
| NAD (nmol min−1 mg−1) | NADP (nmol min−1 mg−1) | NAD/NADP | ||||
| Phenylalanine* | 18 | 211 | 108 | 192 | 0.56 | 0.04–0.08 |
| Phenylacetate | 16 | 138 | 15 | 54 | 0.28 | 0.07–0.11 |
| Phenylglyoxylate | 45 | 192 | 20 | 18 | 1.11 | 0.06–0.10 |
| Benzoate* | 11 | 4 | 3 | 4 | 0.75 | 0.16–0.18 |
| Ethylbenzene | 9 | <2 | 8 | 11 | 0.73 | 0.09–0.12 |
| Benzaldehyde | 20 | 10 | 21 | 32 | 0.66 | 0.04–0.06 |
| Benzyl alcohol | 27 | 15 | 54 | 70 | 0.77 | 0.06–0.10 |
Standard deviations of the values from Phe cultures are given in Fig. 2b; those from other cultures amounted to <20% of the respective activity values.
*, values for these compounds were averaged from three independent cultures.
Within the ranges of observed growth rates (μ), the upper limits represent growth under optimal conditions, whereas imbalanced refeeding of the cultures or other handling problems resulted in lowered μ values within the indicated ranges.
In contrast to PGOR, AOR activity was observed in almost all cell batches grown on any of the substrates tested, exhibiting rather variable values in different batches of A. aromaticum cells grown on the same substrate. Specific AOR activities of cell extracts of Phe-degrading cultures could be as low as 1.5 nmol min−1 mg−1 (12) and reached up to 30 nmol min−1 mg−1. Cultures grown on other intermediates of Phe degradation such as PA, as well as control cells from cultures degrading benzoate, ethylbenzene, benzaldehyde, or benzyl alcohol, generally exhibited lower specific AOR activities, whereas the highest specific AOR activities with more than 45 nmol min−1 mg−1 were recorded in cells grown on phenylglyoxylate (see Table 1 and Fig. 1 for additional details).
FIG 1.
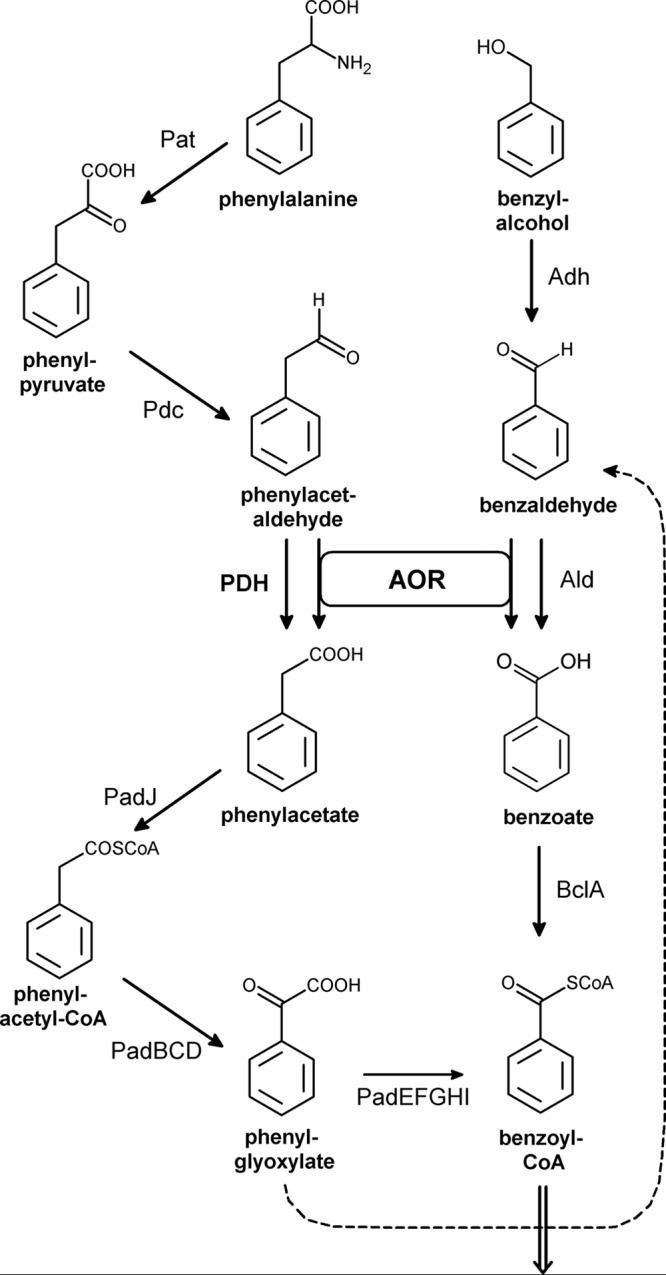
Anaerobic degradation pathways of phenylalanine and benzyl alcohol in A. aromaticum. The enzymes and corresponding genes (in parentheses) are as follows: Pat, phenylalanine aminotransferase (ebA596); Pdc, phenylpyruvate decarboxylase (ebA6545); AOR, phenylacetaldehyde:ferredoxin oxidoreductase (ebA5005); PDH, phenylacetaldehyde dehydrogenase (ebA4954); PadJ, phenylacetate-CoA ligase (ebA5402); PadBCD, phenylacetyl-CoA:acceptor oxidoreductase (ebA5393, ebA5395, and ebA5396); PadEFGHI, phenylglyoxylate:acceptor oxidoreductase (ebA5397, ebB191, and ebA5399 to ebA5401); Adh, benzyl alcohol dehydrogenase (ebA3118 or ebA4623); Ald, benzaldehyde dehydrogenase (ebA5642); BclA, benzoate-CoA ligase (ebA5301). The double arrow indicates further degradation of benzoyl-CoA via aromatic ring reduction and β-oxidation.
Since phenylacetaldehyde dehydrogenase (PDH) was found to be highly specific for PAld, it is appropriate that the pdh gene is expressed when PAld is produced, e.g., as intermediate of Phe degradation but not in cells grown on intermediates further downstream, such as PA or phenylglyoxylate. Indeed, the highest PAld-oxidizing activities were found in Phe-grown cells and can be attributed to PDH based on the characteristic ratio of NAD and NADP-coupling of 0.56 (Table 1) (12). In cells grown on any other aromatic substrates, the PAld oxidizing activities were lower, and the derived NAD/NADP activity ratios suggest the presence of other enzymes than PDH, which couple either to NAD or NADP. Elevated levels of PAld oxidizing activities recorded in cells grown on benzaldehyde or benzyl alcohol are probably due to unspecific turnover by a distinct benzaldehyde dehydrogenase, which has been proposed to be the product of the gene ebA5642 (ald), based on proteomic data (5). Only low-level activities of PAld oxidation were measured in cells grown on benzoate and ethylbenzene which are metabolized without aldehyde intermediates.
Effects of deleting the pdh gene on Phe metabolism.
We further investigated the relative roles of PDH and AOR in oxidizing PAld by constructing a pdh deletion strain from the streptomycin-resistant (Smr) A. aromaticum strain SR7 (30). We first confirmed the wild-type character of strain SR7 with regard to Phe metabolism by establishing identical growth behavior with the wild-type strain EbN1 on the relevant substrates Phe and benzoate, respectively (data not shown) and by identifying the mutation leading to streptomycin-resistance as a single base substitution in the rpsL gene. This changes Lys42 (AAG) to Arg42 (AGG) in ribosomal protein S12, representing a previously known mutation leading to high resistance without interfering in metabolic reactions (31, 32).
