

Abstract
A single ruthenium complex catalyzes two discrete transformations resulting in the net conversion of an acetylenic pyrrole and alcohols to products of carbonyl anti-(α-amino)allylation. An initial catalytic process enables isomerization of an alkyne to a kinetically more reactive allene. A second catalytic process promotes alcohol-to-allene hydrogen transfer to form an aldehyde-allylruthenium pair that engages in regio- and diastereoselective carbonyl addition. A related reductive coupling conducted of aldehydes mediated by 2-propanol also is described. The present catalytic processes represent rare examples of the use of alkynes as nucleophilic allylmetal precursors.
Graphical abstract
Classical methods for carbonyl and imine addition rely on the use of preformed organometallic reagents.1 Merging the characteristics of transfer hydrogenation and carbonyl addition, we have developed a broad, new family of catalytic C-C couplings that directly convert lower alcohols to higher alcohols in the absence of stoichiometric metals.2 In these processes, π-unsaturated reactants serve as equivalents to non-stabilized carbanions. For example, while molar quantities of allylmetal reagents are typically exploited in carbonyl additions to form homoallylic alcohols,3 we find that such products are accessible through the catalytic coupling of primary alcohols (or methanol4f,j) with allenes.4 In these processes, alcohol dehydrogenation triggers allene hydrometalation to generate aldehyde-allylmetal pairs, which upon carbonyl addition and subsequent protonolysis of the resulting ruthenium alkoxide delivers the homoallylic alcohols. The net transformation – a formal insertion of the more substituted allene π-bond into the carbinol C-H bond – is byproduct-free.
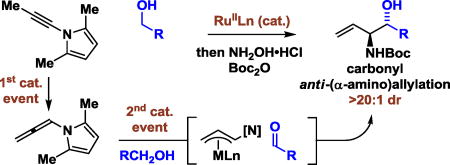
Based on this concept, an alternative to classical asymmetric carbonyl (α-amino)allylations mediated by (aminoallyl)diisopinocampheylboranes was sought (Figure 1).5 Using N-substituted-allenes as allyl donors, carbonyl anti-(α-amino)allylation could be achieved under the conditions of ruthenium catalysis via 2-propanol mediated reductive coupling6a or through redox-neutral coupling to primary alcohols.6b Good yields and anti-diastereoselectivities were observed, but the allenamide required both p-nitrobenzenesulfonyl and 2,4-dimethoxybenzyl protecting groups at nitrogen, which impeded elaboration of the coupling products. A potentially more desirable allyl donor is found in N-allenyl-2,5-dimethylpyrrole iso-1, as the pyrrole moiety is readily converted to the primary amine upon treatment with hydroxylamine hydrochloride.7 However, N-allenyl-2,5-dimethylpyrrole iso-1 did not engage in efficient transfer hydrogenative carbonyl addition due to its thermal instability and high kinetic reactivity under the conditions of ruthenium catalysis.
Figure 1.

Classical carbonyl (α-amino)allylation and ruthenium tandem-catalysis for carbonyl anti-(α-amino)allylation via alkyne-alcohol hydrogen auto-transfer.
Recently, we found that alkynes may serve as a reservoir for allenes, which are kinetically more reactive, under the conditions of ruthenium catalysis, enabling transfer hydrogenative coupling to form homoallylic alcohols.8,9,10 These processes represent examples of “tandem catalysis,” as a single metal complex mediates two discrete catalytic events: alkyne-to-allene isomerization and allene-carbonyl C-C coupling via hydrogen auto-transfer.11 Consequently, given the instability of allene iso-1 to the conditions of transfer hydrogenative coupling, the possibility of exploiting the corresponding acetylenic compound N-propynyl-2,5- dimethylpyrrole 1 as a latent form of allene iso-1 was explored (Figure 1). In fulfillment of this objective, we herewith report the diastereoselective ruthenium catalyzed coupling of acetylenic pyrrole 1 with diverse primary alcohols 2a–2u to form products of carbonyl anti-(α-amino)allylation 3a–3u and related 2-propanol mediated aldehyde reductive couplings.
In an initial set of experiments, para-bromobenzyl alcohol 2a (100 mol %) was exposed to acetylenic pyrrole 1 (300 mol %) in the presence of HClRu(CO)(PPh3)3 (5 mol %) and various chelating bis(phosphine) ligands (5 mol %). As previously observed in certain ruthenium catalyzed allene-alcohol C-C couplings,[4e,6b] the ruthenium complex modified by dippf, bis(diisopropylphosphino)ferrocene, was uniquely effective. By simply exposing acetylenic pyrrole 1 and para-bromobenzyl alcohol 2a to the dippf-modified ruthenium catalyst at 125 °C in dioxane solvent (0.5 M), the desired product of carbonyl (α-amino)allylation 3a was formed with complete anti-diastereoselectivity in 83% yield after isolation by silica gel chromatography. On 1 mmol scale, a 74% yield of 3a was obtained. These conditions were applied to the coupling of acetylenic pyrrole 1 with diverse primary alcohols 2a–2u (Scheme 1). Benzylic alcohols 2a–2h and heterobenzylic alcohols 2i–2m were converted to the respective products of carbonyl anti-(α-amino)allylation 3a–3m. Allylic alcohols 2n and 2o and aliphatic alcohols 2p–2u also could be transformed to the protected vicinal amino alcohols 3n–3u, respectively. For adducts 3a–3u, complete anti-diastereoselectivity was observed in each case. The syn-diastereomer was not observed upon 1H NMR analysis of the crude reaction product in the formation of 3a on 1 mmol scale. The principal side reaction, which was observed in the formation of 3i, 3k and 3n, involved alcohol dehydrogenation-olefin isomerization to form the corresponding α,β-unsaturated ketones. However, such over-oxidation could be suppressed upon introduction of 2-propanol. Finally, beyond redox-neutral coupling from the alcohol oxidation level, carbonyl anti-(α-amino)allylation also can be achieved through 2-propanol-mediated reductive coupling (eq 1).
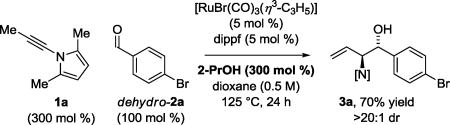 |
(1) |
Scheme 1.

