

SUMMARY
Brown adipose tissue (BAT) activation and subcutaneous white fat browning are essential components of the thermogenic response to cold stimulus in mammals. microRNAs have been shown to regulate both processes in cis. Here, we identify miR-32 as a BAT-specific super-enhancer-associated miRNA in mice that is selectively expressed in BAT and further upregulated during coldexposure. Inhibiting miR-32 invivo led to impaired cold tolerance, decreased BAT thermogenesis, and compromised white fat browning as a result of reduced serum FGF21 levels. Further examination showed that miR-32 directly represses its target gene Tob1, thereby activating p38 MAP kinase signaling to drive FGF21 expression and secretion from BAT. BAT-specific miR-32 overexpression led to increased BAT thermogenesis and serum FGF21 levels, which further promotes white fat browning in trans. Our results suggested miR-32 and Tob1 as modulators of FGF21 signaling that can be manipulated for therapeutic benefit against obesity and metabolic syndrome.
In Brief
Increasing energy expenditure through non-shivering thermogenesis can be exploited for the treatment of metabolic syndromes. Ng et al. found that, in brown fat, miR-32 inhibits Tob1 and thereby activates p38/MAPK signaling and promotes brown fat activity and FGF21 secretion, which trans-activates white fat browning to enhance non-shivering thermogenesis upon cold exposure.
INTRODUCTION
Obesity and the associated metabolic syndromes have become increasingly prevalent. Such a trend represents a major public health issue that has greatly increased the interest in thermogenic fat cells such as brown and beige adipocytes. Beige adipocytes, similar to brown adipocytes, also express a high level of uncoupling protein 1 (UCP1) and can emerge within inguinal white adipose tissue (iWAT, a typical scWAT) in response to prolonged cold exposure or activation of β-adrenergic receptors (Harms and Seale, 2013; Wu et al., 2012, 2013). The emergence of beige adipocytes in white fat depots is termed the “browning” of white fat (Harms and Seale, 2013; Wu et al., 2013). Brown and beige adipocytes contain large amounts of mitochondria and highly express UCP1, which uncouples respiration and dissipates chemical energy as heat, hence increasing energy expenditure and counteracting hypothermia, obesity, and diabetes (Bartelt et al., 2011). Recent studies have indicated that, contrary to previous findings, adult humans contain significant deposits of UCP1-positive brown/beige fat (Cypess et al., 2009). These findings led to renewed interest in brown adipogenesis and its physiological role in adult humans.
The thermogenic program in mammals is under tight control of transcription factors and coactivators (Wu et al., 2013) via various signaling pathways. The p38/MAPK signaling pathway, downstream of β-adrenergic receptors and cyclic AMP (cAMP) activation, plays an essential role in BAT thermogenic response (Cao et al., 2004; Robidoux et al., 2005). Activation of the p38/MAPK pathway leads to the phosphorylation and activation of ATF2 (Cao et al., 2004), thus promoting transcription of downstream target genes such as PGC1α, UCP1, and FGF21 (Robidoux et al., 2005). Conversely, repressors of p38/MAPK signaling such as Tob1, a tumor suppressor, silence the pathway under normal conditions (Jiao et al., 2012; Sun et al., 2013). In addition to the transcriptional regulators, several secreted factors such as FGF21 and Irisin were also shown to promote the browning of subcutaneous white fat (Fisher et al., 2012; Hondares et al., 2011; Lee et al., 2014a), demonstrating the crosstalk and trans-regulation among different metabolic organs.
Furthermore, microRNAs have been recently found to represent another crucial regulatory layer that interacts with transcriptional control mechanisms to regulate metabolic homeostasis (Trajkovski and Lodish, 2013). In particular, several miRNAs have recently emerged as important regulators of either brown or beige adipocyte differentiation and function (Chen et al., 2013; Kim et al., 2014; Liu and Kuang, 2013; Mori et al., 2012; Sun et al., 2011; Trajkovski et al., 2012; Yin et al., 2013). However, few studies have investigated the role of BAT-selective miRNAs in regulating thermogenesis via white fat browning.
Recently, bioinformatic analysis has been increasingly applied to the identification of regulators controlling adipogenic and thermogenic programs. Among various in silico approaches, one that is particularly powerful is the so-called super-enhancer association analysis. Super-enhancers are large genomic regions containing a high density of transcription factor binding, and they are associated with genes crucial for specifying cell identity and function (Hnisz et al., 2013; Whyte et al., 2013). They can be defined by the occupancy of mediator and cell-type-specific transcription factors, as well as chromatin marks such as histone H3K27ac and H3K4me1 (Whyte et al., 2013). Through the super-enhancer association analysis, we have recently identified RREB1 and PIM1 as important factors promoting brown adipogenesis (Brunmeir et al., 2016), while KLF11 was previously identified as an essential regulator of human white fat browning (Loft et al., 2015).
In search for miRNAs important for BAT function, we utilized histone H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) data to define super-enhancers in various tissues and cell lines and subsequently identified 19 miRNAs associated with BAT-specific super-enhancers. miR-32 was the third hit on the list after miR-193b and miR-365, which have been identified previously (Sun et al., 2011). A recent study suggested that miR-32 is involved in lipid metabolism (Shin et al., 2012), but its role in regulating BAT function has not been explored. We found that miR-32 is significantly upregulated in BAT during cold exposure, which further promotes BAT thermogenesis and iWAT browning.
RESULTS
miR-32 Is Associated with a BAT-Specific Super-Enhancer and Is Upregulated during Cold Stress
As we hypothesized that miRNAs associated with BAT-specific super-enhancers may regulate BAT identity and function, we performed super-enhancer analysis on H3K27ac ChIP-seq data in various tissues from ENCODE database using Rank Ordering of Super-Enhancers (ROSE) (Bernstein et al., 2012; Lovén et al., 2013; Whyte et al., 2013). Through this analysis, we found a total of 78 miRNAs that are located within 50 kb from BAT super-enhancers, and they were defined as BAT super-enhancer-associated miRNAs (Table S1). To further identify miRNAs specifically associated with BAT super-enhancers, we ranked these miRNAs by their distance from epididymal WAT (eWAT) super-enhancers, furthest first (Figure 1A). The identification of miR-193b and miR-365-1, the top two hits from our analysis, as well as miR-196a-2 and miR-455 that have previously been shown to play important roles in BAT differentiation or WAT browning (Mori et al., 2012; Sun et al., 2011; Zhang et al., 2015), demonstrated the validity of our approach. miR-32, which was shown to play a role in lipid metabolism (Shin et al., 2012), ranked the third in our list (Figure 1A), and its gene is located approximately 601 kb away from the nearest eWAT super-enhancer. Hence, it was selected for further studies to examine its role in thermogenesis. As shown in Figure 1B, miR-32 is associated with a BAT-specific super-enhancer, which is not present in other tissues such as eWAT, liver, and kidney. qRT-PCR analysis further confirmed that miR-32 expression was significantly higher in BAT compared to other tissues (Figure 1C). In addition, miR-32 expression in BAT was upregulated by 3-fold after 7 days of cold stimulation (Figure 1D), suggesting that miR-32 may regulate the thermogenic response.
Figure 1. miR-32 Associates with BAT-Specific Super-Enhancer and Expresses Selectively in BAT in Response to Cold Stress.

(A) miR-32 is the third highest ranked miRNA based on its distance from eWAT super-enhancers among all miRNAs associated with BAT super-enhancers.
(B) The super-enhancer associated with miR-32 is found only in BAT but not in other tissues or cell lines. Tracks depict H3K27ac ChIP-seq signal used for super-enhancer identification from various tissues and cell lines downloaded from ENCODE as density of mapped reads. Bottom track depicts Ensembl gene annotations. Red bar represents the miR-32-associated super-enhancer in BAT.
(C) miR-32 is highly expressed in BAT as compared with other mouse tissues (n = 3).
(D) miR-32 expression is significantly upregulated in BAT after 7 days’ cold exposure (n = 5).
Data represent mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Table S1.
miR-32-ASO Treatment Does Not Lead to Observable Changes in Whole-Body Metabolic Profile, Tissue Morphology, or Gene Expression under Normal Physiological Conditions
To investigate the role of miR-32 in metabolic regulation, we inhibited miR-32 in vivo via injection of miR-32 anti-sense oligonucleotide (miR-32-ASO). A single 25-mg/kg dose of miR-32-ASO administered via intraperitoneal (i.p.) injection resulted in almost complete inhibition of miR-32 in multiple tissues including BAT, eWAT, liver, iWAT, and skeletal muscle until 7 days post-injection (Figure S1A). However, under room temperature (24°C) housing conditions, miR-32 inhibition did not result in significant changes to core body temperature, energy expenditure, food intake, or ambulatory movement (Figures S1B–S1H). Furthermore, H&E staining showed that the morphology of the five tissues mentioned above was not significantly different (Figure S1I). qRT-PCR analysis of molecular markers of brown adipocytes such as UCP1, PGC1α, and Cidea showed that expression of these genes was not significantly changed in all the tissues examined (Figures S1J–S1N). Adipogenic markers such as Adiponectin (AdipoQ), PPARγ2, and all other markers tested also did not show any significant changes in the tissues examined (Figures S1J–S1N), suggesting that miR-32 may not play a major role in metabolic homeostasis and BAT function under room temperature housing conditions.