The pdh gene was completely deleted and replaced by a gentamicin resistance gene in two selection steps based on the sacB counterselection system, yielding the Δpdh deletion strain (see Materials and Methods). The accurate replacement of pdh was verified by specific PCR assays as described in methods. The deletion strain was originally designed to enforce the usage of AOR for metabolizing Phe but also proved useful to assess the contributions of PDH and AOR to Phe metabolism by comparing its properties to those of the corresponding wild-type variant, strain SR7. The Δpdh strain still grew anaerobically under denitrifying conditions on Phe as sole source of carbon and energy in tungstate-supplemented media, initially at a slightly lower growth rate compared to the wild-type strain (Fig. 2a). No activities of either NAD or NADP-coupled PDH isoenzymes were detected in any of the cell batches recovered from these cultures, confirming the identity of the mutant but also suggesting that the PDH correlated with Phe metabolism in A. aromaticum is not replaced by other dehydrogenases using PAld as the substrate. In contrast, specific activities of AOR were significantly and reproducibly higher than those found in the wild-type strain, which never exceeded 30 nmol min−1 mg−1. In comparison, specific AOR activities were already increased in the first Phe-degrading cell batch (46 nmol min−1 mg−1) and further increased to about 150 nmol min−1 mg−1 in cell batches after 10 rounds of reinoculation (all cultures were grown to an optical density at 578 nm [OD578] of >1). Over a cultivation period of more than 18 months, no significant PAld dehydrogenase activities coupled to NAD or NADP were detected in Phe cultures, whereas AOR activities remained on a high level (in average 140 nmol min−1 mg−1) (Fig. 2b), and optimal growth rates matched with those for the wild-type strain on Phe (Fig. 2a).
FIG 2.

(a) Growth of A. aromaticum SR7 (black) and the Δpdh strain in minimal medium on Phe in the presence (diamonds) and absence of tungstate (−W) (circles). Nonadapted cultures (first inoculation on Phe-containing medium) are shown with green, and adapted cultures are shown with red. The growth curves of the adapted strains were recorded after 10 and 9 cycles of reinoculation, respectively. Calculated initial growth rates (μ) of the respective cultures: strain SR7, 0.08 h−1; Δpdh strain (nonadapted), 0.04 h−1; Δpdh strain (adapted), 0.05 h−1; Δpdh −W strain (nonadapted), 0.02 h−1; and Δpdh −W strain (adapted), 0.06 h−1. The average optimal growth rates for wild-type cultures were 0.077 h−1 (±0.018) for adapted cultures of the Δpdh strain in the presence or absence of tungstate 0.074 (±0.016) h−1 and 0.069 (±0.012) h−1, respectively (each calculated from six cultures). (b) Specific enzyme activities of AOR (red), phenylglyoxylate oxidoreductase (PGOR) (green), and PAld dehydrogenases coupled to NAD (dark blue) or NADP (light blue) in different cultures grown anaerobically on Phe. A. aromaticum wild-type cells (Wt) were grown in standard medium, cells of the Δpdh strain in standard medium (Δpdh) and medium lacking tungstate (Δpdh −W). The activities shown reflect the average values and standard deviations of at least three independent cultures. The values of the Δpdh strain reflect those of the respective adapted strains after >20 reinoculation cycles on W-containing or W-free medium, respectively.
In order to find out whether the increased aor expression levels in the adapted culture of the Δpdh strain are caused by a mutation, the aor genes, including their upstream regions (>350 bp), were amplified and sequenced using primer pair 13 and 14 (see Table 4). Indeed, a specific single-base-pair exchange (G→T) was identified 139 bp upstream of the start codon of the first gene of the aor operon in the adapted culture of the Δpdh strain, whereas no mutation was observed in nonadapted cells. This mutation may affect a potential operator sequence correlated to an apparent σ54-dependent promoter site 71 bp upstream of the first translational start codon (see Fig. 6). In contrast to some related species and strains, A. aromaticum does not contain a closely linked gene for a potential regulator in the vicinity of the aor genes, which may be predicted to be involved in their regulation (see below).
TABLE 4.
Oligonucleotides used in this study for the construction of the pdh deletion strain, its verification by PCR, and control sequencing of rpsL, the aor operon, and aldB
| No. | Primer | Sequence (5′–3′)a |
|---|---|---|
| 1 | rpsL_for | TGGGGCGCCTCGGTATAATGG |
| 2 | rpsL_rev | TTAAGCTTTCTTCGGGCGCTTCGCG |
| 3 | pdh_ur_for | AATCTAGAGACACCGCTTTCACGATGTCC |
| 4 | pdh_ur_rev | GTGCCTTCATCCGTTTCCACGGTGTGCGTCCATCTCGTCTCCTTCACGCTTGCATTGAAG |
| 5 | pdh_dr_for | TTTGATATCGACCCAAGTACCGCCACCTAACGGGGCTCCGCGGCTGGCCGGATGGAGGTG |
| 6 | pdh_dr_rev | AATCTAGAAACTTCATCCTGTCGGAGATG |
| 7 | pdh_GAT_for | CTTCAATGCAAGCGTGAAGGAGACGAGATGGACGCACACCGTGGAAACGGATGAAGGCAC |
| 8 | pdh_GAT_rev | CACCTCCATCCGGCCAGCCGCGGAGCCCCGTTAGGTGGCGGTACTTGGGTCGATATCAAA |
| 9 | control_pdh-up_f | GAAGAACTCGTCGAGCACG |
| 10 | control_pdh-dn_r | CGAAGGAACTGGTGCTGGC |
| 11 | control_pdh_f | ATCCAGCGACAGGGGAAGTGTTCG |
| 12 | control_pdh_r | GTGTATTCCTCGAGCCCGTAGCG |
| 13 | aorP_for | GCATAGTTTCGCCGCTGC |
| 14 | aorP_rev | GGCGGGGTCAATGTGAAGC |
| 15 | aldB-up_f | AGGATCGATTCAGGCACGCTGC |
| 16 | aldB-dn_r | GTTCGATTTCCTTTGGATGCGGG |
Underlining indicates incorporated restriction sites or inverse complementary sequences.
FIG 6.

Putative promoter regions of pdh (a), aor (b), and aldB (c) compared between A. aromaticum and related bacteria. Where intervening sequences have been shortened, the number of omitted bases is indicated between slashes. The putative (−12/−24) promoters are indicated by boxes, the expected transcriptional starts are highlighted in dark gray, and possible conserved operator regions are indicated in light gray. The asterisk in panel b indicates the site of the observed mutation (G→T) within the aor upstream region in the adapted Δpdh strain. Arrows indicate potential dyad symmetries. Strains: EbN1, A. aromaticum; PA01, Azoarcus sp. strain PA01; Tamino, Thauera aminoaromatica; Tphen, Thauera phenylacetica; KH32, Azoarcus sp. strain KH32C; Atoluc, Azoarcus toluclasticus; CIB, Azoarcus sp. strain CIB; Tchl, Thauera chlorobenzoica; Cama, Chromobacterium amazonense. Unfortunately, the similarity of the respective DNA sequences of A. aromaticum and strain PA01 is too high to reasonably predict other conserved elements beyond the promoter boxes.
Growth and enzyme activities in tungstate-free medium.
