

Abstract
N-methyl-D-aspartate receptors (NMDARs) play important roles in brain development and neurological disease. We report two individuals with similar dominant de novo GRIN1 mutations (c.1858 G>A and c.1858 G>C; both p.G620R). Both individuals presented at birth with developmental delay and hypotonia associated with behavioral abnormalities and stereotypical movements. Recombinant NMDARs containing the mutant GluN1-G620R together with either GluN2A or GluN2B were evaluated for changes in their trafficking to the plasma membrane and their electrophysiological properties. GluN1-G620R/GluN2A complexes showed a mild reduction in trafficking, a ~ 2-fold decrease in glutamate and glycine potency, a strong decrease in sensitivity to Mg2+ block, and a significant reduction of current responses to a maximal effective concentration of agonists. GluN1-G620R/GluN2B complexes showed significantly reduced delivery of protein to the cell surface associated with similarly altered electrophysiology. These results indicate these individuals may have suffered neurodevelopmental deficits as a result of the decreased presence of GluN1-G620R/GluN2B complexes on the neuronal surface during embryonic brain development and reduced current responses of GluN1-G620R-containing NMDARs after birth. These cases emphasize the importance of comprehensive functional characterization of de novo mutations and illustrates how a combination of several distinct features of NMDAR expression, trafficking and function can be present and influence phenotype.
INTRODUCTION
N-methyl-D-aspartate receptors (NMDARs) are ligand-gated cation channels that mediate a slow, Ca2+-permeable component of excitatory synaptic transmission in the central nervous system. NMDARs are heterotetrameric complexes comprised of two GluN1 subunits (encoded by GRIN1) in combination with two GluN2 subunits (types A–D, encoded by GRIN2A-D) or some combination of GluN2 and GluN3 subunits (types A and B, encoded by GRIN3A-B).1 While GluN1 is expressed throughout one’s lifetime, the GluN2 and GluN3 subunits differ in their expression pattern during brain development, which results in the differential temporal expression of NMDAR subtypes.1 For example, the GluN2B subunit is highly expressed during the embryonic and early postnatal periods, whereas GluN2A expression occurs soon after birth.
All NMDAR subunits contain four domains: an extracellular amino terminal domain that regulates ion channel opening and response-time course, a bi-lobed extracellular agonist-binding domain (ABD), a transmembrane domain (TM) with four hydrophobic segments (M1-4) and an intracellular carboxyl terminal domain (CTD), which participates in localization and intracellular signaling.1 Activation of NMDARs involves several regulatory mechanisms. The primary mechanism requires occupation of two glycine-binding sites on GluN1 and two glutamate binding sites on GluN2, this combination of binding is thought to trigger the opening of a cation-selective pore that mediates an inward current leading to neuronal depolarization and subsequent increase in intracellular Ca2+ concentration.2 NMDARs are also negatively regulated in several ways, including in a voltage-dependent manner by extracellular Mg2+ (noted to bind deep within the pore) and tonic inhibition by extracellular protons (present at physiological pH), as well as endogenous extracellular Zn2+.1,3
NMDARs play important roles in normal brain development and function, such as synaptic plasticity, neural development, learning and memory.1 As a result of this, NMDAR dysfunction has been associated with several neurological disorders including Parkinson, Alzheimer and Huntington diseases.1,4 Furthermore, mutations in NMDAR subunits have been associated with a variety of epilepsy syndromes (for example, Rolandic epilepsy and electrical status epilepticus of sleep), as well as several neurodevelopmental disorders that may or may not be associated with epilepsy.5–18 Interestingly, many of the mutations associated with these disorders are de novo and have a dominant negative effect on receptor function by reducing or eliminating the response of NMDARs via changes in their electrophysiological behavior.5,6,11–13,16,18 These changes can increase or decrease NMDAR activity, as well as altering the NMDAR response-time course. These effects can subsequently disturb neuronal function and differentially change the neurophysiology of the affected circuits. For example, several GRIN2A mutations have been associated with increased NMDAR-mediated charge transfer, which could increase neuronal excitability and potentially promote neuronal death.7–13,17,19 The phenotypes associated with this hyperexcitability include disorders along the epilepsy-aphasia spectrum, as well as other epileptic encephalopathies associated with severe neurodevelopmental delays.5,11,12 To date, many of these pathologic syndromes have been associated with GluN2A or GluN2B. However, additional data has emerged that suggests that mutations in GluN1 subunits can also play a role in these types of disorders.6,16,20,21
Recently, several GRIN1 mutations have been associated with neurodevelopmental phenotypes. Epilepsy is often present in these patients, and their seizures can be either primary generalized or focal in onset.6,16,20,21 In one case, a missense mutation (p.Y647S) in the M3 TM of GluN1 was identified in a girl with developmental delay and infantile spasms;20 while another variant involving the pre-M1 segment (p.S560dup) was associated with severe developmental delay and complex partial epilepsy.6 Four other de novo GRIN1 missense mutations were reported in four patients exhibiting epilepsies that were either focal or primary generalized in onset. These patients also had developmental delay, hypotonia, facial dysmorphisms, stereotypical hand movements and oculogyric crises.16,21 Three of these mutations were located in TMs, while the other was located in the extracellular loop near transmembrane helix 1 (p.D552E; p.M641I; p.N650K; and p.G815R).21 In addition to these examples, several other variants have been reported that were associated with diverse developmental and epileptic phenotypes.16 Many of these latter variants underwent electrophysiological investigation; however, genuine phenotype–genotype correlations were difficult to establish.16
We report two individuals with similar dominant de novo GRIN1 mutations resulting in the same amino acid substitution: a 12-year-old male (Proband-1: c.1858 G>A; p.G620R) and a 20-year-old female (Proband-2: c.1858 G>C; p.G620R). Both presented at birth with developmental delay and hypotonia associated with cognitive disability, but without epilepsy. Investigations with recombinant human NMDARs possessing the GluN1-G620R mutant along with either GluN2B or GluN2A revealed abnormal activity. When GluN1-G620R was complexed with either GluN2A or GluN2B subunits and evaluated electrophysiologically, there was a decreased potency for glutamate, glycine and magnesium, and a significant reduction of the current responses. Interestingly, in addition to its altered electrophysiological activity, GluN1-G620R co-expression with GluN2B was also noted to have reduced trafficking of this specific NMDAR to the plasma membrane. These data highlight the importance of functional characterization of NMDARs by revealing the complex effects that GRIN1 mutations can have on multiple levels of their function, and suggest that changes in the function and trafficking of specific NMDAR subtypes underlie these individuals’ similar developmental phenotypes.
