

 Selective activation of diol groups by a borinic acid catalyst enables the synthesis of strictly linear polyesters from glycerol, a trifunctional monomer.
Selective activation of diol groups by a borinic acid catalyst enables the synthesis of strictly linear polyesters from glycerol, a trifunctional monomer.
Abstract
Diarylborinic acids catalyze the formation of linear polyesters from glycerol, a trifunctional, carbohydrate-based monomer. The selective activation of 1,2-diols over isolated alcohols by the organoboron catalyst results in polymers that are essentially free of branching or cross-linking and possess a high fraction of 1,3-enchained glycerol units, as assessed by 1H and 13C NMR spectroscopy. The ability to generate well-defined polyester architectures from glycerol is significant in light of the numerous applications of such macromolecules, particularly in the biomedical area. Isomerization, post-polymerization functionalization and controlled cross-linking reactions of the obtained linear poly(glycerol esters) are demonstrated.
Introduction
Bio-based polymers are attracting attention as potential alternatives to macromolecules generated from petrochemical-derived feedstocks. 1–8 Carbohydrates are the most abundant renewable chemical resource on the planet, and possess several desirable features for applications in polymer chemistry, including safety, low cost, structural diversity and synthetic versatility. 9–11 However, the high density of functional groups—specifically, hydroxy (OH) groups—characteristic of carbohydrates poses a challenge to the synthesis of structurally homogeneous macromolecules from these monomers. One solution to this problem is to selectively block or remove OH groups from sugars to generate derivatives that can be used in polymerizations. 12–14 An alternative, and less well-explored strategy, would use catalysis to selectively activate sugar derivatives towards polymerization at particular positions. Here, we describe a successful realization of this second approach, demonstrating that diarylborinic acids (Ar2BOH) are able to catalyze the selective formation of linear polyesters from glycerol, a carbohydrate-derived, trifunctional monomer.
This work builds on our group's finding that diarylborinic acid-derived catalysts are able to activate carbohydrates towards regioselective acylation, sulfonylation, alkylation and glycosylation reactions. 15–19 Experimental and computational studies suggested that these transformations proceed through the formation of cyclic, tetracoordinate borinic esters at cis-1,2- or 1,3-diol motifs.16 We aimed to exploit this type of catalytic reactivity to achieve selective polymerization reactions of carbohydrate derivatives, and selected glycerol-derived polyesters as the targets of our proof-of-concept study (Scheme 1).
Scheme 1. Proposed synthesis of linear, 1,3-linked glycerol polyesters using a diarylborinic acid (Ar2BOH) catalyst. X represents an alkylene or arylene group.

Glycerol, the simplest of the sugar alcohols, is inexpensive and abundant, and is widely used as an additive in foods, pharmaceuticals and personal care products.20 Biodegradable polyesters derived from glycerol have been investigated intensively for biomedical applications, 21,22 including as tissue engineering scaffolds, 23,24 surgical adhesives,25 nanoparticles for drug delivery, 26,27 and drug carrier implants.28 The most commonly employed protocol for the synthesis of such macromolecules—the thermal polycondensation of glycerol with dicarboxylic acids in the absence of solvent—results in branching and cross-linking due to the formation of triacylglycerol motifs. 29,30 The extent of cross-linking and the properties of the obtained polyesters are sensitive to the experimental conditions (particularly the rate of glycerol evaporation upon curing at high temperature and low pressure), 31,32 and on the monomer feed ratio. 