The observed increased specific activities of AOR in cells of the Δpdh strain grown on Phe suggest that AOR compensates for the missing PDH activity. Because AOR activity of A. aromaticum depends on a sufficient supply of tungstate in the growth medium (12), the requirement of AOR for Phe metabolism in the Δpdh strain was tested by growth studies in tungstate-depleted medium. Surprisingly, the cells still grew with Phe as the sole carbon source in these tests, albeit at severely reduced growth rates and with a long lag phase (Fig. 2a). This effect is not shared with the wild-type strain, which shows the same growth parameters on Phe in the presence or absence of tungstate (12). Growth rates (μ) of 0.02 to 0.03 h−1 were determined for freshly established cultures of the Δpdh strain, compared to 0.05 to 0.08 h−1 in regular medium. This clearly corroborated the importance of AOR for Phe metabolism in the pdh deletion strain, but suggested yet another possible way to complement the loss of the pdh gene. When the Δpdh strain was continuously reinoculated under the same conditions, we again observed adaptation to faster growth. After nine cycles of reinoculation, growth rates and biomass yields reached an apparently stable optimum state, which closely approximated the values of wild-type cells (Fig. 2a).
The specific activities of PDH and AOR isoenzymes were tested in the cell extracts of several cultures of the Δpdh strain during the adaptation to tungstate-independent growth on Phe. No PAld oxidizing activity coupled to either benzylviologen or NADP was detected in any cell extract, verifying the absence of AOR (due to W-depletion) and PDH (due to pdh gene deletion) and indicating that yet another enzyme must support growth. Indeed, increasing activity of an NAD-linked PAld dehydrogenase was detected over the course of the reinoculation series, reaching specific activities of 20 to 60 nmol min−1 mg−1 in cell extracts after nine reinoculations and persisting at this level over 20 or more reinoculations (Fig. 2b). Because NADP was not reduced as alternative electron acceptor, PDH has apparently been replaced by an alternative aldehyde dehydrogenase, which also needed higher PAld concentrations for optimal activity in the enzyme assays than applicable for PDH (see methods). Although lower than the original PDH or AOR activities, the measured activities of NAD-linked PAld oxidation were still consistent with the observed growth rates. Moreover, specific phenylglyoxylate oxidoreductase (PGOR) activities reached the same level in adapted cultures as those from wild-type cells, indicating effective ongoing PA degradation (Fig. 3).
FIG 3.

(a) Enrichment of NAD-coupled PAld dehydrogenase activity from the Δpdh strain grown in the absence of tungstate. Proteins were electrophoretically separated on a 12% (wt/vol) SDS-PAGE gel and stained with Coomassie brilliant blue. Each lane except lane 6 contained 30 μg of protein. Lanes: 1 and 7, molecular mass marker; 2, cell extract of strain SR7; 3, cell extract of the Δpdh strain grown in standard medium; 4: cell extract of the Δpdh strain grown without W; 5, active fraction from DEAE-Sepharose; and 6, active fraction from CHT-I (15 μg). Arrows 1 and 2 mark the two highly expressed proteins AldB* and AdhB in the Δpdh strain in the absence of W. (b) Enzyme kinetics of AldB* with acetaldehyde (red), glutardialdehyde (magenta), propionaldehyde (blue), and PAld (green). The kinetic parameters are listed in Table 3.
Broad-range aldehyde dehydrogenase compensating for the absence of PDH and AOR.
To identify the aldehyde dehydrogenase compensating for PDH and AOR, the Phe-adapted Δpdh strain was grown anaerobically on Phe without tungstate. The harvested cells (18 g [wet mass]) were lysed, and NAD-linked PAld dehydrogenase was chromatographically enriched in two steps (see Materials and Methods). The measured PAld dehydrogenase activities eluted as single peaks from both columns, indicating the presence of only one major enzyme in the extract, and we obtained a 7-fold enrichment of the enzyme at 24% yield (Table 2). Analysis of the fractions via SDS-PAGE demonstrated a reasonable purity of the preparation, which represented a highly enriched protein of 55 kDa but still contained several minor bands of contaminations (Fig. 3a). A strongly induced protein comigrating with the enriched subunit was already observed in the extract of Phe-adapted cells of the Δpdh strain in the absence of tungstate, compared to those grown with tungstate (Fig. 3a). To identify the newly induced enzyme, the band containing the enriched subunit was cut from a polyacrylamide gel and analyzed by mass spectrometry of tryptic fragments. The majority of the recorded fragments correlated the enriched 55-kDa protein to the product of the aldB gene (ebA4625 [open reading frame designation]) from the genome of A. aromaticum. This protein has previously been annotated as an aldehyde dehydrogenase, showing 93% identity to an assumed acetaldehyde dehydrogenase of Thauera chlorobenzoica (33, 34) or 80% amino acid identity to an NAD-dependent chloroacetaldehyde dehydrogenase of Xanthobacter autotrophicus (35). The aldB gene is part of an apparent operon with a gene coding for a Fe-dependent alcohol dehydrogenase (adhB, ebA4623) located immediately downstream. The adhB gene codes for a 39 kDa protein, which matches the size of another prominent band induced in Phe-adapted cells of the Δpdh strain grown without tungstate (Fig. 3a, lane 4), and missing in cells supplied with tungstate. The band was analyzed by mass spectrometry of tryptic fragments, identifying it as gene product of adhB. The complete aldB gene, including 350 bp upstream of its translation start codon, was amplified and sequenced using primer pair 15 and 16 (see Table 4) to analyze for potential mutations correlated with the observed adaptation to growth on Phe. Using Phe-adapted cells of the Δpdh strain grown in the absence of tungstate after either 6 or 18 months of reinoculations, we detected a single-base-pair exchange within the aldB coding region. Conversely, no mutations were recorded in the aldB gene in control cells, either of Phe-adapted cultures of the same strain in the presence of tungstate or of wild-type strain SR7. The mutation is localized at position 1379 of the aldB gene (A→G) and leads to a Y460C mutation in the amino acid sequence, which may result in increasing the activity of AldB to oxidize PAld. The structure of AldB was modeled by the Swiss-model server (https://swissmodel.expasy.org), revealing a predicted position of the mutated Tyr460 residue directly adjacent to the conserved catalytic cysteine residue of AldB (Cys299) (Fig. 4). No mutations were observed in the 5′-region of aldB, suggesting that the apparently increased aldB expression may be caused by a mutation in a (as-yet-unknown) regulatory protein.
TABLE 2.
Specific activities and enrichment of PAld oxidizing activity (NAD) from the A. aromaticum EbN1 Δpdh strain grown in tungstate-depleted medium on Phe
| Step | Protein (mg) | Activity (U) | Yield (%) | Sp act (PAld)a (nmol min−1 mg−1) | Enrichment |
|---|---|---|---|---|---|
| Soluble cell extract | 1,050 | 21.6 | 100 | 21 | 1 |
| DEAE-Sepharose | 226 | 6.8 | 31 | 30 | 1.4 |
| Hydroxyapatite (CHT-I) | 37 | 5.2 | 24 | 143 | 7 |
That is, the maximum specific activity started with 400 μM PAld.
FIG 4.

Structural models of the active sites of AldB (a) and AldB* (b) showing the predicted locations of theY460C mutation adjacent to the catalytic Cys299 residue. The carbon atoms of Cys299 are indicated in red, and those of Tyr460 in AldB (a) or Cys460 in AldB* (b) are indicated in green. Distances are given in angstroms (generated using PyMOL v1.10).
Biochemical characterization of AldB.