MATERIALS AND METHODS
Consent and study approvals
Research protocols were approved through the institutional review board, and the families gave informed consent (CSMC IRB protocol Pro00037131). All in vitro studies in this paper were conducted according to the guidelines of Emory University.
Genetic evaluation
Genomic DNA was extracted from blood and exome sequencing was performed as per previous protocols (GeneDx, Gaithersburg, MD, USA; Baylor College of Medicine, Houston, TX, USA).22,23
Electrophysiology evaluation
We utilized cDNA for wild-type human NMDA receptor GluN1-1a (hereafter GluN1; GenBank: NP_015566), GluN2A (GenBank: NP_000824) and GluN2B (GenBank: NP_000825) subunits subcloned into the plasmid vector pCI-neo (Promega, Madison, WI, USA). The mutant GluN1 was generated by site-directed mutagenesis using the QuikChange protocol (Stratagene, La Jolla, CA, USA). Synthesis and injection of cRNA into Xenopus laevis oocytes (Ecocyte Bio Science, Austin TX, USA), as well as two-electrode voltage-clamp recordings from oocytes were performed as previously described.24 The recording solution contained (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, 0.01 EDTA (23 °C, pH 7.4 unless otherwise stated). The membrane potential was held at − 40 mV for all two-electrode voltage-clamp recordings unless otherwise stated. The agonist concentration-response curves were fitted with:
| (1) |
The concentration-response data for Mg2+ inhibition and Food and Drug Administration (FDA)-approved NMDA receptor blockers was fitted with:
| (2) |
where N is the Hill slope, EC50 is the concentration of the agonist that produces a half-maximal effect, IC50 is the concentration of the inhibitor that produces a half-maximal effect and minimum is the degree of residual inhibition at a saturating concentration of the antagonist.
HEK293 cells (ATCC CRL-1573) were transiently transfected with plasmid cDNAs encoding wild-type human GluN1/GluN2A, or GluN1/GluN2B, or the mutant GluN1-G620R/GluN2A, or GluN1-G620R/GluN2B.25 Following 18–24 h transfection, the whole-cell voltage-clamp current recordings were performed with a solution containing (in mM) 150 NaCl, 3 KCl, 10 HEPES, 0.01 EDTA, 0.5 CaCl2 and 11 D-mannitol, with the pH adjusted to 7.4 by addition of NaOH (23 °C). The recording electrodes were prepared using thin-walled filamented borosilicate glass (TW150F-4; World Precision Instruments, Sarasota, FL, USA) using a vertical puller (Narishige P-10, Tokyo, Japan), and filled with the internal solution (in mM: 110 D-gluconic acid, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na2ATP, 0.3 NaGTP adjusted to pH 7.35 with CsOH; the osmolality was adjusted to 300–310 mOsmol kg−1 using CsCl or water). The current responses to external application of glutamate (1000 μM) and glycine (100 μM) were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Sunnyvale, CA, USA) with the holding potential of −60 mV. The current responses were filtered at 8 kHz (−3dB, 8 pole Bessel filter, Frequency Devices, Ottawa, IL, USA) and digitized at 20 kHz using a Digidata 1440A data acquisition system (Molecular Devices).
All data were presented as mean ± s.e.m. Statistical significance was established to P<0.05 by unpaired Student’s t-test. Power to detect a 40% difference was 0.8.
Cell surface biotinylation
The protocol for cell surface biotinylaiton was previously described and several modifications were added.26 HEK293 cells were transfected with human NMDAR cDNAs (GluN1/GluN2A, GluN1-G620R/GluN2A, GluN1/GluN2B and GluN1-G620R/GluN2B) for 24 h with Fugene 6 (Promega, E2691), incubated on ice with 1 mg ml−1 Sulfo-NHS-Biotin (Thermo Scientific, 89881 Waltham, MA, USA) in ice-cold PBS/MgCl2/CaCl2 (1 mM MgCl2 and 0.01 mM CaCl2) for 20 min, rinsed 3 times with 50 mM glycine in tris-buffered saline (TBS) to quench the biotin reactivity, and scraped in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 10 mM Na4P2O7, 50 mM NaF, 1% SDS, 1% Triton X-100 and protease inhibitors). Lysates were sonicated and then centrifuged, and the protein concentration of each supernatant was adjusted to 1 mg ml−1 with Bradford assay. Equal amounts of supernatants were added to Neutravidin beads (Thermo Scientific, 29200), which were then rotated for 1 h at 4 °C. Biotinylated proteins were eluted from the Neutravidin beads with Laemmli Sample Buffer (Bio-Rad, 161-0737, Hercules, CA, USA) containing 200 mM dithiothreitol and evaluated by Western blotting.
Western blotting and statistical analysis
After boiling for 5 min, SDS-polyacrylamide gel electrophoresis (Bio-Rad, 456–8124) was used to separate 10 μl of the total and surface protein fractions for samples of GluN1 co-expressed with GluN2 A, while 5 μl of total protein and 30 μl of the surface protein fraction were separated for samples of GluN1 co-expressed with GluN2B. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, and immunoblotted with following antibodies: mouse anti-GluN1 (BD Pharmingen, San Diego, CA, USA), mouse anti-transferrin receptor (Sigma, St Louis, MO, USA) and mouse anti-tubulin (Sigma). Tubulin was the internal control for total protein expression and monitors the quality of biotinylation experiment as tubulin is only present in the cytoplasmic fraction. Transferrin receptor was a control to show relatively equal surface biotin labeling. Bands were detected with film, and subsaturated bands were quantified with NIH ImageJ. Densitometry was used for chemiluminescence signal quantification, and the ratio of surface-to-total protein level of mutant was normalized to the wild type.