miR-32-ASO-Treated Mice Exhibit Impaired Cold Tolerance and Reduced Energy Expenditure under Cold Stress
Since we observed that miR-32 is upregulated in response to cold stimulation (Figure 1D), we decided to inhibit miR-32 expression and to challenge the mice by placing them at 6°C for 7 days to allow us to examine its effect on both BAT thermogenesis and iWAT browning (Lim et al., 2012). As shown above, 7-day cold exposure increased miR-32 expression in BAT significantly (Figures 1D and 2A). In addition, miR-32 was also upregulated in eWAT, liver, iWAT, and muscle (Figure 2A). Injection of miR-32-ASO 1 day before commencement of the 7-day cold stimulation period led to almost complete inhibition of miR-32 expression when examined at the end of the 7-day cold challenge (Figure 2A). Mice injected with miR-32-ASO had significantly impaired thermogenic response, and their core body temperatures were significantly lower compared to control-ASO-injected mice as soon as 4 hr after being placed at 6°C (Figure 2B). miR-32-ASO-injected mice also had significantly lower core body temperatures compared to control mice throughout the 7-day cold exposure (Figure 2B). Energy expenditure of miR-32-ASO-injected mice were also significantly lower than control-ASO-injected mice during both light and dark periods, further suggesting a defect in cold-induced thermogenesis (Figures 2C and 2D). However, levels of food intake, ambulatory movement, and total mass at the start and end of cold exposure were similar in the two groups of mice (Figures S2A–S2E). Since miR-32 is highly expressed in BAT and is further upregulated when mice are exposed to cold, we postulated that inhibition of miR-32 during cold challenge would have a large impact on BAT thermogenic function. Although H&E staining showed that BAT morphology did not differ significantly between control-ASO- and miR-32-ASO-injected mice (Figure 2E), quantification of the immunostaining against UCP1 revealed that mice injected with miR-32-ASO had significantly lower levels of UCP1 compared to control (Figures 2E and 2F). In addition, qRT-PCR analysis revealed that UCP1 mRNA levels increased more than 2-fold in response to cold stimulation, and miR-32-ASO-injected mice had lower levels of UCP1 expression compared to control mice (Figure 2G). Other BAT and mitochondrial markers such as Cidea, Prdm16, and Cox8b were not significantly changed upon miR-32 inhibition (Figure 2G). Since PGC1α and UCP1 are important in the cold-induced thermogenic response, western blot analysis was carried out to determine the levels of these two proteins in BAT. The data show that UCP1 but not PGC1α expression was significantly reduced in miR-32-ASO-injected mice (Figures 2H and 2I). In addition, no significant changes to tissue morphology and thermogenic gene expression were observed in liver, muscle, or eWAT when comparing control-ASO- or miR-32-ASO-injected mice (Figures S2F–S2I). These results suggest that miR-32 inhibition impaired cold tolerance through blunted BAT’s thermogenic response to cold stimulation and possibly other mechanisms but did not affect liver, eWAT, or skeletal muscle.
Figure 2. Inhibition of miR-32 during Cold Exposure Leads to Significantly Lower Core Body Temperature and Reduced Energy Expenditure in Mice as the Result of Reduced Thermogenic Response in BAT and Beige Cell Emergence in iWAT.

(A) Increases of miR-32 expression in BAT, eWAT, liver, iWAT, and skeletal muscle after cold exposure were severely blunted in miR-32-ASO-treated mice (n = 8) compared to control-ASO mice (n = 7).
(B) miR-32-ASO-treated mice (n = 8) showed lower core body temperatures during cold exposure when compared to control-ASO mice (n = 7).
(C) Total energy expenditure was significantly reduced after 7 days’ cold stress in miR-32-ASO-treated mice (n = 6) as compared with control-ASO mice (n = 6). Energy expenditure was normalized to lean body mass.
(D) Average total energy expenditure was significantly lower in miR-32-ASO-treated mice (n = 6).
(E) BAT tissue of mice kept for 7 days at room temperature (n = 5), at 6°C with injection of control-ASO (n = 7) or miR-32-ASO (n = 8) did not show significant differences in H&E staining (top row). But miR-32-ASO-injected mice had lower UCP1 immunostaining as compared with control-ASO mice after cold exposure (bottom row).
(F) Quantification of UCP1 immunostaining in BAT showed that UCP1 levels were significantly lower in miR-32-ASO-treated mice.
(G) In BAT, mRNA levels of UCP1 were slightly but statistically significantly lower in miR-32-ASO-injected mice, whereas other thermogenic and adipogenic genes were expressed at similar levels. Data were normalized to Cyclophilin A (PPIA).
(H) Protein levels of UCP1 but not PGC1α were lower in BAT from mice injected with miR-32-ASO.
(I) Quantification of relative protein expression using ImageJ showed that protein levels of UCP1 but not PGC1α were significantly lower in BAT from mice injected with miR-32-ASO. Average intensities were normalized to that of Calnexin.
(J) H&E (top row) and UCP1 immunostaining (bottom row) of iWAT showed that the cold-induced emergence of multi-locular, UCP1-positive beige adipocytes was significantly impaired in miR-32-ASO-treated mice.
(K) Quantification of UCP1 immunostaining in iWAT showed that UCP1 levels were significantly lower in miR-32-ASO-treated mice.
(L) mRNA levels of thermogenic genes in iWAT were significantly reduced in miR-32-ASO-treated mice. Data were normalized to PPIA.
(M) Western blots showed that miR-32-ASO treatment significantly reduced UCP1 and PGC1α protein levels in iWAT.
(N) Quantification of relative UCP1 and PGC1α protein levels using ImageJ. Average intensities were normalized to that of Calnexin.
Data represent mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figures S1 and S2.
Systemic Inhibition of miR-32 Significantly Reduces Beige Cell Emergence in iWAT and Blunts the Expression of Thermogenic Genes in iWAT during Cold Stress
Since browning of iWAT has been widely accepted as another important component of thermogenic response to cold stimulation in mice (Lee et al., 2014b; Wu et al., 2012, 2013), we decided to examine whether miR-32 inhibition affected iWAT browning during cold exposure. H&E staining of iWAT showed that cold exposure for 7 days at 6°C was sufficient to induce significant browning of iWAT as observed by the presence of many multi-locular cells within the tissue (Figure 2J). However, iWAT from mice injected with miR-32-ASO had significantly fewer multi-locular cells (Figure 2J). Immunostaining against UCP1 was performed to positively identify beige cells, and miR-32-ASO-injected mice were found to have significantly fewer UCP1-positive cells (Figures 2J and 2K) as compared with control-ASO mice. qRT-PCR analysis showed that, although common adipogenic marker PPARγ2 expression did not change in response to cold stimulation, BAT markers such as Cidea, PPARα, PGC1α, and UCP1 increased significantly when mice were cold stimulated (Figure 2L). Consistent with what was observed in histology, BAT markers were significantly downregulated in miR-32-inhibited mice: Cidea, PPARα, PGC1α, Dio2, Cox8b, and UCP1 were all expressed less than half than control-ASO-injected mice after cold exposure (Figure 2L). Western blot analysis further showed that PGC1α and UCP1 protein levels were much lower in iWAT of miR-32-ASO-injected mice (Figures 2M and 2N). All these results suggest that miR-32 and its rise in response to cold stimulation is important for driving the thermogenic response, and inhibition of miR-32 leads to a significant reduction in beige cell emergence within iWAT.
miR-32 Does Not Promote Beige Cell Emergence within iWAT in a Cell-Autonomous Manner
Since miR-32 expression increases in iWAT during cold exposure (Figure 2A) and systemic inhibition of miR-32, including in iWAT, led to reduced beige cell emergence in iWAT (Figures 2J and 2K), we hypothesized that miR-32 may directly promote beige cell emergence within iWAT. To test this hypothesis, we performed bilateral injection of control-ASO and miR-32-ASO into the left and right iWAT depots of mice, respectively (Liu et al., 2014), and then placed them under cold exposure. Comparing miR-32 expression between right and left iWAT depots showed robust inhibition of miR-32 in the miR-32-ASO-injected depot 7 days after injection, whereas miR-32 levels in both skeletal muscle depots near the injection site remained comparable, suggesting that iWAT-specific miR-32 inhibition was achieved (Figure S3A). However, iWAT-specific inhibition of miR-32 did not lead to a significant reduction in beige cell emergence within the iWAT depot as indicated by expression of UCP1 and PGC1α (Figures S3B–S3D). Moreover, when the expression level of miR-32 was modulated in iWAT stromal vascular fraction (SVF) cells by miR-32-ASO or miR-32 mimic treatment (Figure S5A), the expression of brown selective genes such as UCP1, PGC1α, and FGF21 did not change significantly (Figures S5E and S5G). These results further suggested that miR-32 does not promote the browning of primary iWAT adipocytes in a cell-autonomous manner. In addition, we also evaluated the effects of miR-32 on general adipogenesis of iWAT SVF cells using oil red O staining. As shown in Figure S5C, miR-32 inhibition or overexpression did not exert observable effects on lipid accumulation in iWAT SVF cells after differentiation. In parallel, adipogenic markers such as PPARγ and FABP4 were also unaffected by miR-32 modulation (Figures S5E and S5G). SLC45A3, a gene previously shown to be a target for miR-32 in oligodendrocytes (Shin et al., 2012), was not affected by either miR-32 inhibition or overexpression at both the mRNA and protein levels in iWAT SVF cells (Figures S5E and S5G). In summary, the data suggest that miR-32 does not promote beige cell emergence in a cell-autonomous manner but perhaps through a long-range effect.