33,34 Hyperbranched polyesters have also been accessed from glycerol, using tin oxide catalysts. 35,36 We anticipated that the ability of borinic acids to enhance the rate of acylation of 1,2-diols over isolated alcohols would favor the formation of linear polyesters from glycerol, with a 1,3-linked polymer being preferred due to selective acylation of the less hindered, primary OH groups. The defined content and position of OH groups in this architecture represents an important advantage, since these can be used as sites of functionalization or cross-linking. For example, O-functionalized derivatives of linear poly(glycerol esters) bearing ammonium and phosphate groups have been employed as biomaterials for promotion of neurite growth and for bone tissue engineering. 37,38 Modification of the secondary OH groups with aliphatic acyl moieties has been used to generate macromolecules for drug delivery, with the degree of acylation as well as the polymer molecular weight influencing the degree of drug uptake. 26,27,39 Linear poly(glycerol esters) have also been employed as precursors of graft copolymers. 40,41 The physical properties of glycerol-derived polyesters are also known to be sensitive to their microstructure: for example, the hydrophilicity and glass transition temperature of poly(glycerol adipate) depend on the degree of branching.42 The need for selective access to linear, glycerol-derived polyesters has motivated the development of new synthetic methods. Ring-opening polymerization of glycidyl esters provides macromolecules with low degrees of branching, albeit using synthetic, relatively high-value monomers. 43,44 Lipases have also been used to prepare linear polyesters of glycerol via polycondensation with dicarboxylic acids 45,46 or their bis-vinyl ester derivatives. 47–49 The degrees of polymerization obtained using these enzymatic protocols have been limited due to thermal and shear-induced catalyst degradation, 42,50 issues that could be alleviated using synthetic catalysts. Other potential advantages of using ‘small-molecule’ catalysts in this context include their tolerance of a variety of solvents and reagents, and the possibilities for systematic structural variation.
Results and discussion
Optimization of polymerization conditions
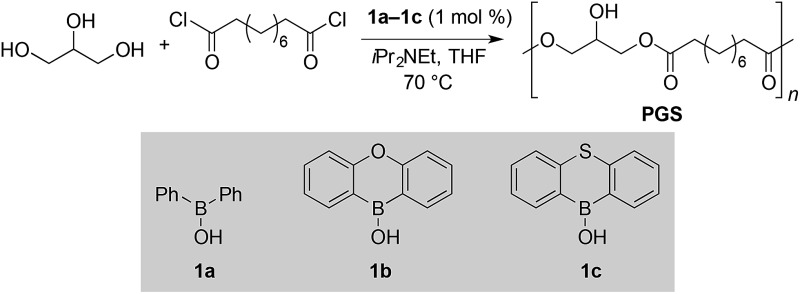
Poly(glycerol sebacate) (PGS), a biodegradable polyester that has been employed extensively for biomedical applications, 51,52 was selected as the target for optimization of reaction conditions. Based on our reported protocol for monoacylation of di- and triols, we used sebacoyl chloride as the diacid-derived monomer, N,N-diisopropylethylamine (iPr2NEt) as base and borinic acids 1a–1c as catalysts (Table 1).‡ The reactions were carried out in tetrahydrofuran (THF) solvent at 70 °C. Because uncatalyzed acylation occurred at an appreciable rate under these conditions, control of polymer microstructure (as judged by the distribution of acylglycerol motifs) was used as the primary criterion for evaluating catalyst performance. In previous studies, 13C nuclear magnetic resonance (NMR) spectroscopy was used to distinguish between mono-, di- and triacylglycerol units in polyesters. 36,43,48,53,54 Accordingly, we were able to assign 13C NMR signals corresponding to these motifs (Fig. 1). The assignments were consistent with literature results, and were further verified by comparisons to spectral data for synthetic model compounds (see the ESI†). Using tris(acetylacetonato)chromium(iii) as a relaxation agent, we obtained 13C NMR spectra that could be integrated to determine relative abundances of the various linkage types. As anticipated, the control PGS generated in the showed a heterogeneous microstructure, with 20% monoacyl-, 74% diacyl- and 5% triacylglycerol motifs. Another control experiment, using DMAP as an acylation catalyst, led to the rapid formation of a gel-like material, presumably reflecting