The statistical difference was determined by paired t-test. The number of independent experiments was represented by n. Samples sizes were determined by a priori power analysis for effect size = 2, power = 0.8 and α = 0.05.
RESULTS
Clinical features
Proband-1 was a 12-year-old male born of a dizygotic twin pregnancy via emergency caesarian section at 35 weeks gestation. Complications included HELLP syndrome and gestational hypothyroidism. The mother noted decreased fetal movements in utero (compared to his twin). Growth parameters were normal. A spontaneous pneumothorax on his first day of life required 2 weeks of endotracheal intubation. His twin was without issues.
Proband-1 was hypotonic and floppy throughout infancy. At ~ 6–7 months, he developed ‘abnormal movements’ of his arms and legs. He started walking at 5 years. His mother noted he had a recent increase in ataxia and falls. He spoke his first words at 2 years of age and by 12 years his vocabulary consisted of 100 spoken words, although he could not put two words together. Mother noted a history of intermittent and episodic regressive aphasic episodes, especially after illness. His mother also noted hyperactivity associated with self-stimulatory and self-injurious behaviors (for example, biting himself). He has never had any eye movement abnormalities or oculogyric crises. He has never had a seizure.
He developed ulcerative colitis at 6 years of age that required total colectomy. He was treated for an ostomy-stoma prolapse and eosinophilic esophagitis the following year. There were no other significant health issues.
Previous testing included a normal electromyography-nerve conduction study and muscle biopsy at 2 years, as well as normal magnetic resonance imaging and magnetic resonance spectroscopy of the brain at 8 years. There were no callosal abnormalities or cerebral atrophy. Electroencephalogram at that time was abnormal due to mild diffuse background slowing, without epileptiform features.
The family was non-consanguineous. The mother was of Scottish and Syrian-Arabic descent and the father was of Italian descent. There was no history of neurological disease in the family. Both his twin sister and 13 year-old brother were healthy.
Proband-2 was a 25-year-old female with a similar history of severe intellectual disability. She was born via spontaneous vaginal delivery with normal growth parameters. Her mother also noted decreased fetal movements in utero. She was hypotonic and had difficulty feeding in the postnatal period and early infancy. She had ‘tremors’ as an infant and was diagnosed with ‘cerebral palsy’ at 15 months. She started walking at 2 years, but was very unsteady with ‘slow progress to a normal walk around 4–5 years of age’. Her first words were at 2–3 years, although she has over 500 words now. She makes good eye contact and has good social reciprocity. She was diagnosed with attention deficit hyperactivity disorder. She had surgery to repair strabismus at 5 years of age; otherwise, ophthalmological exams were normal and she was without oculogyric crises.
Previous testing at 12 years of age included an electroencephalogram that was non-epileptiform and a normal magnetic resonance imaging of the brain. At ~ 15 years, she was noted to have spells of disorientation, ataxia and lethargy lasting over several weeks that resolved on their own. She also had tremors and some abnormal stereotypical movements of her arms and legs that were not consistent with a movement disorder. She has never had a seizure. She had numerous episodes of syncope over the past several years without an established etiology, although an echocardiogram revealed mitral valve leaflet bowing without frank prolapse. She had issues with constipation as a child and has had persistent gastro-esophageal reflux. Menarche was at 16 years.
The family was non-consanguineous of Northern European ancestry. There was no history of neurological disease in the family; however, numerous family members were thought to have joint laxity and mitral valve prolapse. Her older sister was healthy.
On examination, Proband-1 had a thin elongated face associated with mild dolichocephaly, thick eyebrows and deep-set hypoteloric eyes (Figure 1a). He had a maxillary overbite, prominent recessed chin, high-arched palate, and showed frequent twisting and protrusion of his tongue. His sternum had an absent xyphoid tip.
Figure 1.
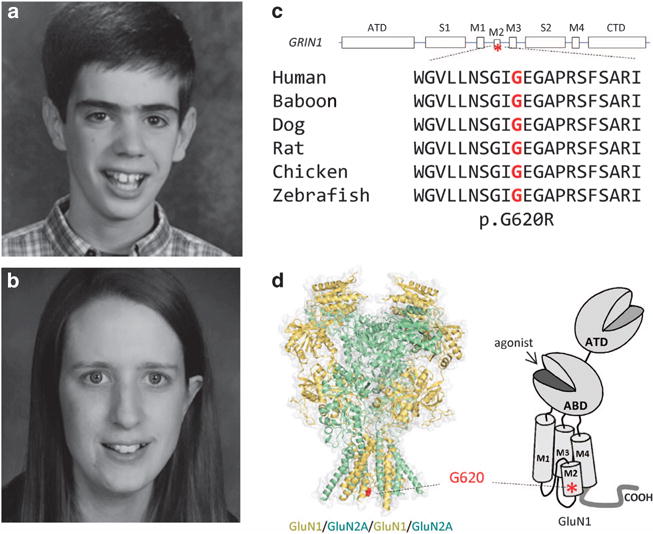
Identification of a GRIN1 missense mutation in patients with developmental delay. (a and b) Facial appearance of Probands 1 and 2. (c) A linear schematic representation of the GluN1 subunit. The position of the mutation is indicated by an asterisk. Gly620 is conserved across vertebral species. ATD, Amino terminal domain; CTD, carboxy terminal domain; M1-4, transmembrane domains (TMs) 1–4; S1 and S2, agonist-binding domains. (d) Homology model of the GluN1/GluN2A receptor built from the GluN2B crystallographic data39 and shown as space fill. The position of the alteration p.G620R is indicated by the red color in region of TM M2.