The enriched AldB-Y460C variant (here designated AldB*) was used to establish the principal biochemical properties of the enzyme. As indicated from the data from cell extracts, AldB* exclusively accepted NAD as electron acceptor, whereas no activity was observed with NADP. An apparent Km value of 280 μM (standard error [SE] = 93 μM) and an apparent Vmax of 90 mU mg−1 (SE = 8 mU mg−1) was established for NAD, using 100 μM PAld as the substrate. Moreover, the enzyme exhibited a rather broad substrate spectrum, which included aromatic aldehydes such as PAld or benzaldehyde, aliphatic aldehydes like acetaldehyde, propionaldehyde or crotonaldehyde, and dialdehydes like glutardialdehyde (Table 3). The only tested aldehyde that was hardly converted by AldB* was formaldehyde. The enzyme exhibited pronounced lag phases after starting the reactions, which differed for various substrates (less pronounced with aliphatic than with aromatic aldehydes) and made assessment and comparison of the activities difficult. We finally obtained reproducible and robust enzyme activity data by modifying the assays to begin with an initial turnover of a low substrate concentration (10 μM) to completion, followed by restarting the enzyme assay with the intended concentrations of substrate. The different substrates tested varied greatly in their turnover rates and catalytic or inhibitory properties. AldB* showed the highest activities with small aliphatic substrates (highest recorded activity with acetaldehyde), whereas the larger substrates showed significantly lower activity values combined with substrate inhibition (Fig. 3b). However, PAld seems to be still better converted than benzaldehyde. The measured catalytic parameters of AldB* for some selected substrates are summarized in Table 3. Data obtained for glutardialdehyde fitted best to a substrate inhibition model but still yielded relatively high standard error values.
TABLE 3.
Apparent kinetic parameters of the oxidation of different aldehydes by AldB* using NAD as the electron acceptora
| Substrate | Vmax (mU mg−1) | Km (μM) | Ki (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | R2 |
|---|---|---|---|---|---|---|
| Acetaldehyde | 360 (±15) | 96 (±13) | 0.33 | 0.0034 | 0.970 | |
| Propionaldehyde | 265 (±15) | 38 (±9) | 0.243 | 0.0064 | 0.916 | |
| Glutardialdehyde | 265 (±67) | 79 (±43) | 1,343 (±1,051) | 0.243 | 0.0031 | 0.862 |
| Phenylacetaldehyde | 77 (±5) | 6.9 (±1.9) | 513 (±127) | 0.071 | 0.0103 | 0.924 |
| Crotonaldehyde | 88 | ND | ND | ND | ND | ND |
| Benzaldehyde | 21 | ND | ND | ND | ND | ND |
| Formaldehyde | 4 | ND | ND | ND | ND | ND |
Values for kcat are given per subunit of AldB* (55 kDa). The R2 values indicate the respective fitting quality; ND, not determined; standard errors are indicated in parentheses where applicable. The relatively bad fitting of the data with glutardialdehyde may be related to the simultaneous inactivation of the enzyme by cross-linking.
Cross-linking experiments with AldB* treated with either glutardialdehyde or dimethyl suberimidate revealed the occurrence of additional bands on SDS-PAGE, which fitted to the expected sizes of the dimer, trimer, and tetramer (data not shown), suggesting a homotetrameric structure of AldB* as usual for the protein family (36). The closely related chloroacetaldehyde dehydrogenase of Xanthobacter autotrophicus was also reported to be a tetramer (35).
Conservation of genes involved in PAld oxidation in related species.
From a bioinformatic analysis of the sequenced genomes of Rhodocyclaceae species closely related to A. aromaticum, it can be stated that only two strains, Azoarcus sp. strain PA01 and Azoarcus sp. strain KH32C, carry genes coding for potential orthologues of PDH (88 to 97% protein sequence identity). However, only the gene of strain PA01 is located in an identical genomic context (Fig. 5a), whereas strain KH32C carries two almost identical copies of the gene at different positions on its chromosome and a large plasmid, respectively. All other sequenced strains contain multiple genes for aldehyde dehydrogenases, but the low sequence conservation (<50% protein sequence identity) precludes any close relation to the biochemical properties of PDH. Interestingly, all potential pdh genes from A. aromaticum and the two Azoarcus strains contain the typical −24/−12 boxes of a potential σ54-promoter in their 5′-regions (see Fig. 6).
FIG 5.

Genomic arrangement of the genes coding for PDH (a), AOR (b), and AldB (c) with their respective putative σ54 promoters in A. aromaticum and related species. Neighboring genes without apparent roles in the metabolic module are shown in gray. nadC, nicotinate-nucleotide diphosphorylase gene; thrC, threonine synthase gene; pdh, phenylacetaldehyde dehydrogenase gene; thrA, homoserine dehydrogenase gene; alaT, aminotransferase gene; arsB, gene for arsenate resistance protein; hyp, gene for hypothetical protein; aorABCDE, putative aor operon; cyt c, cytochrome c550 gene; aorR, transcriptional regulator gene; eut, gene for ethanolamine utilization protein; aldR, transcriptional regulator gene; aldB, aldehyde dehydrogenase gene; adhB, alcohol dehydrogenase gene; ALDH; aldehyde dehydrogenase family protein. *, inserted genes or different genetic context in control strains.
The aor operons are present in identical arrangements in almost all sequenced members of the Rhodocyclaceae and other bacteria. Therefore, we propose to name these genes aorABCDE, with aorB coding for the W-containing subunit similar to archaeal AOR. However, the 5′-flanking sequences of the aor operons share significant similarities only within small groups of related species. The most similar sequence to that of A. aromaticum (and the only one with a conserved genomic context) is again from Azoarcus sp. strain PA01 (Fig. 5b). A closer analysis indicates that both strains may again employ a potential σ54 promoter for expression of the aor genes, although the locations of the −24/−12 boxes are not conserved between the two strains (Fig. 6). The observed potential operator mutation in the adapted mutant is located upstream of the predicted promoter box in a region sharing significant similarity between the two strains, although their spacing to the respective predicted promoter boxes is different (Fig. 6). Remarkably, the genomes of a number of additional related species (Thauera aminoaromatica, T. phenylacetica, Azoarcus sp. strain CIB, A. toluclasticus, and A. tolulyticus) reveal a different consensus sequence in the 5′ flanks of their aor operons, revealing conserved σ54-promoter boxes and the presence of an adjacent gene for a highly conserved σ54-related regulator, for which we propose the name aorR (Fig. 5b). It is probable that this regulator is involved in regulation of the aor operon in these species, but no gene for an orthologous regulator is present in A. aromaticum (closest paralogue at 40% protein sequence identity), leaving it an open question which regulator is responsible for induction of the aor operon in this bacterium.
Finally, the aldB gene product is highly conserved only in the related species T. chlorobenzoica (93% identity). These two strains exhibit a σ54-promoter structure with a conserved −24/−12 box (Fig. 6) and contain a directly adjacent gene coding for a conserved σ54-related regulator, proposed to be named aldR (Fig. 5c). The closest aldehyde dehydrogenase paralogues from either A. aromaticum itself (ebA2242 gene product) or other strains of the Rhodocyclaceae show <75% protein sequence identity to AldB and probably represent other isoenzymes. The aldB operon is disrupting an ethanolamine utilization gene cluster of A. aromaticum (Fig. 5c) which shares high similarity to orthologous genes in Azoarcus strain PA01 (94% identical gene products), which are located at a different genomic locus.
DISCUSSION
Aldehyde:ferredoxin oxidoreductase (AOR) from A. aromaticum EbN1 has originally been proposed to catalyze the oxidation of PAld and to be involved in anaerobic Phe metabolism based on proteomic data (5). Later on, it was found to be not essential for Phe metabolism due to the simultaneous presence of a PAld-specific PDH (12). Based on these observations, PDH has been proposed to represent the main functional enzyme of the pathway, whereas AOR acts in preventing the accumulation of reactive aldehydes (25, 26). Still, analysis of the genomes of closely related strains affiliated to the genera Thauera and Azoarcus indicates that AOR is evolutionary important, because most of these strains contain an identically composed aor operon, whereas only very few contain genes for orthologues of PDH or the alternative enzyme AldB.