miR-32-ASO Treatment Inhibits FGF21 Expression in BAT, Leading to Reduced Serum FGF21 Levels
Although miR-32 was inhibited in multiple tissues (Figure 2A), we hypothesized that its inhibition in BAT, where miR-32 expression is the highest during cold stimulation, may play a major role in suppressing beige cell emergence in iWAT (Figures 2J and 2K) and also lowering expression of thermogenic genes in BAT and iWAT (Figures 2G–2I and 2L–2N). In order for such long-range effects to occur, we postulated that increased miR-32 expression within BAT may promote the secretion of browning factors from BAT into the blood where it can have endocrine signaling effects (Villarroya and Vidal-Puig, 2013). In recent years, FGF21 has emerged as an important browning factor secreted by BAT and other tissues that has pleotropic effects on multiple tissues including browning of iWAT (Chartoumpekis et al., 2011; Fisher et al., 2012; Hondares et al., 2011; Owen et al., 2014). Hence, we performed ELISAs to examine serum levels of FGF21 in order to determine whether miR-32 may indeed regulate serum FGF21 levels. Serum FGF21 levels rose nearly 2-fold in response to cold exposure, but mice treated with miR-32-ASO had significantly lower levels of serum FGF21 (Figure 3A). Although liver has been shown to be a major source of FGF21 at normal physiological conditions, BAT upregulates FGF21 production and secretion to contribute significantly to serum FGF21 levels during cold exposure (Hondares et al., 2011; Villarroya and Vidal-Puig, 2013). qRT-PCR data also confirmed the findings from previous studies with BAT replacing liver as the major source of FGF21 during cold exposure (Figure 3B). Expression of FGF21 increased by 4-fold in BAT while liver FGF21 expression decreased by more than 6-fold in response to cold stimulation (Figure 3B). Other peripheral tissues such as muscle, eWAT, and iWAT were observed to have significantly lower FGF21 expression compared to liver and BAT (Figure 3B). Immunostaining further confirmed the trend of FGF21 expression we observed using qRT-PCR (Figures 3C–3E). FGF21 immunostaining showed that liver had much higher FGF21 expression at room temperature, but this was reversed when mice were placed at 6°C for 7 days (Figures 3C–3E). More importantly, mice injected with miR-32-ASO failed to upregulate FGF21 expression in BAT even though liver FGF21 expression has decreased, thus contributing to lower serum FGF21 levels (Figures 3A and 3C–3E). FGF21 levels in eWAT and muscle remained largely unchanged, although iWAT had increased FGF21 expression due to emergence of beige adipocytes (Figure S3E). However, FGF21 expression in iWAT was not observed to be significantly lower in mice with iWAT-specific or systemic miR-32 inhibition (Figures 3B and S3B–S3E). Western blots performed to examine FGF21 protein levels in BAT and liver showed that BAT FGF21 expression was significantly increased during cold exposure and that miR-32-ASO-injected mice had 5-fold lower FGF21 protein expression (Figures 3F and 3H). Liver FGF21 decreased significantly in response to cold stimulation for both control-ASO- and miR-32-ASO-injected mice (Figures 3G and 3H). Using the brown preadipocyte cell line WT-1, transfections with either miR-32 mimic or miR-32-ASO were able to efficiently modulate miR-32 levels (Figures S4A and S4B) and increase or decrease FGF21 expression at both the mRNA and protein levels, respectively (Figures 3I and 3J). These results strongly suggest that miR-32 functions as an important driver of FGF21 expression in BAT during cold exposure in a cell-autonomous manner, and this could increase serum FGF21 levels and promote beige cell emergence in iWAT.
Figure 3. miR-32-ASO-Injected Mice Have Significantly Lower Serum FGF21 Levels due to Failure of FGF21 Upregulation in BAT under Cold Stress.

(A) Cold-induced increase in serum FGF21 levels was blunted by injection of miR-32-ASO as measured by ELISA (n = 7).
(B) qRT-PCR showed that cold stress decreased FGF21 mRNA expression in liver but increased it in BAT. Injection of miR-32-ASO significantly repressed the increase in FGF21 expression in BAT during cold exposure when normalized to PPIA.
(C) FGF21 immunostaining showed that cold stress decreased FGF21 level in liver but increased it in BAT. Injection of miR-32-ASO significantly repressed the increase in FGF21 expression in BAT during cold exposure.
(D) Quantification of FGF21 immunostaining in BAT showed that FGF21 levels within BAT increased greatly after cold exposure but were significantly lower in miR-32-ASO-treated mice compared to control-ASO-treated mice.
(E) Quantification of FGF21 immunostaining in liver showed that FGF21 levels within liver decreased greatly after cold exposure but were similar between miR-32-ASO-treated mice and control-ASO-treated mice.
(F) Western blot showing cold-induced increases in BAT FGF21 protein levels were blunted by injection of miR-32-ASO. Calnexin served as a loading control.
(G) Western blot showing cold-induced decreases in liver FGF21 protein levels were unaffected by injection of miR-32-ASO. Calnexin served as a loading control.
(H) Quantification of the western blot results in (F) and (G) using ImageJ. Average intensities were normalized to that of Calnexin.
(I) FGF21 mRNA expression in WT-1 cells increased or decreased when transfected with miR-32 mimic or miR-32-ASO, respectively.
(J) Western blotting showed that FGF21 protein levels in WT-1 cells increased or decreased when transfected with miR-32 mimic or miR-32-ASO, respectively.
Data represent mean ± SEM. *p < 0.05 and **p < 0.01. See also Figure S3.
Tob1 Is a Target Gene of miR-32
miRNAs typically function via inhibition of one or multiple target genes (Liu and Olson, 2010; Pillai, 2005). Thus, it is likely that the rise in miR-32 levels during cold stimulation is for repressing the expression of a target gene that is itself repressing FGF21 expression. In silico prediction (Maragkakis et al., 2009) identified multiple hits for miR-32, and Tob1, a well-known tumor suppressor (Helms et al., 2009; Kundu et al., 2012; Lin et al., 2012), was selected as a potential target gene. This was due to Tob1’s ability to repress the p38/MAPK pathway, which is highly activated during cold exposure and can regulate the expression of multiple thermogenic genes including FGF21 (Jiao et al., 2012; Sun et al., 2013). Tob1 was the 11th top hit using in silico prediction software DIANA-microT-CDS (Maragkakis et al., 2009) with a high mirSVR score of −1.3394 (Figure 4A). The program identified a highly probable miR-32 binding site within the 3′ UTR of Tob1 gene, which was also highly conserved in mammals from mice to humans (Figures 4A and 4B). We found that Tob1 mRNA and protein levels decreased or increased in response to transfection of miR-32 mimic or inhibitor in WT-1 cells, respectively (Figures 4C and 4D). Furthermore, transfection of miR-32 mimic or inhibitor led to a corresponding decrease or increase in the activity of a luciferase reporter gene linked to the 3′ UTR of mouse Tob1 gene (Figures 4E and 4F). Moreover, we showed that the Tob1 3′ UTR sequence complimentary to the miR-32 seed sequence was crucial for targeting by miR-32 as mutation of the target sequence in Tob1 3′ UTR abolished the effects of miR-32 mimic and inhibitor on the luciferase activity (Figures 4G and 4H). In summary, these findings suggest that Tob1 is a direct target of miR-32.
Figure 4. miR-32 Directly Targets Tob1 to Promote UCP1, PGC1α, and FGF21 Expression in BAT during Cold Stress by Relieving Phospho-P38-Mediated Activation of ATF2 from Suppression by Tob1.

(A) High mirSVR score and favorable binding energy from bioinformatic analysis predicted that miR-32 directly targets the 3′ UTR of Tob1.
(B) The complementary sequence in the Tob1 3′ UTR and the seed region of miR-32 (red letters) are conserved among mammalian species.
(C) Tob1 mRNA level in WT-1 cells decreased or increased 24 hr after transfection of miR-32 mimic or miR-32-ASO, respectively.
(D) Tob1 protein level decreased or increased in WT-1 cells when transfected with miR-32 mimic or miR-32-ASO, respectively.
(E and F) Activities of a luciferase reporter gene linked to the Tob1 3′ UTR decreased or increased 24 hr after transfection of miR-32 mimic (E) or miR-32-ASO (F), respectively.
(G and H) Site-directed mutagenesis of the Tob1 3′ UTR sequence complimentary to the miR-32 seed sequence abolished the effects of miR-32 mimic (G) and miR-32-ASO (H) transfection on the luciferase activity.
(I) Knockdown of Tob1 by siTob1 suppressed the increase in Tob1 expression and rescued the decrease in FGF21 expression caused by miR-32-ASO transfection.
(J) Western blots showed that siTob1 suppressed the increase in Tob1 expression and rescued the decreases of phospho-p38, phospho-ATF2, and FGF21 expression caused by miR-32-ASO transfection. Numbers indicate protein levels quantified using ImageJ relative to control-ASO.
(K) qRT-PCR showed miR-32-ASO-treated mice failed to fully downregulate Tob1 level in response to cold stimulation.
(L) Western blots showed miR-32-ASO-treated mice fail to downregulate Tob1 levels and activate p38 and ATF2 phosphorylation in BAT in response to cold stimulation.
(M) Quantification of relative protein expression in BAT from the western blots showed in (L) using ImageJ. Average intensities were normalized to that of Calnexin.
(N) Cold-exposure time course showing miR-32 and Tob1 expression in BAT at various time points after cold exposure.
(O) Cold-exposure time course showing FGF21 expression in BAT at various time points after cold exposure.
(P) Cold-exposure time course showing Tob1, UCP1, and FGF21 protein expression in BAT at various time points after cold exposure.
(Q) Cold-exposure time course showing UCP1 protein expression in iWAT at various time points after cold exposure.