a high degree of cross-linking. Conducting the reaction in the presence of diarylborinic acids 1a–1c (1 mol% catalyst loading) had a marked effect, suppressing the formation of mono- and triacylglycerol units and favoring enchainment of glycerol groups through 1,3- rather than 1,2-linkages (roughly 13 : 1 regioselectivity), as illustrated by the 13C NMR spectra overlaid in Fig. 1. That this effect was observed despite an appreciable uncatalyzed reaction under these conditions likely reflects both the significant level of rate acceleration obtained using the borinic acid catalyst 15,16 as well as the steric and electronic deactivation of the secondary OH group in the 1,3-diacylglycerol motif. The polymers obtained from the borinic acid-catalyzed reactions also showed higher number-average molecular weights (M n) and lower dispersities (Đ) than the control polymer, as judged by gel permeation chromatography (GPC) calibrated against poly(methyl methacrylate) standards. The significantly higher fraction of monoacylglycerol end groups in the polymer obtained in the absence of borinic acid (Fig. 1) provides additional evidence for the beneficial effect of catalyst on the degree of polymerization. Differences in performance between diphenylborinic acid and the heterocyclic catalysts 1b and 1c 55 were not dramatic, but thiaboraanthracene-derived catalyst was found to be optimal, giving a number-average degree of polymerization of roughly 74 (M n = 19.2 kDa, entry 4) and a high proportion of 1,3-enchained units. This combination of high degree of polymerization, relatively low dispersity and lack of detectable branching or cross-linking has not been obtained previously, either by direct polycondensation of glycerol or by indirect methods via glycidyl esters.
Table 1. Catalyst optimization for the synthesis of linear poly(glycerol sebacate) a .
| |||||
| Entry | Catalyst | % 1,2 b | % triacylglycerol b | M n c (kDa) | Đ c |
| 1 | None | 11 | 5 | 8.7 | 1.7 |
| 2 | 1a | 7 | <1 | 12.5 | 1.3 |
| 3 | 1b | 6 | <1 | 11.9 | 1.2 |
| 4 | 1c | 6 | <1 | 19.2 | 1.2 |
aBased on analysis of polymers obtained after an aqueous workup, without further purification or fractionation.
bDetermined by quantitative 13C NMR spectroscopy.
cDetermined by GPC.
Fig. 1. 13C NMR spectra of poly(glycerol sebacate) (signals corresponding to glycerol-derived carbons) obtained in the absence of catalyst, and using 1 mol% of catalysts 1a–1c.

Scope of catalyst-controlled selective polycondensation of glycerol
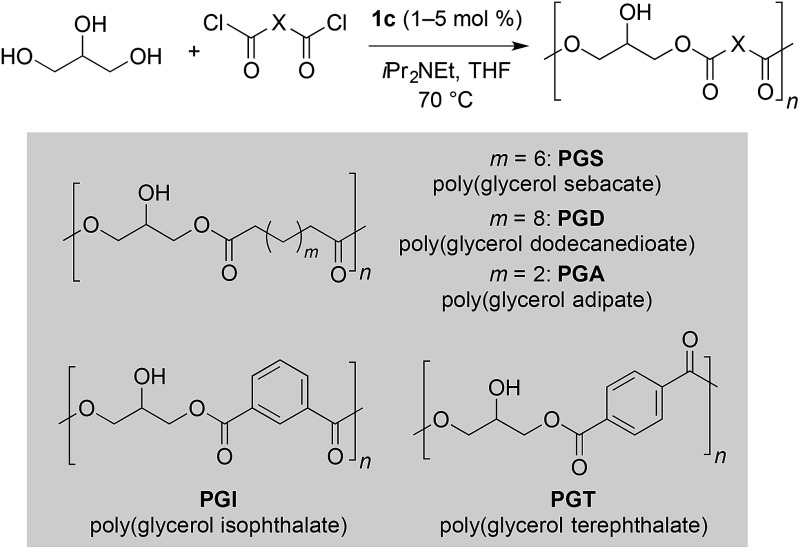
The optimized protocol using catalyst 1c proved to be robust upon scale-up, and could be employed to generate gram quantities of linear PGS (Table 2, entry 1). After purification by precipitation, the obtained polymer was soluble in a range of nonpolar solvents and showed sharp, well-resolved peaks in its 1H NMR spectrum in d 6-DMSO, suggesting a well-defined connectivity and low degree of cross-linking. In these regards, its properties are consistent with those reported for linear PGS generated by ring-opening polymerization of the corresponding diglycidyl ester,43 although the level of branching determined for the present polymer is lower (<1% vs. approximately 10%). The protocol was further extended to polymerizations of other aliphatic and aromatic diacyl chlorides with glycerol (Table 2, entries 2–5). Each of the obtained polymers was free of triacylglycerol units, as judged by quantitative 13C NMR spectroscopy (see the ESI†). The catalyst loading required to generate polymers having this low degree of branching and cross-linking ranged from 1 mol% to 5 mol%, depending on the structure of the diacid chloride component. The presence of one free OH group per polymer repeat unit was further verified by subjecting each product to an excess of acetic anhydride in pyridine, and characterizing the resulting, per-acetylated polyesters by 1H NMR spectroscopy (see the ESI†).