Our experiments confirmed the hypothesis that AOR is mainly involved in aldehyde detoxification, whereas PDH is the major physiological enzyme used for Phe degradation. Various levels of AOR activities were found in cells grown on different intermediates of Phe catabolism or on various other aromatic substrates unrelated to the Phe metabolic pathway, while PDH was only present in Phe-degrading cells and already absent in those grown on PA (or any of the other tested substrates). The control assays for PGOR confirmed its presence in cells grown on Phe or any further intermediates of the PA metabolic pathway, suggesting independent regulatory systems involved in inducing expression of pdh, the aor genes or the PA metabolic genes. The observed AOR activities correlate with the probability of generating potentially toxic aldehyde intermediates in the respective metabolic pathways, for example as side reactions of 2-oxoacid oxidoreductase like PGOR (26) or as an intermediate in benzyl alcohol degradation (Fig. 1), which is supposed to be converted to benzoate by a separate pathway catalyzed by a specific benzyl alcohol dehydrogenase and a benzaldehyde dehydrogenase. From proteomic studies, these enzymes are most likely correlated to the genes ebA3118 and ebA5642, respectively (2, 3, 5). Interestingly, a second induced alcohol dehydrogenase in benzyl alcohol-grown cells was identified as a gene product of ebA4623 (adhB), which forms an apparent operon with aldB and has also been found as highly overproduced protein in our study. The low activities of PAld dehydrogenase(s) detected under various other growth conditions appear to be correlated to low pressure for aldehyde detoxification and may be attributed to other aldehyde dehydrogenases than PDH, based on the deviating activity ratios measured with NAD and NADP. Potential candidates for these enzymes are the products of at least 17 additional genes coding for aldehyde dehydrogenases in the genome of A. aromaticum (2).
In conclusion, our observations suggest that AOR is normally produced in very small amounts sufficient for aldehyde detoxification, which strongly impairs efforts to purify and characterize the enzyme (12). However, we were able to construct a mutant strain of A. aromaticum using AOR as the exclusive metabolic enzyme for anaerobic Phe degradation by deleting the pdh gene. This strain underwent an apparent genetic adaptation to produce 5-fold more AOR, which may be caused by a single-base mutation in a putative operator element. This mutation may affect the binding sites of an activator protein required for σ54-dependent transcription initiation (upstream activating sequence, though unusually close to the promoter), of a DNA-bending protein like integration host factor, or of an additional repressor protein involved in regulation of the aor operon (37). Some related Thauera and Azoarcus strains indeed contain a gene coding for a σ54-related activator (aorR) adjacent to their aor operons which is probably involved in their regulation, but A. aromaticum does not carry an orthologous gene and therefore must regulate aor expression differently. Regarding the broad substrate range of AOR, its synthesis may be expected to be induced with many substrates generating aldehyde intermediates during their degradation pathways (e.g., p-cresol, alcohols, or amines).
The surprising retention of growth on Phe of the pdh deletion mutant without either PDH or AOR activities in tungstate-depleted medium and its adaptation to regain high growth rates was caused by a vastly increased amount of an NAD-dependent aldehyde dehydrogenase, the aldB gene product. Notably, aldB forms an apparent operon with the coinduced adhB gene coding for a Fe-type alcohol dehydrogenase, which has previously been reported as a protein induced in benzyl alcohol-grown cells (3, 5), but the actual metabolic tasks of AldB or AdhB still remain to be determined (38). The apparent absence of a coinduced aldB gene product in benzyl alcohol-grown cells suggests the presence of an internal promoter in the operon, which induces only adhB expression, whereas the expression of both aldB and adhB appears to be induced under the conditions reported here (Fig. 3a). The detected Y460C mutation in AldB of the adapted culture suggests a modulation of the catalytic properties of the enzyme to act as suitable catalyst for PAld oxidation. This is supported by the predicted location of Y460 directly adjacent to the C299 of the active site in a structural model of AldB (Fig. 4), suggesting that the mutation may help in creating the space required to accommodate the aromatic ring of PAld into the substrate-binding site of AldB. The enriched mutant variant AldB* indeed revealed a very broad substrate range from short aliphatic aldehydes to large aromatic aldehydes. The activities and Km values for acetaldehyde and propionaldehyde were similar to those of the closely related chloroacetaldehyde dehydrogenase from Xanthobacter autotrophicus (35), whereas the activity with PAld was rather low and affected by substrate inhibition.
The relatively low activity of AldB* with PAld is probably compensated by the observed high expression of the aldB and adhB genes to obtain sufficient enzyme activity for the observed growth rate, as evident from the relatively low specific activities in cell extracts which were close to the calculated minimum at the observed growth rates. A similar effect of high expression compensating for low affinity has previously been recorded by the similar chloroacetaldehyde dehydrogenase from X. autotrophicus growing on 2-chloroacetaldehyde (35). Like pdh and the aor operon, aldB appears to be expressed from a σ54-type promoter, and a gene for a putative correlated activator protein (aldR) is localized directly adjacent to the aldB gene in the opposite direction (Fig. 5).
A comparison of the three enzyme systems involved in PAld oxidation in A. aromaticum and several closely related species of the Thauera/Azoarcus cluster revealed that only the aor operon is generally conserved. This also applies to many other bacterial species affiliated to the Betaproteobacteria or Firmicutes. All of these species show a similar composition of the operon, consisting of three genes for putative subunits or electron transfer components of AOR (aorABC), a gene for a small protein paralogous to the molybdopterin synthesis protein MoaD, which is potentially involved in AOR maturation (aorD), and a gene for a conserved small hypothetical protein (aorE). Several members of the Rhodocyclaceae contain highly conserved σ54-type promoters upstream of their aor operons and also carry a directly adjacent gene for a σ54-related regulator, AorR (Fig. 5 and 6). However, A. aromaticum and several other Rhodocyclaceae species have apparently lost the gene for this regulator, along with the associated conserved σ54-type promoter, and have instead developed new potential σ54-type promoter sites within a distance of 100 bp (Fig. 6). This hypothetical scenario gains particular credibility from comparing the highly similar upstream sequences of the aor operons of A. aromaticum EbN1 and Azoarcus sp. strain PA01. Both strains lack the aorR gene, and the genome context of their aor operon is different than in aorR-containing strains but conserved between the two strains. In both strains, the former consensus σ54-type promoter is still recognizable in a sequence alignment but has been inactivated by deletion of one base between the −24 and −12 boxes (data not shown), while each of the two strains has apparently evolved a new σ54-type promoter in the adjacent sequence (Fig. 6). In spite of the high similarity of the DNA sequences between strains EbN1 and PA01, the new promoter sequences differ and are located at different distances from the translational start of the first genes (Fig. 5 and 6). Therefore, we assume that A. aromaticum has recently lost its dedicated regulatory system for aor expression and remodeled the aor upstream region to respond to a different, but still unknown σ54-type regulator. In addition to the activation process mediated by σ54-type regulator, the operon may be controlled by an additional repressor protein as known for other operons (e.g., ArcAB and Fnr affecting the cydAB operon of Escherichia coli [39]).
Remarkably, the pdh gene, as well as the aldB-adhB operon, also seems to be regulated by σ54-linked regulation systems, judging from the presence of σ54-type promoters in front of both transcriptional units and the presence of a gene for a σ54-related regulator (aldR) in front of the aldB-adhB operon. Unfortunately, these genes are only conserved in very few strains, indicating that many different aldehyde dehydrogenase isoenzymes may be recruited for Phe metabolism even in highly related strains. However, some potential common properties involved in their regulatory systems may already be envisaged: it appears that σ54-type regulatory systems are very prominently used for that purpose, but it will take much more research efforts to correlate the individual genes with their corresponding regulators. The genome of A. aromaticum alone contains nine genes for σ54-type regulators (2) that could be involved in regulating pdh, aor, or the aldB-adhB operon.