Data represent mean ± SEM. *p < 0.05 and **p < 0.01. See also Figures S4 and S5.
miR-32 Promotes FGF21 Expression by Repressing Tob1, Thereby Activating p38/MAPK and ATF2 In Vitro and In Vivo in BAT
Tob1’s ability to repress the p38/MAPK pathway (Sun et al., 2013) suggests that the rise of miR-32 during cold stimulation may serve to activate the thermogenic genes downstream of p38/MAPK. Previous studies have identified ATF2 as an essential component of the signaling cascade downstream of p38/MAPK (Cao et al., 2004; Hondares et al., 2011). These studies showed that ATF2 can bind to and promote the expression of thermogenic genes such as UCP1, PGC1α, and most importantly FGF21 (Cao et al., 2004; Hondares et al., 2011) when activated by phosphorylation. We have shown that in WT-1 cells, overexpression or inhibition of miR-32 led to increased or decreased FGF21 expression, respectively (Figures 3I and 3J). Subsequently, we examined the expression of PGC1α and UCP1 in miR-32-ASO-transfected WT-1 cells. We found miR-32-ASO-treated cells have significantly lower expression of both genes (Figures S4D and S4E). Luciferase assays further showed that miR-32 overexpression or inhibition led to a significant increase or decrease in the activity of a luciferase reporter gene downstream of the FGF21 promoter, respectively (Figures S4F and S4G). A rescue experiment where Tob1 was knocked down using small interfering RNA (siRNA) against Tob1 (siTob1) after miR-32 was inhibited further showed that miR-32′s effect on FGF21 expression was mainly mediated through Tob1 (Figures S4C and S4H). In line with previous studies (Hondares et al., 2011; Robidoux et al., 2005), we performed reporter-gene assays comprising either the UCP1 enhancer or PGC1α promoter inserted upstream of a luciferase gene. Our data also showed that miR-32 overexpression or inhibition increased or decreased the luciferase activity, respectively (Figures S4I–S4L). These results suggest that the effects of miR-32 on promoting PGC1α, UCP1, and FGF21 expression are likely mediated through direct binding of activated ATF2 to the corresponding enhancer and promoter of these genes.
To further confirm that miR-32 acts through Tob1 to drive endogenous FGF21 gene expression, a rescue experiment was conducted whereby Tob1 was knocked down using siTob1 after miR-32-ASO treatment. miR-32-ASO mediated an increase in Tob1 mRNA, and protein expression was successfully blunted by siTob1 (Figures 4I and 4J). Knockdown of Tob1 also rescued the decrease in FGF21 expression observed during miR-32 inhibition (Figures 3I, 3J, 4I, and 4J). Tob1 knockdown also reversed the inhibition on p38/MAPK signaling, and cells treated with siTob1 had increased phosphorylated-p38 and phosphorylated-ATF2 (Figure 4J). To validate our observations in WT-1 cells, we used primary brown adipocytes (BAT SVF cells). When miR-32 was modulated by its ASO or mimic treatment (Figure S5B), the expression of brown selective genes such as UCP1, PGC1α, and FGF21 were significantly affected (Figures S5F and S5H). Using western blotting, we found the inhibition of miR-32 led to increased expression of its target gene Tob1, the suppression of p38/MAPK signaling as manifested by the decreased phosphorylation of p38 and ATF2, and reduced levels of UCP1, PGC1α, and FGF21 (Figures S5F and S5H). On the other hand, the overexpression of miR-32 led to the opposite effects as compared to the effects of miR-32 inhibition in BAT SVF cells. Again, these effects are independent of the general adipogenesis process (Figure S5D) or SLC45A3 expression (Figures S5F and S5H) as no obvious changes were observed in BAT SVF cells. To examine the effects of miR-32 upon β-adrenergic stimulation, we evaluated the expression of brown selective genes UCP1, PGC1α, and FGF21 after norepinephrine (NE) treatment in BAT SVF cells. We found that the significant upregulation of these brown genes was blunted by miR-32 inhibition, while miR-32 overexpression only modestly upregulated the significantly increased expression of UCP1, PGC1α, and FGF21 after NE treatment (Figure S5I). Subsequently, western blot and qRT-PCR were performed on miR-32-ASO-treated mice to confirm the results observed in vitro. Tob1 expression in BAT was indeed lower when mice were cold stimulated and miR-32-ASO prevented this decrease in Tob1 levels, further confirming Tob1 as a direct target of miR-32 in vivo (Figures 4K–4M). Furthermore, phosphorylated-p38 and phosphorylated-ATF2 levels were also decreased in miR-32-ASO-treated mice compared to control-ASO mice that had increased p38/MAPK signaling during cold exposure, suggesting that failure to downregulate Tob1 led to decreased p38/MAPK signaling (Figures 4L and 4M). This inhibition of p38/MAPK signaling by miR-32 inhibition led to reduced FGF21 expression in and secretion from BAT (Figures 3B–3D and 3F), leading to reduced serum FGF21 levels (Figure 3A) and reduced iWAT browning (Figures 2J and 2K). A cold-exposure time course was also performed to further understand the relationship between miR-32, Tob1, and FGF21 expression in BAT as well as UCP1 expression in BAT and iWAT (Figures 4N–4Q). As expected, miR-32 levels are inversely correlated with Tob1 mRNA and protein levels (Figures 4N and 4P), further suggesting that Tob1 is directly repressed by miR-32. Similarly, FGF21 and UCP1 expression in BAT increases as soon as 6 hr after cold exposure (Figures 4O and 4P), which precedes the increase of UCP1 in iWAT (Figures 4P and 4Q). These results support our hypothesis that miR-32 drives expression of FGF21 from BAT as soon as 6 hr after cold exposure, leading to beige cell emergence in iWAT a few days later as shown by UCP1 expression (Figures 4N–4Q).
BAT-Specific Inhibition of miR-32 Reduces BAT Thermogenic Activity, BAT Mass, and FGF21 Expression in BAT, Resulting in Impaired Beige Cell Recruitment in iWAT
BAT-specific inhibition of miR-32 was performed (Liu et al., 2014) in order to determine whether miR-32 exerts its effects on BAT thermogenesis and beige cell emergence mainly through its action in BAT during cold exposure. Injection of miR-32-ASO directly into BAT led to robust, tissue-specific inhibition of miR-32 in BAT without affecting liver miR-32 expression (Figure 5A). Mice with BAT-specific miR-32 inhibition (miR-32-ASO-BS) also showed significantly impaired thermogenic response, and their core body temperatures were significantly lower compared to control-ASO-injected mice (control-ASO-BS) as soon as 4 hr after being placed at 6°C and throughout the 7-day cold exposure (Figure 5B). Energy expenditure of miR-32-ASO-BS mice was significantly lower than control-ASO-BS mice during both light and dark periods, further suggesting impairment in cold-induced thermogenesis (Figures 5C and 5D). BAT tissue mass was also significantly lower in miR-32-ASO-BS mice (Figure 5E), suggesting that miR-32 inhibition led to a reduction in cold-induced BAT mass expansion. However, levels of food intake and ambulatory movement were similar in the two groups of mice (Figures S6A–S6D). As predicted, miR-32-ASO-BS mice had increased Tob1 expression, repressed p38/MAPK signaling, and reduced expression of brown markers such as UCP1, PGC1α, and PPARα as well as FGF21 in BAT (Figures 5F–5H), although protein concentration per milligram of BAT tissue remained unchanged (Figure S6E). miR-32-ASO-BS mice also had reduced FGF21 serum levels (Figure 5I) derived from reduced FGF21 production in BAT (Figures 5F–5H), leading to reduced browning of iWAT as shown by reduced expression of brown markers such as UCP1, PGC1α, PPARα, and Cidea (Figures 5J–5L). These results demonstrate that miR-32 expression within BAT is required for cold-induced BAT thermogenesis, beige cell emergence, and the expression of UCP1, PGC1α, and FGF21.
Figure 5. BAT-Specific Inhibition of miR-32 during Cold Exposure Leads to Significantly Lower Core Body Temperature and Reduced Energy Expenditure in Mice as the Result of Reduced Thermogenic Response in BAT and Beige Cell Emergence in iWAT.

(A) miR-32 expression in BAT but not in liver was blunted in BAT-specific miR-32-ASO-injected mice (n = 8) compared to control-ASO-injected mice (n = 7).
(B) miR-32-ASO-BS mice (n = 8) showed lower core body temperatures during cold exposure when compared to control-ASO-BS mice (n = 7).
(C) Total energy expenditure was significantly reduced after 7 days’ cold stress in miR-32-ASO-BS mice (n = 6) as compared with control-ASO-BS mice (n = 6). Energy expenditure was normalized to lean body mass.
(D) Average total energy expenditure was significantly lower in miR-32-ASO-BS mice (n = 6).
(E) Percentage BAT mass was significantly lower in miR-32-ASO-BS mice (n = 8) compared to control mice (n = 7).
(F) In BAT, mRNA levels of Tob1 were higher in miR-32-ASO-BS mice (n = 8), whereas expression of thermogenic genes was significantly lower in miR-32-ASO-BS mice.
(G) Protein levels of Tob1 were higher in miR-32-ASO-BS mice (n = 8) compared to control mice (n = 7). However, p-p38, p-ATF2, FGF21, UCP1, and PGC1α were lower in miR-32-ASO-BS mice (n = 8). Total p38 and total ATF2 expression were not significantly different between the two groups.