Table 2. Synthesis of glycerol-derived polyesters using 1c as catalyst.
| |||||||
| Entry | Polymer | Mol% 1c | % 1,2 a | % branch a | M n b (kDa) | Đ b | Yield |
| 1 c | PGS | 1 | 7 | <1 | 24.4 | 1.4 | 93% |
| 2 | PGD | 1 | 5 | <1 | 19.6 | 1.1 | 87% |
| 3 d | PGA | 5 | 3 | <1 | 30.0 | 1.4 | 94% |
| 4 | PGI | 2 | 15 | <1 | 10.0 | 1.6 | 90% |
| 5 | PGT | 2 | 13 | <1 | 10.0 | 1.4 | 92% |
aDetermined by quantitative 13C NMR spectroscopy.
bDetermined by GPC.
cExperiment was carried out on 1 g scale.
d2 equiv. of iPr2NEt were employed.
Although predominant 1,3-enchainment of glycerol units was observed across this series of polymers, the level of regioselectivity was variable, with aliphatic diacid chlorides giving higher levels of 1,3-selectivity than isophthaloyl and terephthaloyl chloride. In the case of adipoyl chloride, reducing the loading of base from three equivalents to two equivalents was needed to maintain a high ratio of 1,3- to 1,2-enchained units. This change of conditions reduces the rate of base-catalyzed acyl migration that results in equilibration between the two linkage types (see below).
Limitations of this protocol were encountered when we attempted to prepare the polyesters derived from glycerol and succinic acid or glutaric acid (having two and four methylene groups, respectively). The use of these shorter-chain diacid chlorides resulted in precipitation during the polymerization reactions, and 13C NMR spectroscopy of the isolated material indicated the presence of triacylglycerol units (data not shown). A higher tendency towards branching for the succinoyl and glutaroyl derivatives has also been noted in studies of organotin-catalyzed polycondensations with glycerol.53
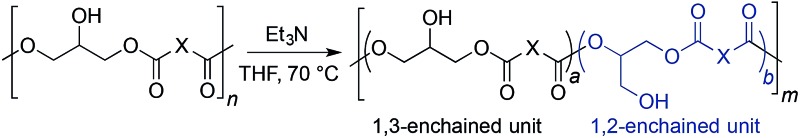
Polyester rearrangement via base-catalyzed ester migration
The relatively high ratios of 1,3- to 1,2-enchained glycerol units obtained under borinic acid catalysis (e.g., Table 2, entries 1–3) appear to reflect kinetic rather than thermodynamic control. To address this point, we subjected each of the obtained polyesters to triethylamine (Et3N, 3 equivalents per monomer unit) in THF at 70 °C, conditions that should favor base-promoted acyl group migration.56 Indeed, the proportion of 1,2-enchained glycerol moieties (as determined by quantitative 13C NMR spectroscopy – Table 3) increased for each of the polymer samples, appearing to reach equilibrium after approximately 12 hours. The increase in the fraction of 1,2-diacylglycerol motifs was not accompanied by an increase in the degree of branching, indicating that the linear polymer architecture was maintained during the isomerization. Similar proportions of 1,2-linkages at equilibrium (roughly 20%) were obtained for the sebacate, dodecanedioate, isophthalate and terephthalate-derived polymers (PGS, PGD, PGI and PGT), while PGA gave 32% of 1,2-diacylglycerol units after isomerization. We speculate that the differences in 1,2-diacylglycerol content of the initially synthesized polymers most likely arise from varying extents of acyl migration under the polymerization conditions. In general, the levels of regioselectivity observed in the polymerizations were lower than those for simple acylations of primary versus secondary OH groups in diol substrates.16 Presumably, acyl migration is more prevalent in the polymerizations because of the higher reaction temperatures employed: borinic acid-catalyzed acylations of diols were generally conducted at room temperature or below. In any case, this ability to use kinetic control to access polymers whose structures can be altered through a subsequent equilibration, while maintaining the linear architecture, is a noteworthy aspect of the catalyst-controlled polymerizations described here.