MATERIALS AND METHODS
Growth of bacteria.
Aromatoleum aromaticum strain EbN1 was grown anaerobically on carbonate-buffered minimal medium using Phe or other aromatic substrates as sole carbon source and nitrate as an electron acceptor, as described previously (40). Substrate (except benzoate) and nitrate were supplied at concentrations of 1 and 4 mM, respectively, cultures on benzoate were carried out at concentrations of 4 and 10 mM. Cultures were discontinuously refed with the same concentrations and incubated at 28°C in stoppered glass bottles without continuous shaking (volume, 0.1 to 2 liters). Growth was monitored by determining the increase in the OD578 and the consumption of nitrate (Quantofix; Macherey-Nagel, Düren, Germany). The standard culture medium for A. aromaticum contained 150 nM Na2MoO4 and 18 nM Na2WO4 of the highest purities available (99.9 and 99.995%, respectively). Tungstate-free medium was prepared in bottles with ultrapure water (conductivity < 0.05 μS/cm).
E. coli strain DH5α was used for plasmid construction and was grown aerobically with continuous shaking at 37°C on Luria-Bertani (LB) medium (1% tryptone, 0.5% yeast extract, 0.8% NaCl). Plasmids were transferred by transformation into chemically competent E. coli cells (41). Antibiotics were added at the following final concentrations: streptomycin, 50 μg ml−1; kanamycin, 30 μg ml−1; and gentamicin, 20 μg ml−1.
In this study, we used a spontaneous Smr mutant of A. aromaticum (strain SR7) (30) for conjugation experiments and as a wild-type control strain. Strain SR7 grows in mineral salt medium containing up to 1,000 μg ml−1 streptomycin sulfate. The rpsL gene encoding the ribosomal protein S12 was amplified and sequenced using the primer pair 1 and 2 (Table 4). For conjugational plasmid transfer, strain SR7 was grown in phosphate-buffered mineral salt medium (30), solidified with 1.5% (wt/vol) agar, if required. As a donor strain for conjugational plasmid transfer, the diaminopimelate (DAP) auxotrophic E. coli strain WM3064, a derivative of strain β2155 (42), was used and grown on LB medium containing DAP (50 μg/ml).
Preparation of cell extracts.
All steps performed with cells or extracts of A. aromaticum were carried out under anoxic conditions. Cells were harvested in the late-exponential growth phase before reaching the stationary phase at OD578 values between 1 and 1.5 by centrifugation at 17,000 × g and 4°C for 20 min (0.1- to 2-liter scale cultures). Sedimented cells were immediately frozen and stored at −80°C. For the preparation of extracts, cells were suspended in one volume of 50 mM 4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid (HEPPS)–KOH buffer (pH 8.0) or in the respective buffer for subsequent chromatographic enzyme enrichment (see below) containing 10% glycerol and 0.05 mg of DNase I per ml. Cell suspensions were disrupted by sonication (volumes < 5 ml) or passed twice through a French pressure cell press. Cell debris and membranes were removed by ultracentrifugation at 100,000 × g and 4°C for 1 h. Supernatants were stored in sealed glass vessels with 10% (vol/vol) glycerol at −80°C until use.
Enzyme activity assays.
Enzyme activities involved in anaerobic degradation of phenylalanine were assayed photometrically in extracts of A. aromaticum cells. These enzymes were phenylacetaldehyde:ferredoxin oxidoreductase (AOR), phenylacetaldehyde dehydrogenase (PDH), and phenylglyoxylate:acceptor oxidoreductase (PGOR). All assays were carried out with 100,000 × g extracts at 25°C and were repeated at least twice.
AOR.
The benzylviologen-dependent oxidation of PAld (AOR activity) was assayed under anaerobic conditions as described previously (25), but using 100 mM Tris-HCl buffer (pH 8.4). The reaction was started by the addition of PAld (2 mM) and monitored at 600 nm to record reduction of benzylviologen (ε = 7,400 M−1 cm−1).
PDH.
The NAD or NADP-dependent oxidation of PAld was measured as described previously (4), but modified as follows: the enzyme was assayed in HEPPS-KOH buffer (pH 8.5) containing 1 mM NAD or NADP, and the reaction was started by addition of a low concentration of the substrate PAld (25 μM), because of the observed inhibition of PDH by higher concentrations of PAld (12). The absorbance was monitored at 365 nm (ε = 3,400 M−1 cm−1).
NAD-dependent aldehyde oxidation by AldB* was measured in the same buffer with NAD (1.5 mM) and differing aldehyde concentrations, 100 μM in the standard assay. The activity assay showed lag phases of various extents after adding the substrate but produced suitable results if the enzyme was first exposed to small substrate concentration of 10 μM. The assay was restarted with the intended substrate concentrations immediately after the initial phase had to run to completion, yielding reproducible activity values within the first 2 min after restart.
Phenylglyoxylate:acceptor oxidoreductase (PadEFGHI).
Benzylviologen- and coenzyme A-dependent oxidation of phenylglyoxylate was assayed under anaerobic conditions as described previously (28). The reaction was monitored photometrically by determining the increase of absorbance of reduced benzylviologen at a wavelength of 600 nm.
Construction of a chromosomal pdh deletion mutant.
The pdh gene was replaced by a gentamicin resistance gene by homologous recombination as reported previously (30). Two flanking regions of 1 kb upstream and downstream of the pdh gene were amplified by PCR using the primer pairs 13 and 4 and 5 and 6, respectively (Table 4), and a gentamicin resistance gene from plasmid pBBR1MCS-5 (43) was amplified by primer pair 7 and 8. The flanking regions of pdh were fused to the gentamicin resistance cassette by overlapping fusion PCR using primer pair 3 and 6. The resulting PCR product was cloned into the suicide vector pK19mobsacB (44) using an XbaI restriction site to get plasmid pK19mobsacB-Δpdh. The vector was transferred into A. aromaticum EbN1 by conjugational plasmid transfer as described previously (30), taking E. coli WM3064 (auxotrophic for DAP) as the donor strain (45). Transconjugants were obtained after incubation of the mixtures on minimal medium agar plates (40, 46) containing 5 mM benzoate as sole carbon source, 10 mM nitrate, and 50 μg/ml kanamycin in an anaerobic chamber (N2 atmosphere) at 28°C for at least 7 days. Colonies were purified by replating on new agar plates, and the obtained clones were checked for the absence of E. coli donor cells by plating on LB agar containing DAP and sequencing the 16S rRNA genes as negative controls. The resulting insertion mutant, A. aromaticum SR7(pK19mobsacB-Δpdh), was then cultivated over two transfers in liquid culture without antibiotics and plated on NM agar containing benzoate and 5% sucrose to select for loss of the plasmid. The obtained deletion strain, A. aromaticum SR7_Δpdh, was tested to be gentamicin resistant and kanamycin sensitive and was verified by specific PCRs using primer pairs 9 and 10 (Table 4) (Δpdh strain, 3.298-kb product; wild type, 3.931 kb) and 11 and 12 (Δpdh strain, no product; wild type, 1.32 kb), respectively.
Enrichment of AldB from the Δpdh strain.