(H) Quantification of relative protein expression using ImageJ showed that protein level of Tob1 was upregulated, but p-p38, p-ATF2, FGF21, UCP1, and PGC1α were significantly lower in BAT of miR-32-ASO-BS mice (n = 8) compared to control mice (n = 7). Total p38 and total ATF2 expression were comparable between the two groups. Average intensities were normalized to that of Calnexin.
(I) Serum FGF21 levels were decreased in miR-32-ASO-BS mice (n = 8) compared to control mice (n = 7).
(J) mRNA levels of thermogenic genes in iWAT were significantly reduced in miR-32-ASO-BS mice. Data were normalized to PPIA.
(K) Immunoblots showed that miR-32-ASO-BS mice (n = 8) had significantly reduced UCP1 and PGC1α protein levels in iWAT compared to control-ASO-BS mice (n = 7).
(L) Quantification of relative UCP1 and PGC1α protein levels using ImageJ. Average intensities were normalized to that of Calnexin.
Data represent mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S6.
BAT-Specific Overexpression of miR-32 Increases BAT Thermogenic Activity, BAT Mass, and FGF21 Expression in BAT, Leading to Enhanced Beige Cell Emergence in iWAT
To determine whether increased miR-32 within BAT would lead to increased thermogenic activity and beige cell emergence, BAT-specific overexpression of miR-32 was performed. Injecting miR-32-expressing adeno-associated virus (miR-32-AAV) with adipose-specific capsid protein directly into BAT (Liu et al., 2014) led to robust, tissue-specific increase in miR-32 expression in BAT of up to 40-fold without affecting liver or iWAT miR-32 expression (Figure 6A). Mice that have undergone BAT-specific injection of miR-32-AAV (miR-32-AAV-BS) showed increased thermogenic response, and their core body temperatures were significantly higher compared to control-AAV-injected mice (control-AAV-BS) as soon as 4 hr after being placed at 6°C and throughout the 7-day cold exposure (Figure 6B). Energy expenditure of miR-32-AAV-BS mice were also significantly higher than control-AAV-BS mice during both light and dark periods, further suggesting a robust activation in cold-induced thermogenesis above normal levels (Figures 6C and 6D). BAT tissue mass was also significantly higher in miR-32-AAV-BS mice (Figure 6E), therefore suggesting that miR-32 promotes cold-induced BAT mass expansion. However, levels of food intake and ambulatory movement were similar in the two groups of mice (Figures S6F–S6I). As predicted, miR-32-AAV-BS mice had reduced Tob1 expression, activated p38/MAPK signaling, and increased expression of brown markers such as UCP1, PGC1α, and PPARα as well as FGF21 in BAT (Figures 6F–6H), although protein concentration per milligram of BAT tissue remained unchanged (Figure S6J). miR-32-AAV-BS mice also had increased FGF21 serum levels (Figure 6I), leading to enhanced browning of iWAT as shown by significantly upregulated expression of brown markers such as UCP1, PGC1α, PPARα, and Cidea (Figures 6J–6L). These results demonstrate that increasing miR-32 within BAT can drive cold-induced thermogenesis by increasing the expression of UCP1, PGC1α, PPARα, and other thermogenic genes as well as promote beige cell emergence by increasing serum FGF21 levels.
Figure 6. BAT-Specific Overexpression of miR-32 during Cold Exposure Leads to Significantly Higher Core Body Temperature and Increased Energy Expenditure in Mice as the Result of Increased Thermogenic Response in BAT and Beige Cell Emergence in iWAT.

(A) miR-32 expression in BAT but not in liver or iWAT was increased in miR-32-AAV-BS mice (n = 8) compared to control-AAV-BS mice (n = 7).
(B) miR-32-AAV-BS mice (n = 8) showed higher core body temperatures during cold exposure when compared to control-AAV-BS mice (n = 7).
(C) Total energy expenditure was significantly increased after 7 days’ cold stress in miR-32-AAV-BS mice (n = 6) as compared with control-AAV-BS mice (n = 6). Energy expenditure was normalized to lean body mass.
(D) Average total energy expenditure was significantly higher in miR-32-AAV-BS mice (n = 6).
(E) Percentage BAT mass was significantly higher in miR-32-AAV-BS mice (n = 8) compared to control-AAV-BS mice (n = 7).
(F) In BAT, mRNA levels of Tob1 were lower in miR-32-AAV-BS mice, whereas expression of thermogenic genes was significantly higher in miR-32-AAV-BS mice.
(G) Protein levels of Tob1 were lower in miR-32-AAV-BS mice (n = 8) compared to control-AAV-BS mice (n = 7). However, p-p38, p-ATF2, FGF21, UCP1, and PGC1α were higher in miR-32-AAV-BS mice (n = 8). Total p38 and total ATF2 expression were not significantly different between the two groups.
(H) Quantification of relative protein expression using ImageJ showed that protein level of Tob1 was reduced, but p-p38, p-ATF2, FGF21, UCP1, and PGC1α were significantly higher in BAT of miR-32-AAV-BS mice compared to control. Total p38 and total ATF2 expression were comparable between the two groups. Average intensities were normalized to that of Calnexin.
(I) Serum FGF21 levels were increased in miR-32-AAV-BS mice (n = 8) compared to control-AAV-BS mice (n = 7).
(J) mRNA levels of thermogenic genes in iWAT were significantly increased in miR-32-AAV-BS mice. Data were normalized to PPIA.
(K) Immunoblots showed that miR-32-AAV-BS mice (n = 8) had significantly increased UCP1 and PGC1α protein levels in iWAT compared to control-AAV-BS mice (n = 7).
(L) Quantification of relative UCP1 and PGC1α protein levels using ImageJ. Average intensities were normalized to that of Calnexin.
Data represent mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S6.
BAT-Specific Ablation of FGF21 Significantly Reduces the Increased BAT Thermogenic Activity, BAT Mass, and Beige Cell Emergence in iWAT Observed from BAT-Specific miR-32 Overexpression
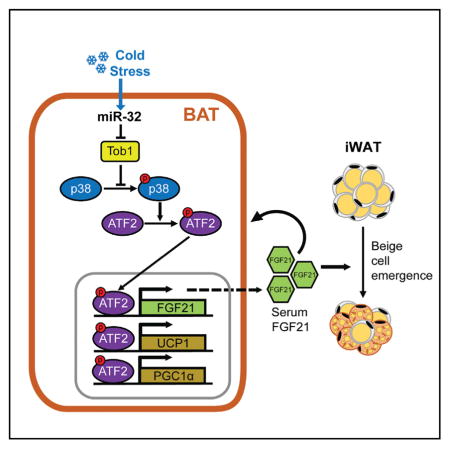
To determine whether FGF21 is responsible for the increased BAT thermogenic activity and beige cell emergence seen during BAT-specific miR-32 overexpression, BAT-specific ablation of FGF21 was performed together with BAT-specific miR-32 over-expression. This was achieved by co-injecting AAV-expressing Cre recombinase together with AAV-overexpressing miR-32 directly into BAT of FGF21 floxed mice. Co-injecting Cre-expressing adeno-associated virus led to robust, tissue-specific ablation of FGF21 in BAT of FGF21 floxed mice withouta affecting liver or iWAT FGF21 expression (Figures 7A–7C). BAT-specific ablation of FGF21 also led to reduced serum FGF21 levels (Figure 7D). miR-32-overexpressing mice that had undergone BAT-specific ablation of FGF21 (miR-32-AAV-BS+Cre) showed reduced thermogenic response, and their core body temperatures were only significantly higher during the first 48 hr of cold exposure compared to control mice that had also undergone BAT-specific FGF21 ablation (control-AAV-BS+Cre) (Figure 7E). Energy expenditure of control-AAV-BS+Cre and miR-32-AAV-BS+Cre mice were also similar during both light and dark periods, further suggesting BAT-specific FGF21 ablation reduced the thermogenic effects of BAT-specific miR-32 overexpression (Figures 7F and 7G). BAT tissue mass was still slightly higher in miR-32-AAV-BS+Cre mice although not significantly (Figure 7H), therefore suggesting that miR-32-mediated BAT mass expansion during cold exposure is partially dependent on FGF21, although multiple other factors may also play a role. Similar to previous experiments, levels of food intake and ambulatory movement between the two groups of mice were not different (Figures S7B–S7E). As predicted, miR-32-AAV-BS+Cre mice had reduced Tob1 expression due to increased miR-32 expression (Figures 7I and S7A). However, BAT-specific ablation of FGF21 led to similar expression of brown markers between the two groups of mice with the exception of UCP1 and PGC1α, which were still significantly higher, suggesting that FGF21 secreted from BAT is partially responsible for the increased expression of thermogenic genes such as UCP1 and PGC1α (Figures 7I–7K). Protein concentration per milligram of BAT tissue remained similar between the two groups of mice (Figure S7F). Increased browning of iWAT due to BAT-specific miR-32 overexpression (Figures 6J–6L) was severely blunted by BAT-specific ablation of FGF21 as demonstrated by diminished upregulation of brown markers such as UCP1, PGC1α, PPARα, and Cidea (Figures 7L–7N). These results demonstrate that BAT-mediated FGF21 expression and secretion are partially responsible for the effects of BAT-specific miR-32 overexpression such as the increased expression of thermogenic genes including UCP1, PGC1α, and FGF21 within BAT and increased beige cell emergence in iWAT. Taken together, our study shows that, during cold stimulation, increased miR-32 expression within BAT represses Tob1 expression to activate p38/MAPK signaling and drive the expression of thermogenic genes and secretion of FGF21; FGF21 secreted from BAT can function in a paracrine fashion to promote further thermogenic gene expression and increased BAT mass as well as function in an endocrine fashion to promote browning of iWAT (Figure 7O).