Table 3. Base-promoted isomerization of glycerol-derived polyesters.
aDetermined by quantitative 13C NMR spectroscopy.
Properties and synthetic transformations of linear poly(glycerol esters)
Differential scanning calorimetry (DSC) was used to identify the thermal transitions of the linear poly(glycerol esters) obtained by borinic acid-catalyzed polycondensation. The glass transition temperatures (T g) of the polymers derived from the aliphatic diacids were relatively low: T g values of –36 °C, –34 °C and 18 °C were determined for PGA, PGS and PGD, respectively. The significant jump in T g for the dodecanedioate-linked polymer relative to PGS is consistent with reported glass transition temperatures for the corresponding cross-linked polyesters. 30,57 PGS and PGD showed additional transitions corresponding to the crystallization and melting of the hydrocarbon domains (see the ESI†). The isophthalate- and terephthalate-derived polymers displayed higher glass transition temperatures than the aliphatic variants: T g values of 58 °C and 66 °C were determined for PGI and PGT, respectively.
An important feature of linear poly(glycerol esters) is the presence of a free OH group at each repeat unit. Post-polymerization transformations of these groups58 provide ways to incorporate additional functionality or to modify the physical properties of the material. Indeed, Wang and co-workers have demonstrated several such transformations of polymers generated by ring-opening of diglycidyl esters. 37,38,43,44 In keeping with these results, carbodiimide-mediated coupling43 of linear PGS with fluorenylmethyoxycarbonyl- (Fmoc)-protected glycine at room temperature in dichloromethane led to quantitative introduction of protected glycine esters at the free OH groups of the polymer, as judged by 1H NMR spectroscopy (Scheme 2).
Scheme 2. Post-polymerization transformations of linear PGS: esterification with Fmoc–glycine and crosslinking with hexamethylene diisocyanate. DIC and DMAP denote N,N-diisopropylcarbodiimide and 4-(dimethylamino)pyridine, respectively.

In a similar way, controlled cross-linking of linear PGS was accomplished by treatment with hexamethylene diisocyanate (HDI) to create urethane linkages.59 By varying the loading of HDI from 0.5 to 20 mol% per polymer OH group, a series of cross-linked PGS derivatives was generated. The presence of urethane groups was established by attenuated total reflectance FTIR spectroscopy. In particular, the intensity of the urethane C O stretch at 1540 cm–1 was found to increase linearly with the loading of HDI employed in the cross-linking reaction (see the ESI†). Higher loadings of HDI also resulted in increased apparent number-average molecular weights, as judged by GPC. Catalyst-controlled preparation of linear poly(glycerol esters), followed by introduction of a precise level of cross-linking in a post-polymerization step, may enable fine-tuning of the physical, mechanical and biodegradation properties of these materials in ways that are useful for their diverse applications.