A PAld oxidizing dehydrogenase activity (NAD) was enriched from the A. aromaticum Δpdh strain grown anaerobically on Phe in tungstate-free minimal medium. Cells from a 5-liter culture were harvested, dissolved in buffer A (100 mM Tris-HCl [pH 8.0], 10% glycerol), and disrupted with a French pressure cell press. Using the cell extract after ultracentrifugation (1 h, 100,000 × g), an enrichment of the NAD-coupled PAld oxidizing activity was performed by two separation steps on an Äkta Pure FPLC system. Cell extract was first loaded to an anion exchange column (DEAE-Sepharose_26/12) equilibrated with buffer A and developed by a linear gradient of increasing NaCl concentration (buffer B is buffer A plus 1 M NaCl). The PAld oxidizing activity eluted at approximately 200 mM NaCl. The most active fractions were combined, rebuffered to pH 6.8 (buffer C is 5 mM MES-KOH [pH 6.8] plus 1 mM CaCl2) using a HiPrep_26/10 desalting column (GE Healthcare), and applied to a hydroxyapatite column (CHT-I) equilibrated with buffer C. The column was developed with a linear gradient of increasing potassium phosphate concentrations (employing buffer D [5 mM MES-KOH plus 400 mM potassium phosphate; pH 6.8]), and the enzyme eluted at 170 mM potassium phosphate. Active fractions were supplied with 10% glycerol and frozen at −80°C until further use. The enrichment was documented on a 12% (wt/vol) SDS-polyacrylamide gel stained with Coomassie brilliant blue.
Other methods.
Chromosomal DNA was prepared as described previously (47). The protein concentration was determined according to Bradford (48) using bovine serum albumin as a standard. Proteins were separated by discontinuous SDS-PAGE (49). Cross-linking of AldB was performed as described previously (50) by incubating with either dimethyl suberimidate or glutardialdehyde as cross-linking agents at pH 8.5 (200 mM triethanolamine) and room temperature. The cross-linked polypeptides were analyzed by SDS-PAGE. The identities of proteins separated by SDS-PAGE were determined by mass spectrometry of tryptic fragments using a 4800 Proteomics Analyzer (MDS Sciex, Concord, Ontario, Canada). MS data were evaluated against an in-house database using Mascot embedded into GPS explorer software (MDS Sciex).
ACKNOWLEDGMENTS
This study was supported by grants from the Deutsche Forschungsgemeinschaft (priority program 1927) and the SYNMIKRO LOEWE Centre, Marburg, Germany.
We acknowledge R. Rabus for providing A. aromaticum strain SR7.
REFERENCES
- 1.Heider J, Fuchs G. 1997. Anaerobic metabolism of aromatic compounds. Eur J Biochem 243:577–596. doi: 10.1111/j.1432-1033.1997.00577.x. [DOI] [PubMed] [Google Scholar]
- 2.Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R. 2005. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36. doi: 10.1007/s00203-004-0742-9. [DOI] [PubMed] [Google Scholar]
- 3.Rabus R, Trautwein K, Wöhlbrand L. 2014. Towards habitat-oriented systems biology of “Aromatoleum aromaticum” EbN1. Appl Microbiol Biotechnol 98:3371–3388. doi: 10.1007/s00253-013-5466-9. [DOI] [PubMed] [Google Scholar]
- 4.Schneider S, Mohamed ME-S, Fuchs G. 1997. Anaerobic metabolism of l-phenylalanine via benzoyl-CoA in the denitrifying bacterium Thauera aromatica. Arch Microbiol 168:310–320. doi: 10.1007/s002030050504. [DOI] [PubMed] [Google Scholar]
- 5.Wöhlbrand L, Kallerhoff B, Lange D, Hufnagel P, Thiermann J, Reinhardt R, Rabus R. 2007. Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1. Proteomics 7:2222–2239. doi: 10.1002/pmic.200600987. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs G, Boll M, Heider J. 2011. Microbial degradation of aromatic compounds: from one strategy to four. Nat Rev Microbiol 9:803–816. doi: 10.1038/nrmicro2652. [DOI] [PubMed] [Google Scholar]
- 7.Aklujkar M, Risso C, Smith J, Beaulieu D, Dubay R, Giloteaux L, DiBurro K, Holmes D. 2014. Anaerobic degradation of aromatic amino acids by the hyperthermophilic archaeon Ferroglobus placidus. Microbiol 160:2694–2709. doi: 10.1099/mic.0.083261-0. [DOI] [PubMed] [Google Scholar]
- 8.Wöhlbrand L, Jacob JH, Kube M, Mussmann M, Jarling R, Beck A, Amann R, Wilkes H, Reinhardt R, Rabus R. 2013. Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ Microbiol 15:1334–1355. doi: 10.1111/j.1462-2920.2012.02885.x. [DOI] [PubMed] [Google Scholar]
- 9.Heider J, Boll M, Breese K, Breinig S, Ebenau-Jehle C, Feil U, Gad'on N, Laempe D, Leuthner B, Mohamed ME-S, Schneider S, Burchhardt G, Fuchs G. 1998. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch Microbiol 170:120–131. doi: 10.1007/s002030050623. [DOI] [PubMed] [Google Scholar]
- 10.Dangel W, Brackmann R, Lack A, Mohamed M, Koch J, Oswald B, Seyfried B, Tschech A, Fuchs G. 1991. Differential expression of enzyme activities initiating anoxic metabolism of various aromatic compounds via benzoyl-CoA. Arch Microbiol 155:256–262. doi: 10.1007/BF00252209. [DOI] [Google Scholar]
- 11.Mohamed ME-S, Fuchs G. 1993. Purification and characterization of phenylacetate-coenzyme A ligase from a denitrifying Pseudomonas sp., an enzyme involved in the anaerobic degradation of phenylacetate. Arch Microbiol 159:554–562. doi: 10.1007/BF00249035. [DOI] [PubMed] [Google Scholar]
- 12.Debnar-Daumler C, Seubert A, Schmitt G, Heider J. 2014. Simultaneous involvement of a tungsten-containing aldehyde:ferredoxin oxidoreductase and a phenylacetaldehyde dehydrogenase in anaerobic phenylalanine metabolism. J Bacteriol 196:483–492. doi: 10.1128/JB.00980-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee S-K, Fuchs G. 1999. Phenylacetyl-CoA:acceptor oxidoreductase, a membrane-bound molybdenum-iron-sulfur enzyme involved in anaerobic metabolism of phenylalanine in the denitrifying bacterium Thauera aromatica. Eur J Biochem 262:507–515. doi: 10.1046/j.1432-1327.1999.00399.x. [DOI] [PubMed] [Google Scholar]
- 14.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NCJ. 2003. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Mol Biol 10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 15.Mukund S, Adams MWW. 1991. The novel tungsten-iron-sulfur protein of the hyperthermophilic archaebacterium, Pyrococcus furiosus, is an aldehyde ferredoxin oxidoreductase. Evidence for its participation in a unique glycolytic pathway. J Biol Chem 266:14208–14216. [PubMed] [Google Scholar]
- 16.Kletzin A, Mukund S, Kelley-Crouse TL, Chan MK, Rees DC, Adams MWW. 1995. Molecular characterization of the genes encoding the tungsten-containing aldehyde ferredoxin oxidoreductase from Pyrococcus furiosus and formaldehyde ferredoxin oxidoreductase from Thermococcus litoralis. J Bacteriol 177:4817–4819. doi: 10.1128/jb.177.16.4817-4819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukund S, Adams MWW. 1995. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 270:8389–8392. doi: 10.1074/jbc.270.15.8389. [DOI] [PubMed] [Google Scholar]
- 18.Mukund S, Adams MWW. 1993. Characterization of a novel tungsten-containing formaldehyde ferredoxin oxidoreductase from the hyperthermophilic archaeon, Thermococcus litoralis: a role for tungsten in peptide catabolism. J Biol Chem 268:13592–13600. [PubMed] [Google Scholar]
- 19.Johnson MK, Rees DC, Adams MW. 1996. Tungstoenzymes. Chem Rev 96:2817–2840. doi: 10.1021/cr950063d. [DOI] [PubMed] [Google Scholar]