Figure 7. BAT-Specific Ablation of FGF21 Significantly Reduces the Increased BAT Thermogenic Activity, BAT Mass, and Beige Cell Emergence in iWAT Observed from BAT-Specific miR-32 Overexpression.

(A) FGF21 mRNA expression in BAT but not in liver or iWAT was ablated in miR-32-AAV-BS+Cre mice (n = 6) and control-AAV-BS+Cre mice (n = 6).
(B) FGF21 protein expression in BAT but not in liver or iWAT was ablated in miR-32-AAV-BS+Cre mice (n = 6) and control-AAV-BS+Cre mice (n = 6).
(C) Quantification of relative protein expression using ImageJ showed that protein level of FGF21 in BAT but not in liver or iWAT was ablated in miR-32-AAV-BS+Cre mice (n = 6) and control-AAV-BS+Cre mice (n = 6).
(D) Serum FGF21 levels were decreased in miR-32-AAV-BS+Cre mice (n = 6) and control-AAV-BS+Cre mice (n = 6) compared to wild-type mice (n = 4).
(E) miR-32-AAV-BS+Cre mice (n = 6) showed higher core body temperatures only during first 48 hr of cold exposure when compared to control-AAV-BS+Cre mice (n = 6).
(F) Total energy expenditure was similar after 7 days’ cold stress in miR-32-AAV-BS+Cre mice (n = 6) as compared with control-AAV-BS+Cre mice (n = 6). Energy expenditure was normalized to lean body mass.
(G) Average total energy expenditure was slightly higher in miR-32-AAV-BS+Cre mice (n = 6) than control-AAV-BS+Cre mice (n = 6) but not statistically significant.
(H) Percentage BAT mass was slightly higher in miR-32-AAV-BS+Cre mice (n = 6) compared to control-AAV-BS+Cre mice (n = 6).
(I) In BAT, mRNA levels of Tob1 were lower in miR-32-AAV-BS+Cre mice (n = 6) compared to control-AAV-BS+Cre mice (n = 6), whereas expression of several thermogenic genes including UCP1 was higher in miR-32-AAV-BS+Cre mice.
(J) Protein levels of PGC1α and UCP1 were higher in miR-32-AAV-BS+Cre mice (n = 6) compared to control-AAV-BS+Cre mice (n = 6).
(K) Quantification of relative protein expression using ImageJ showed that protein levels of PGC1 and UCP1 were higher in miR-32-AAV-BS+Cre mice (n = 6) compared to control-AAV-BS+Cre mice (n = 6). Average intensities were normalized to that of Calnexin.
(L) mRNA levels of thermogenic genes in iWAT were similar in both groups of mice (both n = 6). Data were normalized to PPIA.
(M) Immunoblots showed that miR-32-AAV-BS+Cre mice (n = 6) had similar UCP1 and PGC1α protein levels in iWAT compared to control-AAV-BS+Cre mice (n = 6).
(N) Quantification of relative UCP1 and PGC1α protein levels using ImageJ. Average intensities were normalized to that of Calnexin.
(O) Proposed mechanism by which miR-32 promotes BAT thermogenesis by inhibiting Tob1, activating p38/MAPK signaling and driving UCP1, PGC1α, and FGF21 expression in BAT. The BAT secreted FGF21 functions in a paracrine fashion to promote further thermogenic gene expression in BAT as well as in an endocrine fashion to promote iWAT browning.
Data represent mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S7.
DISCUSSION
In recent years, microRNAs have emerged as a safe and efficacious therapeutic reagent that are currently being tested in clinical trials against a myriad of diseases (van Rooij et al., 2012). microRNAs that selectively promote energy expenditure through BAT thermogenesis or browning of iWAT are of significant interest as potential therapeutics for anti-obesity treatments due to BAT and beige fat’s ability to metabolize vast amounts of glucose and lipids in proportion to their tissue mass (Bartelt et al., 2011; Harms and Seale, 2013). We identified and characterized miR-32 as a BAT-selective miRNA that plays a critical role in regulating cold tolerance and energy expenditure.
FGF21, an important endocrine and paracrine signaling molecule, is normally produced by the liver and acts as a metabolic regulator that controls ketogenesis, insulin sensitivity, and glucose homeostasis (Hondares et al., 2011; Itoh, 2014; Murata et al., 2011; Owen et al., 2014). However, we and others show that during cold-induced thermogenesis, the β-adrenergic pathway is stimulated causing BAT to significantly upregulate and secrete FGF21 into systemic circulation (Chartoumpekis et al., 2011; Fisher et al., 2012; Hondares et al., 2011; Lee et al., 2014a). Meanwhile, we and others also show that liver FGF21 expression is significantly reduced, making BAT the main source of FGF21 production (Fisher et al., 2012; Hondares et al., 2011) under cold stress. In BAT, stimulation of the β-adrenergic receptors activates the p38/MAPK pathway, leading to ATF2 phosphorylation and binding to the FGF21 promoter driving its expression (Cao et al., 2004; Hondares et al., 2011). FGF21 is purported to induce mitochondrial uncoupling respiration and glucose oxidation (Chartoumpekis et al., 2011; Owen et al., 2014), thereby producing more heat. More importantly, our results show that BAT-derived FGF21 can promote browning of iWAT and enhance BAT thermogenesis during cold exposure similar to other studies (Chartoumpekis et al., 2011; Fisher et al., 2012; Itoh, 2014; Lee et al., 2014a). Our study in which BAT-specific ablation of FGF21 was performed uniquely shows that BAT is both a main source and target of FGF21 during cold-induced thermogenesis complementing previous studies (Fisher et al., 2012; Hondares et al., 2011). Our results further suggest that upregulation of serum FGF21 levels caused by increased FGF21 secretion from BAT as a result of cold stimulation could lead to increased thermogenic gene expression, total energy expenditure, and beige cell emergence within iWAT, thus highlighting BAT-derived FGF21’s therapeutic potential against obesity and other metabolic diseases.
Our findings show that miR-32 differs from previously identified miRNAs (Mori et al., 2012; Sun et al., 2011; Trajkovski and Lodish, 2013; Trajkovski et al., 2012; Yin et al., 2013). miR-193-365, miR-133, miR-155, and miR-196a all act in cis by promoting drivers or inhibiting repressors of BAT differentiation. miR-32 not only acts in cis to promote BAT thermogenesis and BAT mass, but can also act in trans to promote FGF21 secretion from BAT, leading to further endocrine effects such as browning of iWAT, although it is not capable of directly driving beige cell emergence cell autonomously. Furthermore, miR-32 is able to directly activate the BAT thermogenic program by repressing Tob1 and activating p38/MAPK signaling, leading to upregulation of thermogenic markers such as PGC1α and UCP1, which may partially explain why its inhibition led to impaired core-body-temperature regulation and reduced energy expenditure. miR-32’s proximal location to a BAT-specific super-enhancer, high expression, and further upregulation during cold exposure within BAT to repress its target gene Tob1 imply that both miR-32 and Tob1 might be part of a highly conserved regulatory network crucial to thermogenesis and lipid metabolism, which warrants further investigation. Future studies on upstream regulation of miR-32 expression by the proximal super-enhancer, other regulatory elements, and transcription factors may allow us to further our understanding about thermogenic gene regulation. Our data on BAT-specific miR-32 overexpression leading to increased thermogenesis and energy expenditure strongly suggest that using miR-32 mimetics or Tob1 inhibitors as therapeutic reagents against obesity and other metabolic diseases should be further explored (Itoh, 2014; Murata et al., 2011; Owen et al., 2014). In summary, our study demonstrates the importance of the miR-32-Tob1-FGF21 pathway in regulating BAT/beige adipocyte function and development, which can be exploited for development of therapeutic agents against obesity and metabolic syndrome.
EXPERIMENTAL PROCEDURES
Cell Culture
The brown preadipocyte cell line WT-1 (Klein et al., 1999) was used for in vitro experiments. BAT and iWAT SVF cells were prepared as described previously (Brunmeir et al., 2016). Inhibition of miR-32 was performed using either 40 or 80 nM of mIRCURY locked nucleic acid (LNA) microRNA Inhibitors (Exiqon). For overexpression studies, either 40 or 80 nM of miRIDIAN microRNA mimics of control or miR-32 (Dharmacon) was used. Knockdown of endogenous Tob1 was performed using 40 nM of ON-TARGETplus siRNA against Tob1 from Dharmacon. All experiments were performed thrice.
Transfection upon reaching 70%–80% cell confluency was performed uniformly across all experiments presented 24 hr after seeding cells. Cells were transfected for 24 hr using Lipofectamine 2000, with a 3-hr treatment of NE prior to cell harvest. Addition of NE was to simulate cold conditions.
Control-ASO and miR-32-ASO Generation and Injection
Both anti-sense oligonucleotides were purchased from Exiqon. The sequences of control-ASO and miR-32-ASO are 5′-ACGTCTATACGCCCA-3′ and 5′-TTAGTAATGTGCAAT-3′, respectively. Both oligonucleotides were purified by reverse-phase high-performance liquid chromatography and lyophilized. For i.p. injections, lyophilized oligonucleotides were resuspended in saline to a final concentration of 30 μg/μL. A dose of 25 mg/kg body weight oligonucleotide in a total volume of 100 μL saline was introduced to mice via i.p. injection.