Conclusions
The results of the present study have illustrated how catalysts capable of regioselective OH group activation can be used to achieve control of microstructure in polyester synthesis. The ability to generate well-defined polyester architectures from glycerol is significant in light of the numerous applications of such macromolecules, particularly in the biomedical area. More broadly, this work points towards new ways to employ carbohydrate derivatives as feedstocks for polymer chemistry. Ongoing work in our laboratory is aimed at extending this approach to other polymer classes, and at generating structurally unprecedented types of polyesters based on more complex polyols or carbohydrate derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
This work was funded by NSERC (Discovery Grants and Canada Research Chairs programs), the Canada Foundation for Innovation (Projects #17545 and #19119) and the Ontario Ministry of Research and Innovation.
Footnotes
‡While 1a–1c exist as μ-oxo-linked (Ar2B–O–BAr2) dimers in the solid state, the catalyst loadings shown in Table 1 reflect the active, monomeric form.
References
- Gross R. A., Kalra B. Science. 2002;297:803–807. doi: 10.1126/science.297.5582.803. [DOI] [PubMed] [Google Scholar]
- Mecking S. Angew. Chem., Int. Ed. 2004;311:484–489. [Google Scholar]
- Ragauskas A. J., Williams C. K., Davison B. H., Britovsek G., Cairney J., Eckert C. A., Frederich Jr W. J., Hallett J. P., Leak D. J., Liotta C. L., Mielenz J. R., Murphy R., Templer R., Tschaplinski T. Science. 2006;311:484–489. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]
- Meier M. A. R., Metzger O., Schubert U. S. Chem. Soc. Rev. 2007;36:1788–1802. doi: 10.1039/b703294c. [DOI] [PubMed] [Google Scholar]
- Coates G. W., Hillmyer M. A. Macromolecules. 2009;42:7987–7989. [Google Scholar]
- Ulery B. D., Nair L. S., Laurencin C. T. J. Polym. Sci., Part B: Polym. Phys. 2011;49:832–864. doi: 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschan M. J., Brulé E., Haquette P., Thomas C. M. Polym. Chem. 2012;3:836–851. [Google Scholar]
- Gandini A., Lacerda T. M., Carvalho A. J. F., Trovatti E. Chem. Rev. 2016;116:1637–1669. doi: 10.1021/acs.chemrev.5b00264. [DOI] [PubMed] [Google Scholar]
- Engineered Carbohydrate-Based Materials for Biomedical Applications, ed. R. Narain, John Wiley & Sons, Inc., Hoboken, New Jersey, 2011. [Google Scholar]
- Galbis J. A., de Gracia García-Martín M., Violante de Paz M., Galbis E. Chem. Rev. 2016;116:1600–1636. doi: 10.1021/acs.chemrev.5b00242. [DOI] [PubMed] [Google Scholar]
- Wang D., Dordick J. S., Linhardt R. J. Chem. Mater. 2002;14:3232–3244. [Google Scholar]
- Fenouillot F., Rousseau A., Colomines G., Saint-Loup R., Pascault J.-P. Prog. Polym. Sci. 2010;35:578–622. [Google Scholar]
- Tang M., White A. J. P., Stevens M. M., Williams C. K. Chem. Commun. 2009:941–943. doi: 10.1039/b817658b. [DOI] [PubMed] [Google Scholar]
- Liu Y., Reineke T. M. J. Am. Chem. Soc. 2005;127:3004–3015. doi: 10.1021/ja0436446. [DOI] [PubMed] [Google Scholar]