- 20.Roy R, Adams MWW. 2002. Characterization of a fourth tungsten-containing enzyme from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 184:6952–6956. doi: 10.1128/JB.184.24.6952-6956.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bevers LE, Bol E, Hagedoorn P-L, Hagen WR. 2005. WOR5, a novel tungsten-containing aldehyde oxidoreductase from Pyrococcus furiosus with a broad substrate specificity. J Bacteriol 187:7056–7061. doi: 10.1128/JB.187.20.7056-7061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White H, Strobl G, Feicht R, Simon H. 1989. Carboxylic acid reductase: a new tungsten enzyme catalyses the reduction of non-activated carboxylic acids to aldehydes. Eur J Biochem 184:89–96. doi: 10.1111/j.1432-1033.1989.tb14993.x. [DOI] [PubMed] [Google Scholar]
- 23.Weinert T, Huwiler SG, Kung JW, Weidenweber S, Hellwig P, Stark H-J, Biskup T, Weber S, Cotelesage JJ, George GN, Ermler U, Boll M. 2015. Structural basis of enzymatic benzene ring reduction. Nat Chem Biol 11:586–591. doi: 10.1038/nchembio.1849. [DOI] [PubMed] [Google Scholar]
- 24.Kletzin A, Adams MW. 1996. Tungsten in biological systems. FEMS Microbiol Rev 18:5–63. doi: 10.1111/j.1574-6976.1996.tb00226.x. [DOI] [PubMed] [Google Scholar]
- 25.Heider J, Ma K, Adams MW. 1995. Purification, characterization, and metabolic function of tungsten-containing aldehyde ferredoxin oxidoreductase from the hyperthermophilic and proteolytic archaeon Thermococcus strain ES-1. J Bacteriol 177:4757–4764. doi: 10.1128/jb.177.16.4757-4764.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma K, Hutchins A, Sung S-JS, Adams MW. 1997. Pyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon, Pyrococcus furiosus, functions as a CoA-dependent pyruvate decarboxylase. Proc Natl Acad Sci U S A 94:9608–9613. doi: 10.1073/pnas.94.18.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan M, Mukund S, Kletzin A, Adams M, Rees D. 1995. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 267:1463–1469. doi: 10.1126/science.7878465. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch W, Schägger H, Fuchs G. 1998. Phenylglyoxylate:NAD+ oxidoreductase (CoA benzoylating), a new enzyme of anaerobic phenylalanine metabolism in the denitrifying bacterium Azoarcus evansii. Eur J Biochem 251:907–915. doi: 10.1046/j.1432-1327.1998.2510907.x. [DOI] [PubMed] [Google Scholar]
- 29.Schneider S, Fuchs G. 1998. Phenylacetyl-CoA:acceptor oxidoreductase, a new α-oxidizing enzyme that produces phenylglyoxylate: assay, membrane localization, and differential production in Thauera aromatica. Arch Microbiol 169:509–516. doi: 10.1007/s002030050604. [DOI] [PubMed] [Google Scholar]
- 30.Wöhlbrand L, Rabus R. 2009. Development of a genetic system for the denitrifying bacterium Aromatoleum aromaticum strain EbN1. J Mol Microbiol Biotechnol 17:41–52. doi: 10.1159/000159194. [DOI] [PubMed] [Google Scholar]
- 31.Funatsu G, Wittmann HG. 1972. Ribosomal proteins. XXXIII. Location of amino acid replacements in protein S12 isolated from Escherichia coli mutants resistant to streptomycin. J Mol Biol 68:547–550. [DOI] [PubMed] [Google Scholar]
- 32.Gregory ST, Cate JHD, Dahlberg AE. 2001. Streptomycin-resistant and streptomycin-dependent mutants of the extreme thermophile Thermus thermophilus. J Mol Biol 309:333–338. doi: 10.1006/jmbi.2001.4676. [DOI] [PubMed] [Google Scholar]
- 33.Song B, Palleroni NJ, Häggblom MM. 2000. Description of strain 3CB-1, a genomovar of Thauera aromatica, capable of degrading 3-chlorobenzoate coupled to nitrate reduction. Intl J Syst Evol Microbiol 50:551–558. doi: 10.1099/00207713-50-2-551. [DOI] [PubMed] [Google Scholar]
- 34.Song B, Palleroni NJ, Kerkhof LJ, Häggblom MM. 2001. Characterization of halobenzoate-degrading, denitrifying Azoarcus and Thauera isolates and description of Thauera chlorobenzoica sp. nov. Int J Syst Evol Microbiol 51:589–602. doi: 10.1099/00207713-51-2-589. [DOI] [PubMed] [Google Scholar]
- 35.van der Ploeg J, Smidt MP, Landa AS, Janssen DB. 1994. Identification of chloroacetaldehyde dehydrogenase involved in 1,2-dichloroethane degradation. Appl Environ Microbiol 60:1599–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perozich J, Nicholas H, Wang B-C, Lindahl R, Hempel J. 1999. Relationships within the aldehyde dehydrogenase extended family. Prot Sci 8:137–146. doi: 10.1110/ps.8.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collado-Vides J, Magasanik B, Gralla JD. 1991. Control site location and transcriptional regulation in Escherichia coli. Microbiol Rev 55:371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carmona M, Zamarro MT, Blázquez B, Durante-Rodríguez G, Juárez JF, Valderrama JA, Barragán MJL, García JL, Díaz E. 2009. Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol Mol Biol Rev 73:71–133. doi: 10.1128/MMBR.00021-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotter PA, Melville SB, Albrecht JA, Gunsalus RP. 1997. Aerobic regulation of cytochrome d oxidase (cydAB) operon expression in Escherichia coli: roles of Fnr and ArcA in repression and activation. Mol Microbiol 25:605–615. doi: 10.1046/j.1365-2958.1997.5031860.x. [DOI] [PubMed] [Google Scholar]
- 40.Rabus R, Widdel F. 1995. Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch Microbiol 163:96–103. doi: 10.1007/BF00381782. [DOI] [PubMed] [Google Scholar]
- 41.Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28. doi: 10.1016/0378-1119(90)90336-P. [DOI] [PubMed] [Google Scholar]
- 42.Dehio C, Meyer M. 1997. Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J Bacteriol 179:538–540. doi: 10.1128/jb.179.2.538-540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovach ME, Elzer PH, Steven Hill D, Robertson GT, Farris MA, Roop RM II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 44.Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multipurpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 45.Muhr E, Leicht O, González Sierra S, Thanbichler M, Heider J. 2016. A fluorescent bioreporter for acetophenone and 1-phenylethanol derived from a specifically induced catabolic operon. Front Microbiol 6:1561. doi: 10.3389/fmicb.2015.01561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Widdel F, Bak F. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p 3352–3378. In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer KH (ed), The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications. Springer, New York, NY. [Google Scholar]
- 47.Wilson K. 2001. Preparation of genomic DNA from bacteria, p 2.4.1–2.4.5 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Struhl K (ed), Current protocols in molecular biology. John Wiley & Sons, Inc, Hoboken, NJ. [DOI] [PubMed] [Google Scholar]
- 48.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 49.Laemmli V. 1970. Determination of protein molecular weight in polyacrylamide gels. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 50.Davies GE, Stark GR. 1970. Use of dimethyl suberimidate, a cross-linking reagent, in studying the subunit structure of oligomeric proteins. Proc Natl Acad Sci U S A 66:651–656. doi: 10.1073/pnas.66.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]