Animal Model
Male C57BL6/J mice aged 8–10 weeks were used for in vivo experiments. Mice were divided into three categories: uninjected, wild-type mice housed at room temperature (wild-type), and mice housed at 6°C–7°C over 7 days that were injected intraperitoneally with single-dose (25 mg/kg) control-ASO or miR-32-ASO 24 hr prior to cold exposure. BAT- and iWAT-specific injection of miR-32-ASO (125 μg/kg), miR-32-AAV (1 × 108 viral particles for BAT and 1 × 109 viral particles for iWAT), and CMV-iCre (5 × 108 viral particles) were performed as shown elsewhere (Liu et al., 2014). Male FGF21 floxed mice aged 8–10 weeks were obtained from Jackson Laboratory. For all animal experiments, n ≥ 3 mice for each group, and similar results were observed from at least three independent experiments. Upon the start of the cold experiment, mice were singly housed and kept on normal chow diet. All procedures involving mice were performed according to an approved protocol (IACUC#130838) from the Institutional Animal Care and Use Committee of A*STAR.
Indirect Calorimetry and Core-Body-Temperature Measurement
Mice were individually housed and maintained at 24°C under a 12:12-hr light-dark cycle in a 12-cage equal flow Oxymax Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments). Total energy expenditure, physical activity, and food intake were simultaneously measured for each mouse as described elsewhere (Li et al., 2014; Lou et al., 2011). The core body temperature was measured at stipulated timings with a probe thermometer (Advance Technology) at a constant depth as described elsewhere (Kim et al., 2014).
rAAV and CMV-iCre Vector Construction and Packaging
The recombinant adeno-associated virus (rAAV) plasmid contains a vector expression cassette consisting of the CMV enhancer and chicken β-actin (CBA) promoter, woodchuck post-transcriptional regulatory element (WPRE), and bovine growth hormone (bGH) poly(A) flanked by AAV2 inverted terminal repeats. Transgenes miR-32 (Mus musculus) and non-specific control sequence were inserted into the multiple cloning sites between the CBA promoter and WPRE sequence. The engineered hybrid serotype Rec2 vectors were packaged and purified as described elsewhere (Liu et al., 2014). CMV-iCre was purchased from Vector Labs and used according to the manufacturer’s published protocols.
Dual-Luciferase Assay
FGF21 promoter (−1,497 to +5 bp) (Badman et al., 2007; Hondares et al., 2011) and PGC1α promoter (−170 to +68 bp) (Cao et al., 2004) were amplified from mouse genomic DNA and inserted into the vector pGL3 (Promega). UCP1 enhancer (−2,530 to −2,310 bp) (Cao et al., 2004) was also amplified and inserted into psiCHECK-2 plasmid (Promega). The 3′ UTR region of Tob1 was amplified from BAT cDNA and inserted into pMIRGLO (Promega). The target sequence of miR-32 within Tob1 3′ UTR was mutated from TGTGCAAT to TGTTACCT by site-directed mutagenesis using Infusion-HD Cloning System from Clontech as per the manufacturer’s protocol. WT-1 cells were transfected with 30 ng of plasmid per well (24-well plate) for 24 hr. Dual-Luciferase assay (Promega) was then performed according to the manufacturer’s protocol.
RNA Extraction and Real-Time qPCR
For WT-1 cell line, cells were washed twice with PBS and then lysed with 750 μL of Trizol per sample. For tissue samples, chunks of approximately 3 × 3 × 3-mm tissue material were homogenized in 750 μL of Trizol per sample. RNA extraction was then carried out using miRNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. Reverse transcription was performed using iScript Reverse Transcription Supermix (Bio-Rad) or Exiqon Universal miRNA RT kit according to the manufacturer’s protocol. Real-time PCR was performed and analyzed using published protocols (Ng et al., 2012). Primer sequences used in this study are shown in Table S2.
ELISA
Thermo Scientific 96-well, clear, flat-bottom plates (Nunc) were coated overnight at 4°C with a mouse monoclonal anti-FGF21 antibody (MAB25371, R&D Systems) at 1:1,500 dilution in Na2CO3/NaHCO3 buffer at pH 9.6. After washing plates twice with PBS, 1% BSA in PBS was used to block the plate for 1 hr at 4°C. Upon decanting the blocking buffer, 10 μL of rabbit anti-FGF21 antibody (ab64857, Abcam), 10 μL of blood serum diluted 10-fold in PBS, and 30 μL of 1% BSA in PBS were added to each well. This was incubated at room temperature for 2.5 hr. Each sample was triplicated. After incubation, wells were washed twice with PBS plus 0.1% Tween 20 and 1 M NaCl, twice with PBS plus 0.1% Tween 20 (phosphate buffered saline tween-20 [PBST]), and twice again with PBS. Secondary antibody anti-rabbit horseradish peroxidase (HRP) was added at 1:2,000 dilution and incubated for 30 min at room temperature. Wells were next washed thrice with PBST and thrice with deionized water. O-phenylenediamine dihydrochloride (OPD) tablets from Life Technologies were dissolved in equal parts of Life Technologies’ Stable Peroxide Substrate Buffer and Milli-Q water. 100 μL of the OPD mixture was added into each well to produce a colorimetric reaction. This reaction was quenched after 3- to 5-min incubation at room temperature using 5% sulfuric acid. Absorbance levels of each well was read at 492 nm using Bio-Rad xMark Microplate Spectrophotometer.
H&E and Immunostaining
Paraffin-embedded tissue samples were sectioned and stained using H&E or immunohistochemistry (IHC) kit (Abcam ab80437) as per the manufacturer’s instructions. For immunostaining, the rabbit anti-UCP1 (ab10983, Abcam) and rabbit anti-FGF21 (ab64857, Abcam) were used at 1:100 dilutions. Antigen retrieval was performed by placing slides in 95°C citrate buffer (pH 6) for 10 min. IHC was performed using Expose Kit (Abcam) according to the manufacturer’s protocol. Quantification of IHC-stained images (three to five images per group were used) were performed as previously shown using IHC profiler Plugin for ImageJ software (Varghese et al., 2014).
Western Blots
Western blots were performed for both cell-culture and tissue samples. For WT-1 cells, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer for 30 min while shaking at approximately 300 rpm. The snap-frozen tissue samples were homogenized in TLC-γ buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EGTA, and Complete Protease Inhibitor Cocktail [Roche Applied Science]). After homogenization, tissue extracts were centrifuged at 14,000 × g at 4°C, and the supernatants were collected and sonicated. Protein concentration was quantified using Bradford Reagent (Bio-Rad) according to the manufacturer’s protocol. 20 μg protein per well was loaded on 4%–12% SDS-PAGE gradient gels (Life Tech). After electrophoresis, gels were electro-transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (Sigma-Aldrich; TBST) and incubated with primary antibodies in 5% skim milk in TBST overnight at 4°C. In addition to the antibodies listed above, rabbit anti-PGC1α (ab106814, Abcam), rabbit anti-Tob1 (ab48826, Abcam), rabbit anti-phospho-p38 (#4511, Cell Signaling Technology), rabbit anti-p38 (#8690, Cell Signaling Technology), rabbit anti-phospho-ATF2 (#9221, Cell Signaling Technology), rabbit anti-ATF2 (#9226, Cell Signaling Technology), rabbit anti-PPARγ (#2443, Cell Signaling Technology), mouse anti-SLC45A3 (sc-393069, Santa Cruz Biotechnology), and rabbit anti-Calnexin (H-70, Santa Cruz Biotechnology) antibodies were used (all at 1:1,000 dilution). After three washes with TBST, membranes were incubated with the secondary antibody goat anti-rabbit-HRP (Pierce) at 1:5,000 dilution in 5% skim milk in TBST and developed with ECL or ECLplus Western Chemiluminescent HRP substrate (Pierce). Signal intensities were quantified and normalized to Calnexin using ImageJ.
Oil Red O Staining
Oil red O staining was performed as described previously (Brunmeir et al., 2016).
Super-Enhancer Identification and Visualization
Super-enhancers were identified using ROSE: rank ordering of super-enhancer from the Young Lab (Hnisz et al., 2013; Whyte et al., 2013). The H3K27ac BAM files of embryonic stem cells, mouse embryonic fibroblast (MEF), bone-marrow-derived macrophage, cerebellum, cortex, bone marrow, spleen, testis, placenta, kidney, liver, and BAT were obtained from ENCODE ftp server ftp://hgdownload.cse.ucsc.edu/goldenPath/mm9/encodeDCC/wgEncodeLicrHistone/, and GFF files were converted from the corresponding broadpeak files deposited in the server. For C2C12 myotube and epididymal WAT, we downloaded the raw sequencing reads from GEO accession GSM822520 and GSM1561376, respectively. After that, we mapped the reads to mm9 genome using bowtie with parameter (-m 5–best -n 2 -l 28 -B 1 -S -p 7) to generate the BAM files and then ran model-based analysis of ChIP-seq (MACS) using default settings without input (as they are not provided) to generate the enriched peak files. The enriched bed files were then converted to GFF format for use with ROSE. After obtaining the relevant BAM files and GFF files, we proceeded to run ROSE with its default settings to obtain the list of super-enhancers. A cutoff of 50 kb was set to identify microRNAs associated with any BAT super-enhancers. For visualization, BigWig files were generated from the acquired BAM files using makeTagDirectory and makeBigWig.pl from a suite of next-generation sequencing tools provided in HOMER (Heinz et al., 2010). The Wash U Epigenetics browser (Zhou and Wang, 2012) was used to visualize the histone marks and super-enhancers.
Statistics
Statistical significance was determined with two-tailed Student’s t test. A p value less than 0.05 was considered significant.