He was alert, but hyperactive. He made appropriate eye contact and followed only a few commands. He was sensitive to sounds and had mild hyperekplexia. He had decreased bulk throughout, with mildly increased appendicular tone and normal strength. He had frequent stereotypical movements of his arms and legs throughout the exam. There was no action or resting tremor. His reflexes were brisk (legs greater than arms). There was mild dysmetria when reaching for objects and fine repetitive movements had reduced amplitude and speed. He had a wide-based stance and ambulated with exaggerated slapping of his feet.
Proband-2 was awake and alert during her examination. She exhibited some mild anxiety. She was macrocephalic (as were her parents) with a thin elongated face associated with prominent nose and deep-set eyes (Figure 1b). She had a marfanoid body habitus associated with hyperextension and hyperflexibility of the knees, elbows and her thin long fingers. She had good tone and strength. There were no reports of hyperreflexia or dysmetria. Her gait was normal. She had scoliosis (~25 degrees).
Exome sequencing
Exome sequencing identified de novo heterozygous variants (Proband-1: c.1858 G>A; p.G620R; Proband-2: c.1858 G>C; p.G620R) in exon 13 of the GRIN1 gene. These variants have not been previously reported in ExAC; although, Proband-2’s variant had been reported in one other individual.16 The residue lies within the second transmembrane segment (TM2) of the GluN1 protein and is conserved across several species (Figures 1c and d). The substitution of a large polar arginine in place of a small non-polar glycine has been predicted to alter protein structure and function.16
Functional analysis of mutant GluN1 receptor
NMDARs possessing wild-type or mutant GluN1-G620R subunits were co-expressed with wild-type GluN2A or GluN2B subunits. The electrophysiological properties of these complexes were compared across several pharmacological parameters. Half-maximal current responses (EC50) were determined by measuring the response to a range of glutamate and glycine concentrations (Figures 2a and b). GluN1-G620R/GluN2A NMDARs showed a 1.8-fold increase in the EC50 value (decreased potency) for glutamate (4.2 ± 0.48 μM vs 7.5 ± 0.40 μM) and a 1.9-fold increase in the EC50 value for glycine (1.0 ± 0.06 μM vs 1.9 ± 0.10 μM; Figures 2c and d; see Table 1). Agonist potency for GluN1-G620R/GluN2B was similarly decreased, with the glutamate EC50 increased by 2.9-fold (from 0.80 μM in wild type to 2.3 μM), and glycine EC50 increased by 1.8-fold (from 0.27 μM in wild type to 0.49 μM; Figures 2e and f; see Table 1). These data suggest that the GluN1-G620R mutation caused a significant decrease in the potency of glutamate and glycine in both GluN1-G620R/GluN2A and GluN1-G620R/GluN2B NMDARs.
Figure 2.
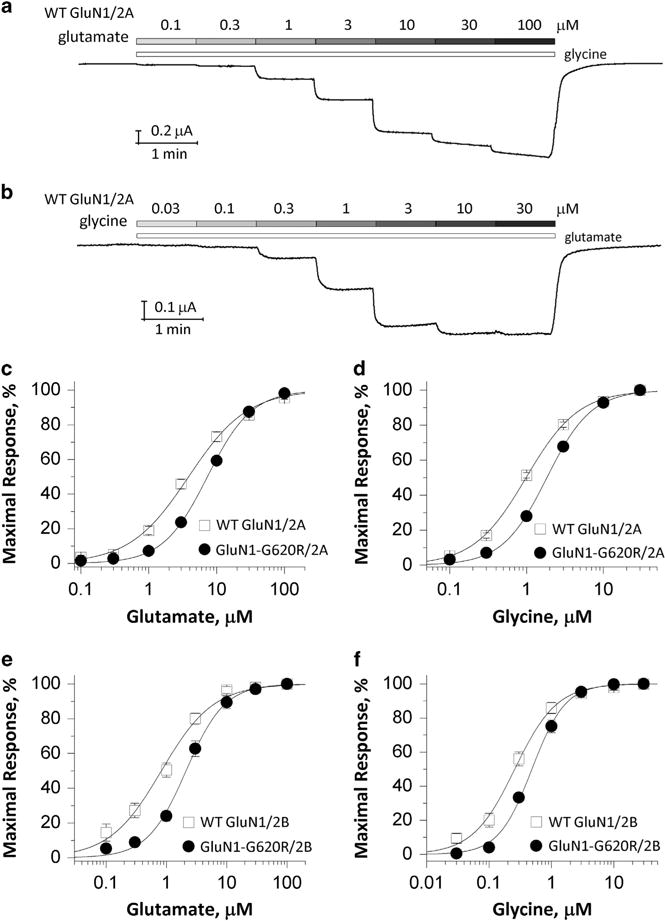
GluN1-G620R changes agonist potency. (a and b) Representative two-electrode voltage-clamp recordings obtained from oocytes expressing wild-type GluN1/GluN2A receptors in which the currents were evoked by increasing concentrations (μM) of glutamate (a, in the presence of 100 μM glycine) and glycine (b, in the presence of 100 μM glutamate) at the holding potential of − 40 mV. (c–f) Composite concentration-response curves determined by two-electrode voltage-clamp (TEVC) recordings from Xenopus oocytes are shown for wild-type GluN1 or GluN1-G620R co-expressed with GluN2A (c and d) or GluN2B (e and f). Glutamate (c and e) and glycine (d and f) concentration-effect curves showed that the GluN1-G620R mutant has a reduced agonist potency (increased EC50 values, see Table 1) for both agonists.
Table 1.