- Lee D., Taylor M. S. J. Am. Chem. Soc. 2011;133:3724–3727. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]
- Lee D., Williamson C. L., Chan L., Taylor M. S. J. Am. Chem. Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- Chan L., Taylor M. S. Org. Lett. 2011;13:3090–3093. doi: 10.1021/ol200990e. [DOI] [PubMed] [Google Scholar]
- Gouliaras C., Lee D., Chan L., Taylor M. S. J. Am. Chem. Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]
- D'Angelo K. A., Taylor M. S. J. Am. Chem. Soc. 2016;138:11058–11066. doi: 10.1021/jacs.6b06943. [DOI] [PubMed] [Google Scholar]
- Top Value Added Chemicals from Biomass, ed. T. Werpy and G. Petersen, US Department of Energy, Oak Ridge, Tennessee, 2004, vol. 1. [Google Scholar]
- Rai R., Tallawi M., Grigore A., Boccaccini A. R. Prog. Polym. Sci. 2012;37:1051–1078. [Google Scholar]
- Zhang H., Grinstaff M. W. Macromol. Rapid Commun. 2014;35:1906–1924. doi: 10.1002/marc.201400389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaky S. H., Lee K., Gao J., Jensen A., Close J., Wang Y., Almarza A. J., Sfeir C. Tissue Eng., Part A. 2014;20:45–53. doi: 10.1089/ten.TEA.2013.0172. [DOI] [PubMed] [Google Scholar]
- Chen Q., Bismarck A., Hansen U., Junaid S., Tran M. Q., Harding S. E., Ali N. N., Boccaccini A. R. Biomaterials. 2008;29:47–57. doi: 10.1016/j.biomaterials.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Lang N., Pereira M. J., Lee Y., Ingeborg F., Vasilyev N. V., Feins E. N., Ablasser K., O'Cearbhaill E. D., Xu C., Fabozzo A., Padera R., Wasserman S., Freudenthal F., Ferreira L. S., Langer R., Karp J. M., del Nido P. J. Sci. Transl. Med. 2015;6:1–19. doi: 10.1126/scitranslmed.3006557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallinteri P., Higgins S., Hutcheon G. A., Pourc C. B. S., Garnett M. C. Biomacromolecules. 2005;6:1885–1894. doi: 10.1021/bm049200j. [DOI] [PubMed] [Google Scholar]
- Weiss V. M., Naolou T., Hause G., Kuntsche J., Kressler J., Mäder K. J. Controlled Release. 2012;158:156–164. doi: 10.1016/j.jconrel.2011.09.077. [DOI] [PubMed] [Google Scholar]
- Sun Z., Chen C., Sun M., Ai C., Lu X., Zheng Y.-F., Yang B.-F., Dong D.-L. Biomaterials. 2009;30:5209–5214. doi: 10.1016/j.biomaterials.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Kiyotsukuri T., Kanaboshi M., Tsutsumi N. Polym. Int. 1994;33:1–8. [Google Scholar]
- Wang Y., Ameer G. A., Sheppard B. J., Langer R. Nat. Biotechnol. 2002;20:602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- Li Y., Cook W. D., Moorhoff C., Huang W.-C., Chen Q.-Z. Polym. Int. 2012;62:534–547. [Google Scholar]
- Li X., Hong A. T., Naskar N., Chung H. Biomacromolecules. 2015;16:1525–1533. doi: 10.1021/acs.biomac.5b00018. [DOI] [PubMed] [Google Scholar]
- Liu Q., Tian M., Ding T., Shi R., Zhang L. J. Appl. Polym. Sci. 2005;98:2033–2041. [Google Scholar]
- Liu Q., Tian M., Ding T., Shi R., Feng Y., Zhang L., Chen D., Tian W. J. Appl. Polym. Sci. 2007;103:1412–1419. [Google Scholar]
- Stumbé J.-F., Bruchmann B. Macromol. Rapid Commun. 2004;25:921–924. [Google Scholar]
- Zhang T., Howell B. A., Dumitrascu A., Martin S. J., Smith P. B. Polymer. 2014;55:5065–5072. [Google Scholar]