Supplementary Material
Highlights.
miR-32 is associated with BAT-specific super-enhancer and is highly expressed in BAT
miR-32 is induced by cold, and its inhibition leads to impaired thermogenesis
miR-32 directly inhibits Tob1 to activate p38/MAPK signaling to induce FGF21 in BAT
miR-32 promotes BAT function and trans-activates white fat browning through FGF21
Acknowledgments
We thank Drs. Kai Ge and Yu-Hua Tseng for kindly providing the brown preadipocyte cell line WT-1. This work was supported by intramural funding from the Agency for Science, Technology and Research (A*STAR) of Singapore to F.X. and in part by the Biomedical Research Council Young Investigator Grant of A*STAR to R.N. and National Institutes of Health Grants CA163640, CA166590, and AG041250 to L.C.
Footnotes
AUTHOR CONTRIBUTIONS
R.N. and F.X. conceived and designed the experiments. R.N., N.A.H., Q.Z., C.C., H.L., and Y.F. performed experiments. R.N., N.A.H., and C.C. analyzed the data. L.C., W.H., and W.S. provided equipment and reagents. R.N. and F.X. wrote the manuscript.
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.04.035.
References
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPAR-alpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, Wu J, Peng X, Kim SY, Julien SG, Zhang Q, Xie W, Xu F. Comparative transcriptomic and epigenomic analyses reveal new regulators of murine brown adipogenesis. PLoS Genet. 2016 doi: 10.1371/journal.pgen.1006474. Published online December 6, 2016. http://dx.doi.org/10.1371/journal.pgen. [DOI] [PMC free article] [PubMed]
- Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med. 2011;17:736–740. doi: 10.2119/molmed.2011.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Siegel F, Kipschull S, Haas B, Fröhlich H, Meister G, Pfeifer A. miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat Commun. 2013;4:1769. doi: 10.1038/ncomms2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, Spiegelman BM. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, Seale P. Brown and beige fat: Development, function and therapeutic potential. Nat Med. 2013;19:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms MW, Kemming D, Contag CH, Pospisil H, Bartkowiak K, Wang A, Chang SY, Buerger H, Brandt BH. TOB1 is regulated by EGF-dependent HER2 and EGFR signaling, is highly phosphorylated, and indicates poor prognosis in node-negative breast cancer. Cancer Res. 2009;69:5049–5056. doi: 10.1158/0008-5472.CAN-08-4154. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, Villarroya F. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem. 2011;286:12983–12990. doi: 10.1074/jbc.M110.215889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N. FGF21 as a hepatokine, adipokine, and myokine in metabolism and diseases. Front Endocrinol (Lausanne) 2014;5:107. doi: 10.3389/fendo.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Sun KK, Zhao L, Xu JY, Wang LL, Fan SJ. Suppression of human lung cancer cell proliferation and metastasis in vitro by the transducer of ErbB-2.1 (TOB1) Acta Pharmacol Sin. 2012;33:250–260. doi: 10.1038/aps.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Cho H, Alexander R, Patterson HC, Gu M, Lo KA, Xu D, Goh VJ, Nguyen LN, Chai X, et al. MicroRNAs are required for the feature maintenance and differentiation of brown adipocytes. Diabetes. 2014;63:4045–4056. doi: 10.2337/db14-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Fasshauer M, Ito M, Lowell BB, Benito M, Kahn CR. beta(3)-adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J Biol Chem. 1999;274:34795–34802. doi: 10.1074/jbc.274.49.34795. [DOI] [PubMed] [Google Scholar]
- Kundu J, Wahab SMR, Kundu JK, Choi YL, Erkin OC, Lee HS, Park SG, Shin YK. Tob1 induces apoptosis and inhibits proliferation, migration and invasion of gastric cancer cells by activating Smad4 and inhibiting β-catenin signaling. Int J Oncol. 2012;41:839–848. doi: 10.3892/ijo.2012.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014a;19:302–309. doi: 10.1016/j.cmet.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Werner CD, Kebebew E, Celi FS. Functional thermogenic beige adipogenesis is inducible in human neck fat. Int J Obes. 2014b;38:170–176. doi: 10.1038/ijo.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wei S, Cheng K, Gounko NV, Ericksen RE, Xu A, Hong W, Han W. BIG3 inhibits insulin granule biogenesis and insulin secretion. EMBO Rep. 2014;15:714–722. doi: 10.1002/embr.201338181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Honek J, Xue Y, Seki T, Cao Z, Andersson P, Yang X, Hosaka K, Cao Y. Cold-induced activation of brown adipose tissue and adipose angiogenesis in mice. Nat Protoc. 2012;7:606–615. doi: 10.1038/nprot.2012.013. [DOI] [PubMed] [Google Scholar]
- Lin S, Zhu Q, Xu Y, Liu H, Zhang J, Xu J, Wang H, Sang Q, Xing Q, Fan J. The role of the TOB1 gene in growth suppression of hepatocellular carcinoma. Oncol Lett. 2012;4:981–987. doi: 10.3892/ol.2012.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Kuang S. miR-133 links to energy balance through targeting Prdm16. J Mol Cell Biol. 2013;5:432–434. doi: 10.1093/jmcb/mjt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–525. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Magee D, Wang C, McMurphy T, Slater A, During M, Cao L. Adipose tissue insulin receptor knockdown via a new primate-derived hybrid recombinant AAV serotype. Mol Ther Methods Clin Dev. 2014 doi: 10.1038/mtm.2013.8. Published online February 5, 2014. http://dx.doi.org/10.1038/mtm.2013.8. [DOI] [PMC free article] [PubMed]
- Loft A, Forss I, Siersbæk MS, Schmidt SF, Larsen ASB, Madsen JGS, Pisani DF, Nielsen R, Aagaard MM, Mathison A, et al. Browning of human adipocytes requires KLF11 and reprogramming of PPARγ superenhancers. Genes Dev. 2015;29:7–22. doi: 10.1101/gad.250829.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou PH, Gustavsson N, Wang Y, Radda GK, Han W. Increased lipolysis and energy expenditure in a mouse model with severely impaired glucagon secretion. PLoS ONE. 2011;6:e26671. doi: 10.1371/journal.pone.0026671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K, et al. DIANA-microT web server: Elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Nakagami H, Rodriguez-Araujo G, Nimura K, Kaneda Y. Essential role for miR-196a in brown adipogenesis of white fat progenitor cells. PLoS Biol. 2012;10:e1001314. doi: 10.1371/journal.pbio.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Konishi M, Itoh N. FGF21 as an endocrine regulator in lipid metabolism: From molecular evolution to physiology and pathophysiology. J Nutr Metab. 2011;2011:981315. doi: 10.1155/2011/981315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng R, Song G, Roll GR, Frandsen NM, Willenbring H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J Clin Invest. 2012;122:1097–1108. doi: 10.1172/JCI46039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, Kliewer SA, Mangelsdorf DJ. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014;20:670–677. doi: 10.1016/j.cmet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS. MicroRNA function: Multiple mechanisms for a tiny RNA? RNA. 2005;11:1753–1761. doi: 10.1261/rna.2248605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robidoux J, Cao W, Quan H, Daniel KW, Moukdar F, Bai X, Floering LM, Collins S. Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38alpha MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes. Mol Cell Biol. 2005;25:5466–5479. doi: 10.1128/MCB.25.13.5466-5479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Howng SY, Ptáček LJ, Fu YH. miR-32 and its target SLC45A3 regulate the lipid metabolism of oligodendrocytes and myelin. Neuroscience. 2012;213:29–37. doi: 10.1016/j.neuroscience.2012.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Xie H, Mori MA, Alexander R, Yuan B, Hattangadi SM, Liu Q, Kahn CR, Lodish HF. Mir193b-365 is essential for brown fat differentiation. Nat Cell Biol. 2011;13:958–965. doi: 10.1038/ncb2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun KK, Zhong N, Yang Y, Zhao L, Jiao Y. Enhanced radio-sensitivity of NSCLC cells by transducer of erbB2.1 (TOB1) through modulation of the MAPK/ERK pathway. Oncol Rep. 2013;29:2385–2391. doi: 10.3892/or.2013.2403. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Lodish H. MicroRNA networks regulate development of brown adipocytes. Trends Endocrinol Metab. 2013;24:442–450. doi: 10.1016/j.tem.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovski M, Ahmed K, Esau CC, Stoffel M. MyomiR-133 regulates brown fat differentiation through Prdm16. Nat Cell Biol. 2012;14:1330–1335. doi: 10.1038/ncb2612. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012;110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- Varghese F, Bukhari AB, Malhotra R, De A, Ruifrok A, Johnston D., Jr IHC Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE. 2014;9:e96801. doi: 10.1371/journal.pone.0096801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroya F, Vidal-Puig A. Beyond the sympathetic tone: The new brown fat activators. Cell Metab. 2013;17:638–643. doi: 10.1016/j.cmet.2013.02.020. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Pasut A, Soleimani VD, Bentzinger CF, Antoun G, Thorn S, Seale P, Fernando P, van Ijcken W, Grosveld F, et al. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 2013;17:210–224. doi: 10.1016/j.cmet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Guan M, Townsend KL, Huang TL, An D, Yan X, Xue R, Schulz TJ, Winnay J, Mori M, et al. MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1α signaling network. EMBO Rep. 2015;16:1378–1393. doi: 10.15252/embr.201540837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang T. Using the Wash U epigenome browser to examine genome-wide sequencing data. Curr Protoc Bioinformatics. 2012;Chapter 10(Unit 10.10) doi: 10.1002/0471250953.bi1010s40. http://dx.doi.org/10.1002/0471250953.bi1010s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.