Summary of pharmacological data for GluN1-G620R
| wild-type GluN1/GluN2A | 1-G620R/GluN2A | wild-type GluN1/GluN2B | 1-G620R/GluN2B | |
|---|---|---|---|---|
| Glutamate, EC50, μM (n) | 4.2 ± 0.48 (16) | 7.5 ± 0.40 (13)a | 0.80 ± 0.15 (9) | 2.3 ± 0.30 (8)a |
| Glycine, EC50, μM (n) | 1.0 ± 0.06 (16) | 1.9 ± 0.10 (13)a | 0.27 ± 0.04 (6) | 0.49 ± 0.02 (5)a |
| Mg2+, IC50, μM (n)b | 25 ± 2.3 (10) | 446 ± 265 (13)a | 22 ± 2.5 (6) | >1000a |
| Proton, % (n)c | 47 ± 2.7 (12) | 25 ± 1.4 (11)a | 16 ± 1.2 (6) | 9.6 ± 0.29 (5)a |
| Zinc, IC50, nM (n) | 24 ± 2.6 (5) | 9.2 ± 0.28 (8)a | – | – |
| % inhibition by zincd | 53 ± 2.3% (5) | 77 ± 1.6% (8)a | – | – |
The data are expressed as mean ± s.e.m. (n).
P<0.01, compared to the corresponding wild type, unpaired t-test; agonist potency was compared using logEC50 or logIC50 values.
Holding potential of −60 mV.
Percentage current remaining at pH 6.8 compared with the pH 7.6.
At 300 nM of zinc.
For all comparisons, number of samples was chosen a priori to give power to detect a 40% change of 0.8.
We also evaluated the effect of GluN1-G620R on sensitivity of NMDARs to several endogenous extracellular negative modulators (Mg2+, Zn2+ and protons).1 When co-expressed with GluN2A, the GluN1-G620R showed a significant 18-fold increase in the IC50 values (decreased potency) for magnesium at a holding potential of −60 mV (25 ± 2.3 μM for wild-type GluN1/GluN2A2A vs 446 ± 26 uM for 1-G620R/2A; Figure 3a; see Table 1). Co-expression of GluN1-G620R with GluN2B strongly reduced Mg2+ inhibition to an extent such that we could not determine the IC50 value; we observed current responses that were 106 ± 7% (n = 12) of control in the presence of 1 mM extracellular Mg2+ (Figure 3b; see Table 1). The apparent potentiation on GluN1-G620R/GluN2B receptors by 1 mM Mg2+ might occur through extracellular Mg2+ acting at the spermine site,27 unmasked via the removal of Mg2+-induced voltage-dependent channel block by the GluN1-G620R mutation.
Figure 3.
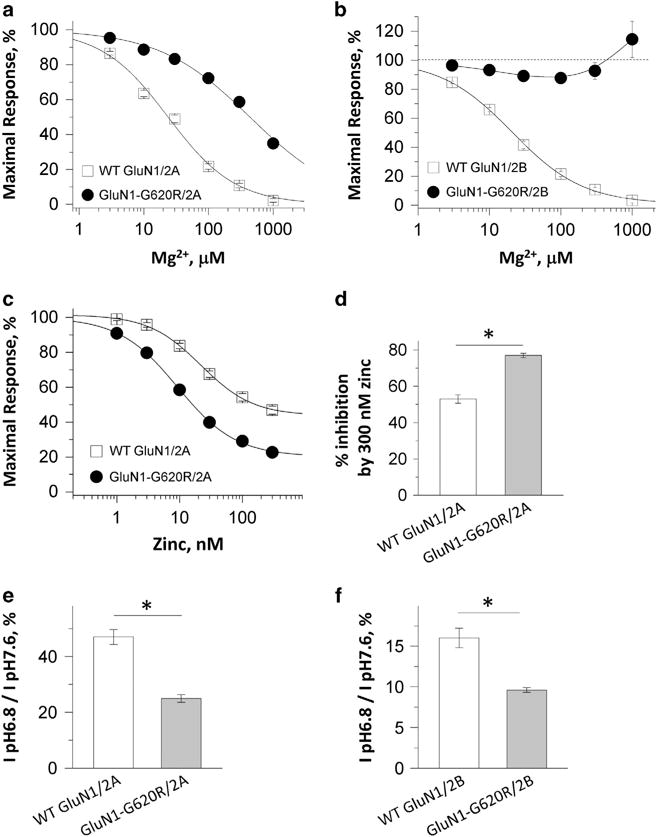
GluN1-G620R alters sensitivity to negative modulators. (a and b) Concentration-response curves for wild-type GluN1 and GluN1-G620R co-expressed with GluN2A (a) and GluN2B (b) receptors at holding potential −60 mV revealed decreased inhibition by extracellular Mg2+. (c and d) Composite inhibitory concentration-response curves for zinc at holding potential − 20 mV showing an enhanced inhibition by zinc, with a decreased IC50 value (c) and an increased percentage inhibition at 300 nM zinc (d). (e and f) Decreased percentage of current at pH 6.8 compared to the pH 7.6 in the mutant GluN1-G620R/2A receptors (e) and GluN1-G620R/2B receptors (f). *P<0.01, unpaired t-test.
When co-expressed with GluN2A, the GluN1-G620R mutation showed enhanced zinc sensitivity due to decreased IC50 values (24 ± 2.6 nM for wild-type GluN1/GluN2A vs 9.2 ± 0.28 nM for GluN1-G620R/GluN2A) and increased degree of inhibition at Zn2+ concentrations that saturate the high-affinity site (53 ± 2.3% inhibition for wild-type GluN1/GluN2A vs 77 ± 1.6% inhibition for GluN1-G620R/GluN2A; Figures 3c and d; see Table 1). The GluN1-G620R mutation showed enhanced proton sensitivity when co-expressed with GluN2A, with the current recorded at pH 6.8 as a percent of that recorded at 7.6 being 47 ± 2.3% for wild-type GluN1/GluN2A and 25 ± 1.4% for GluN1-G620R/GluN2A (Figure 3e; see Table 1). For GluN2B, the current measured at pH 6.8, as a percent of that recorded at pH 7.6, was 16 ± 1.2% for wild-type GluN1/GluN2B and 9.6 ± 0.29% for GluN1-G620R/GluN2B (Figure 3f; see Table 1). These results suggest that the GluN1-G620R substitution in the two probands had a mixed effect on the sensitivity to negative modulators, with reduced Mg2+ inhibition but enhanced zinc and proton inhibition.