- Huang P., Bi X., Gao J., Sun L., Wang S., Chen S., Fan X., You Z., Wang Y. J. Mater. Chem. B. 2016;4:2090–2101. doi: 10.1039/c5tb02542g. [DOI] [PubMed] [Google Scholar]
- Wang S., Jeffries E., Gao J., Sun L., You Z., Wang Y. ACS Appl. Mater. Interfaces. 2016;8:9590–9599. doi: 10.1021/acsami.5b12379. [DOI] [PubMed] [Google Scholar]
- Puri S., Kallinteri P., Higgins S., Hutcheon G. A., Garnett M. G. J. Controlled Release. 2008;125:59–76. doi: 10.1016/j.jconrel.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn D., Pulst M., Naolou T., Busse K., Balko J., Kressler J. J. Polym. Sci., Part B: Polym. Phys. 2013;51:1581–1591. [Google Scholar]
- Jbeily M., Naolou T., Bibal M., Amado E., Kressler J. Polym. Int. 2014;63:894–901. [Google Scholar]
- Taresco V., Creasey R. G., Kenon J., Mantovani G., Alexander C., Burley J. C., Garnett M. C. Polymer. 2016;89:41–49. doi: 10.1002/pola.28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Cao H., Gao J., Shin P. H., Day B. W., Wang Y. Biomaterials. 2010;31:3129–3138. doi: 10.1016/j.biomaterials.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Wang Y. Adv. Funct. Mater. 2012;22:2812–2820. [Google Scholar]
- Kumar A., Kulshrestha A. S., Gao W., Gross R. A. Macromolecules. 2003;36:8219–8221. [Google Scholar]
- Iglesias L. E., Fukuyama Y., Nonami H., Erra-Balsells R., Baldessari A. Biotechnol. Tech. 1999;13:923–926. [Google Scholar]
- Kline B. J., Beckman E. J., Russell A. J. J. Am. Chem. Soc. 1998;120:9475–9480. [Google Scholar]
- Uyama H., Inada K., Kobayashi S. Macromol. Rapid Commun. 1999;20:171–174. [Google Scholar]
- Uyama H., Inada K., Kobayashi S. Macromol. Biosci. 2001;1:40–44. [Google Scholar]
- Korupp C., Weberskirch R., Müller J. J., Liese A., Hilterhaus L. Org. Process Res. Dev. 2010;14:1118–1124. [Google Scholar]
- Wang Y., Kim Y. M., Langer R. J. Biomed. Mater. Res., Part A. 2003;66:192–197. doi: 10.1002/jbm.a.10534. [DOI] [PubMed] [Google Scholar]
- Loh X. J., Karim A. A., Owh C. J. Mater. Chem. B. 2015;3:7641–7652. doi: 10.1039/c5tb01048a. [DOI] [PubMed] [Google Scholar]
- Wyatt V. T., Strahan G. D. Polymers. 2012;4:396–407. [Google Scholar]
- Li Y., Cook W. D., Moorhoff C., Huang W.-C., Chen Q.-Z. Polym. Int. 2013;62:534–547. [Google Scholar]
- Dimitrijević E., Taylor M. S. Chem. Sci. 2013;4:3298–3303. [Google Scholar]
- Roslund M. U., Aitio O., Wärna J., Maaheimo H., Murzin D. Y., Leino R. J. Am. Chem. Soc. 2008;130:8769–8772. doi: 10.1021/ja801177s. [DOI] [PubMed] [Google Scholar]
- Migneco F., Huang Y.-C., Birla R. K., Hollister S. J. Biomaterials. 2009;30:6479–6484. doi: 10.1016/j.biomaterials.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga D. E., Petrie T. A., Kumar A., Weck M., García A. J., Collard D. M. Biomacromolecules. 2008;9:2056–2062. doi: 10.1021/bm800292z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey J., Xu H., Shen J., Thevenot P., Gondi S. R., Nguyen K. T., Sumerlin B. S., Tang L., Yang J. Biomaterials. 2008;29:4637–4649. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.