GluN1-G620R mutant affects receptor trafficking and current response in mammalian cells
We assessed recombinant NMDAR protein expression and localization in transiently transfected HEK293 cells by measuring both total protein and surface localized protein by western blot. There was no significant difference in the ratio of surface-to-total protein expression of the mutant GluN1-G620R/GluN2A compared to wild-type GluN1/GluN2A (13 ± 15%; P = 0.43, paired t-test; Figures 4a and c). In contrast, the ratio of surface-to-total protein was significantly decreased by 60% when the mutant GluN1-G620R was co-expressed with GluN2B and compared to wild-type GluN1/GluN2B (P = 0.0001, paired t-test; Figures 4b and c). These data reveal that GluN1-G620R can disrupt the forward trafficking of some NMDAR subtypes. We also measured and compared the current response of the GluN1-G620R mutant to the wild-type receptors from transiently transfected HEK293 cells. Consistent with the trafficking data, cells expressing the G620R mutant and GluN2B showed a dramatic reduction in current amplitude (Figure 4e; GluN1-G620R/GluN2B: 2.0 ± 0.65 pA/pF, n = 6 vs wild-type GluN1/GluN2B: 71 ± 27 pA/pF, n = 5; P = 0.02, unpaired t-test) when co-expressed with GluN2B subunits. Surprisingly, the mutant also showed a significantly decreased current amplitude when co-expressed GluN2A subunit (Figure 4d; 1-G620R/GluN2A: 33 ± 8.3 pA/pF, n = 13 vs wild-type GluN1/GluN2A: 160 ± 42 pA/pF, n = 7; P = 0.001, unpaired t-test).
Figure 4.
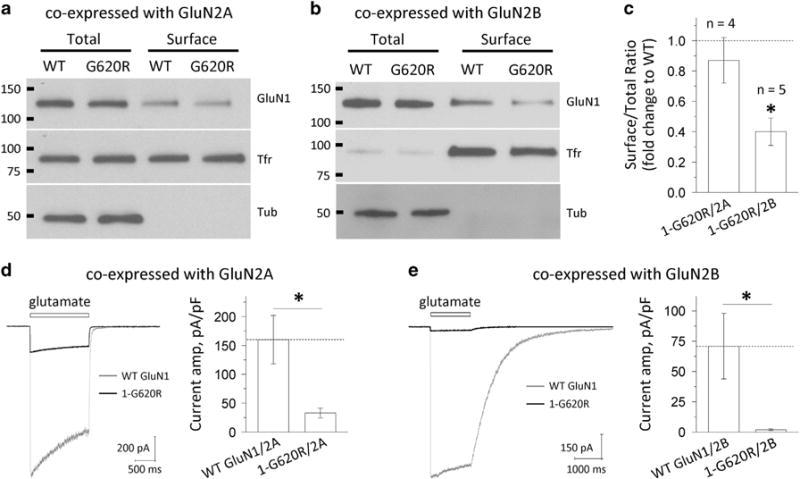
GluN1-G620R alters N-methyl-D-aspartate (NMDA) receptor surface expression and current response. (a and b) The surface proteins (wild-type GluN1 or GluN1-G620R) were expressed in HEK293 cells for 24 h, labeled with biotin and pulled down with avidin beads. Equivalent amounts of wild type and G620R protein samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE; GluN2A: 10 μl of total and surface; GluN2B: 5 μl of total and 30 μl of surface). The expression of NMDARs was assessed by western blotting for GluN1, transferrin receptor (TfR) and tubulin (Tub). Total GluN1 signals were normalized to tubulin signal, which served as a loading control. TfR served as a control to show relatively equal surface labeling. Representative blots are shown for HEK293 cells expressing wild-type GluN1/GluN2A and GluN1-G620R/GluN2A (a), and GluN1/GluN2B and GluN1-G620R/GluN2B (b). (c) Densitometry was used for chemiluminescence signal quantification, and the ratio of surface-to-total protein level of wild-type GluN1 was compared to GluN1-G620R by paired t-tests for GluN2A (P = 0.43, n = 4) and GluN2B (*P = 0.0001, n = 5). Mutant surface-to-total protein ratios were normalized to the wild type (dashed line) and plotted as mean ± s.e.m. (d and e) Current response was recorded by using the whole-cell voltage patch-clamp recordings from transiently transfected HEK293 cells at holding potential of −60 mV. The current amplitude normalized to cell capacitance was significant decreased in the GluN1-G620R mutant when co-expressed with GluN2A (d, 33 pA/pF vs 160 pA/pF for the wild type, n = 7, 13; *P = 0.001) or with GluN2B (e, 2.0 pA/pF vs 71 pA/pF for the wild type, n = 5, 6; *P = 0.02).
DISCUSSION
We report two individuals with de novo mutations in GRIN1 resulting in the G620R substitution. These individuals presented with intellectual disability, motor delays and abnormal stereotypical movements in the absence of seizures. De novo mutations in GRIN1 have recently been identified in several patients with non-syndromic intellectual disabilities, oftentimes associated with epilepsy.6,16,20,21 A 7-year-old boy had previously been reported with the same G620R substitution and similar phenotypic features (although dysmorphism was not reported),16 indicating this mutation may be associated with its own phenotypic presentation. We provide the first analysis of the expression, trafficking and function of the mutant GluN1-G620R subunits in the context of co-expression with either GluN2A or GluN2B. Our data suggest that the proband’s phenotype may have been influenced by the combination of altered protein subtype trafficking and abnormal electrophysiological activity.
Receptor trafficking experiments revealed that the GluN1-G620R mutant, when co-expressed with GluN2B, decreased the forward trafficking of NMDARs to the plasma membrane compared to wild type GluN1. In contrast, surface expression of GluN1-G620R complexed with GluN2A was only slightly decreased when compared to wild-type NMDARs. Because GluN2B is predominantly expressed during fetal development and the neonatal period, these data indicate that decreased trafficking of GluN1-G620R/GluN2B NMDARs may have a negative impact on embryonic neurodevelopment.
In addition to this trafficking defect, electrophysiological analyses indicated that this amino acid substitution also affected NMDAR ion channel activity. The GluN1-G620R variant lies within the second transmembrane (M2) domain associated with a reentrant loop that lines the ion channel pore. Changes in amino acid residues in this region have been shown to alter Mg2+ blockade and calcium permeability.28,29 Specifically, mutations at nearby asparagine residues have been shown to produce a weaker voltage-dependent block by magnesium and lower calcium permeability.28,30,31 These findings support the important role of the amino acid residues in M2 region for regulating flow of ions through NMDA receptors as well as the voltage-dependent block by extracellular Mg2+. Our functional analysis of GluN1-G620R showed a ~ 2-fold decrease in glutamate and glycine binding, and an 18-fold decrease in Mg2+ blockade in the context of GluN2A co-expression. Because GluN1-G620R/GluN2A complex is correctly trafficked to the plasma membrane, we assume its expression and localization at synapses is largely unchanged. Although the reduced Mg2+ inhibition will increase current at resting membrane potentials and will likely dominate at synaptic receptors activated by high concentrations of glutamate, it is also important to consider that its lower potency may also produce a briefer excitatory postsynaptic current (EPSC) time course. Our data showed significantly decreased current responses to a maximal concentration of agonists. At this time, we do not understand the basis of this discrepancy between the near normal receptor trafficking and the reduced current responses; further study with native cells/tissues and animal models is warranted.
In addition, the reduction in Mg2+ block will alter the voltage-dependent aspect of NMDA receptor-mediated currents, potentially interrupting cellular mechanisms of NMDA receptor-dependent synaptic plasticity.32,33 For receptors that contain GluN1-G620R complexed with GluN2B, it is difficult to predict the effects on receptor function. For instance, there will be a reduction in function due to the reduced delivery of protein to the cell surface. If the deficit in trafficking we observed persists in neurons, then this mutation could produce a hypofunction of GluN1-G620R/GluN2B receptor activity, which could play a role in these individuals’ poor developmental progress. Alternatively, when these receptors do reach the surface, they will be more active than WT receptors because there will be essentially no voltage-dependent Mg2+ block.
NMDAR function has been investigated in several transgenic mouse models. Grin1 hypomorphic mice that express GluN1 at 5–10% of normal levels were found to have increased stereotypy and social behaviors.34,35 Alternatively, mice with homozygous GluN1-D481N mutations had a 5-fold reduction in glycine potency and exhibited features including hyper-reactivity, noise sensitivity, stereotypies and self-injurious behaviors.36 Furthermore, another Grin1 mouse that possessed a homozygous R844C substitution was shown to have increased locomotor activity, decreased social interactions and startle responses, as well as abnormal anxiety-like behaviors.37,38 Although these hypofunctional models are not directly analogous to our heterozygous patients, they do suggest that GRIN1 may play a similar role in social and cognitive behaviors in both species.
To date, several patients have been reported with GRIN1 mutations associated with dysmorphic faces, intellectual disability and hyperkinetic movement disorder.16,21 We report two individuals with similar phenotypic presentations who had de novo GluN1-G620R mutations residing in TM2 of the GluN1 subunit. Our probands seem to be unique among patients with GRIN1 mutations due to their ability to ambulate, as well as their more extensive language capabilities. One other individual reported with the G620R substitution was noted to have a similar phenotype (intellectual disability without epilepsy).16 In that report, the GluN1-G620R mutation was thought to produce a ‘nonfunctional’ NMDAR, but was only tested when co-expressed with the GluN2B. This interpretation could have resulted from the trafficking defect that we have report here with GluN1-G620R/GluN2B receptors, since there was no direct electrophysiological evaluation of GluN1-G620R/GluN2A-containing receptors in that report.
Interestingly, the potential differential effects of this mutation on GluN2A- and GluN2B-containing NMDARs suggest that the impediments to these individual’s development might be multi-faceted with temporally distinct components. Embryonically, the probands may have suffered neurological deficits as a result of the decreased presence of GluN1-G620R/GluN2B complexes on the surface of neurons during brain development. After birth, the reduced current responses of both GluN1-G620R/GluN2A and GluN1-G620R/GluN2B (along with its reduced surface expression) complexes, coupled with diminished voltage-dependent block by Mg2+, may have compromised both cellular function, network processing and synaptic plasticity. These cases provide an example that a single amino acid substitution can produce a combination of factors that perturb NMDAR function in multiple ways, which together, can produce a profound genotype-specific phenotype.
Acknowledgments
We are grateful to Kelli Dejohn, Shantel Brown, Carmela Brito, Joanne Baez, Jing Zhang and Barrington Burnett for clinical and technical assistance, and critical analysis. We especially thank the families of our patients for the loving care for their children and cooperation with our work. During this work, WC was supported by the Xiangya-Emory Medical Schools Visiting Student Program; HY was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development, NIH-R01HD082373, by the Emory +Children’s Pediatric Center Seed Grant Program, by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454, by NIH HHSN268201400169P; SFT was supported by NIH-NINDS R01NS036654, R24NS092989 and R01NS092989. TMP and CS were funded by the Cedars-Sinai institutional funding program and the Cedars-Sinai Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
CONFLICT OF INTEREST
SFT is a consultant for Janssen Pharmaceuticals, NeurOp, Inc., and Pfizer Inc, and co-founder of NeurOp Inc. The remaining authors declare no conflict of interest.
Author contributions: SFT, SAS, HY and TMP designed the experiments and wrote the paper. WC, SAS, AT and HY performed the biological experiments and analyzed biological data. CS and TMP collected clinical information and data. CS, JMG, SMK, MT, MA, MM and TMP evaluated the patients, provided clinical assessments and whole-exome sequencing data. All authors discussed the results and implications and commented on the manuscript.
References
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McBain CJ, DiChiara TJ, Kauer JA. Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. J Neurosci. 1994;14:4433–4445. doi: 10.1523/JNEUROSCI.14-07-04433.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 2014;82:279–293. doi: 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 5.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 6.Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvill GL, Regan BM, Yendle SC, O’Roak BJ, Lozovaya N, Bruneau N, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45:1073–1076. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kingwell K. Epilepsy: GRIN2A mutations identified as key genetic drivers of epilepsy-aphasia spectrum disorders. Nat Rev Neurol. 2013;9:541. doi: 10.1038/nrneurol.2013.181. [DOI] [PubMed] [Google Scholar]
- 9.Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. 2013;45:1067–1072. doi: 10.1038/ng.2728. [DOI] [PubMed] [Google Scholar]
- 10.Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;45:1061–1066. doi: 10.1038/ng.2726. [DOI] [PubMed] [Google Scholar]
- 11.Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, et al. GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol. 2014;1:190–198. doi: 10.1002/acn3.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan H, Hansen KB, Zhang J, Pierson TM, Markello TC, Fajardo KV, et al. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat Commun. 2014;5:3251. doi: 10.1038/ncomms4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burnashev N, Szepetowski P. NMDA receptor subunit mutations in neurodevelopmental disorders. Curr Opin Pharmacol. 2015;20:73–82. doi: 10.1016/j.coph.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol. 2015;88:203–217. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu W, Mayer S, Yuan H, Traynelis SF. Human GRIN2B mutations in neurodevelopmental disorders. J Pharmacol Sci. 2016;132:115–121. doi: 10.1016/j.jphs.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemke JR, Geider K, Helbig KL, Heyne HO, Schutz H, Hentschel J, et al. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology. 2016;86:2171–2178. doi: 10.1212/WNL.0000000000002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, et al. GRIN2D recurrent de novo mutation is an autosomal dominant cause of severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet. 2016;S0002-9297:30287–30287. doi: 10.1016/j.ajhg.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, Strong KL, et al. Mechanistic insight into NMDA receptor dysregulation by disease-associated rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet. 2016;99:1261–1280. doi: 10.1016/j.ajhg.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lesca G, Rudolf G, Labalme A, Hirsch E, Arzimanoglou A, Genton P, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. 2012;53:1526–1538. doi: 10.1111/j.1528-1167.2012.03559.x. [DOI] [PubMed] [Google Scholar]
- 20.Epi KC, Epilepsy Phenome/Genome, P. Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohba C, Shiina M, Tohyama J, Haginoya K, Lerman-Sagie T, Okamoto N, et al. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia. 2015;56:841–848. doi: 10.1111/epi.12987. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet. 2015;97:457–464. doi: 10.1016/j.ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med. 2016;18:678–685. doi: 10.1038/gim.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jorgensen L, Vance KM, et al. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Mol Pharmacol. 2013;84:114–127. doi: 10.1124/mol.113.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swanger SA, He YA, Richter JD, Bassell GJ. Dendritic GluN2A synthesis mediates activity-induced NMDA receptor insertion. J Neurosci. 2013;33:8898–8908. doi: 10.1523/JNEUROSCI.0289-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paoletti P, Neyton J, Ascher P. Glycine-independent and subunit-specific potentiation of NMDA responses by extracellular Mg2+ Neuron. 1995;15:1109–1120. doi: 10.1016/0896-6273(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 28.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 29.Mori H, Masaki H, Yamakura T, Mishina M. Identification by mutagenesis of a Mg (2+)-block site of the NMDA receptor channel. Nature. 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- 30.Sharma G, Stevens CF. Interactions between two divalent ion binding sites in N-methyl-D-aspartate receptor channels. Proc Natl Acad Sci USA. 1996;93:14170–14175. doi: 10.1073/pnas.93.24.14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma G, Stevens CF. A mutation that alters magnesium block of N-methyl-D-aspartate receptor channels. Proc Natl Acad Sci USA. 1996;93:9259–9263. doi: 10.1073/pnas.93.17.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato N, Yoshimura H. Reduced Mg2+ block of N-methyl-D-aspartate receptor-mediated synaptic potentials in developing visual cortex. Proc Natl Acad Sci USA. 1993;90:7114–7118. doi: 10.1073/pnas.90.15.7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 35.Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, et al. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav. 2009;8:661–675. doi: 10.1111/j.1601-183X.2009.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballard TM, Pauly-Evers M, Higgins GA, Ouagazzal AM, Mutel V, Borroni E, et al. Severe impairment of NMDA receptor function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J Neurosci. 2002;22:6713–6723. doi: 10.1523/JNEUROSCI.22-15-06713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuse T, Wada Y, Hattori K, Yamada I, Kushida T, Shibukawa Y, et al. Phenotypic characterization of a new Grin1 mutant mouse generated by ENU mutagenesis. Eur J Neurosci. 2010;31:1281–1291. doi: 10.1111/j.1460-9568.2010.07164.x. [DOI] [PubMed] [Google Scholar]
- 38.Umemori J, Takao K, Koshimizu H, Hattori S, Furuse T, Wakana S, et al. ENU-mutagenesis mice with a non-synonymous mutation in Grin1 exhibit abnormal anxiety-like behaviors, impaired fear memory, and decreased acoustic startle response. BMC Res Notes. 2013;6:203. doi: 10.1186/1756-0500-6-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]