Ruthenium catalyzed C-C coupling of alcohols 2a–2u with acetylenic pyrrole 1 to form products of carbonyl anti-(α-amino)allylation 3a–3u.a
aYields are of material isolated by silica gel chromatography. b2-PrOH (200 mol %). c48h. ddioxane (1 M). See Supporting Information for further experimental details.
Having developed efficient conditions for ruthenium catalyzed carbonyl anti-(α-amino)allylation of alcohols 2a–2u, deprotection of the 2,5-dimethylpyrrole moiety7 was explored for representative adducts 3b, 3n and 3q derived from aromatic, allylic and aliphatic alcohols 2b, 2n and 2q, respectively (Scheme 2). In the event, exposure of 3b, 3n and 3q to hydroxylamine hydrochloride in the aqueous ethanol at 100 °C followed by Boc-protection of the resulting primary amine, delivered the corresponding N-Boc-protected anti-vicinal amino alcohols 4b, 4n and 4q in moderate to good yield after isolation by silica gel chromatography. Finally, to illustrate the utility of these compounds as building blocks in chemical synthesis, Boc-protected amino alcohol 4b was converted to the nonproteinogenic α-amino-β-hydroxy amino ester 5b via modified Johnson-Lemieux conditions for alkene oxidative cleavage12 followed by treatment with TMS-diazomethane (eq 2).
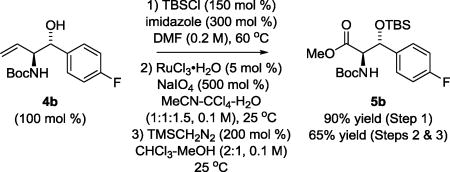 |
(2) |
Scheme 2.

Deprotection of adducts 3b, 3n, and 3q and conversion to corresponding N-Boc-protected amino alcohols 4b, 4n, and 4q.a
aCited yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
To gain insight into the catalytic mechanism, the following competition experiment was performed (Scheme 3). Acetylenic pyrrole 1 was exposed to equimolar quantities of alcohol 2a and aldehyde dehydro-2d under standard conditions employing the ruthenium catalyst generated in situ from HClRu(CO)(PPh3)3 and dippf at 125 °C in dioxane solvent (0.5 M). The C-C coupling products 3a and 3d were produced in a roughly 3:1 ratio. Under the same conditions, but inverting oxidation level by employing equimolar quantities of aldehyde dehydro-2a and alcohol 2d, a nearly identical product ratio of coupling products 3a and 3d was observed. These data are consistent with rapid, reversible primary alcohol dehydrogenation in advance of turnover-limiting carbonyl addition.13
Scheme 3.

Competition experiments corroborating rapid, reversible dehydrogenation with respect to C-C coupling.a
aCited yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
In summary, we report diastereoselective ruthenium catalyzed couplings of acetylenic pyrrole 1 with primary alcohols 2a–2u to form products of carbonyl anti-(α-amino)allylation 3a–3u and related 2-propanol-mediated reductive couplings. These processes occur by way of two discrete catalytic events: alkyne-to-allene isomerization and allene-carbonyl C-C coupling via hydrogen auto-transfer. As demonstrated by formation of N-Boc-protected amino alcohols 4b, 4n and 4q, deprotection of the 2,5-dimethylpyrrole moiety occurs readily upon exposure to hydroxylamine hydrochloride in the aqueous ethanol.7 These studies along with prior work from our laboratory demonstrate that reactions traditionally employing stoichiometric carbanions may now be conducted catalytically in the absence of stoichiometric metals via alcohol-mediated hydrogen transfer.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445) and the UT Austin Center for Green Chemistry and Catalysis for partial support of this research.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). Single crystal X-ray diffraction data for compound 4b. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Knochel P, Molander GA. Comprehensive Organic Synthesis. 2. 1 and 2 Elsevier; Oxford: 2014. [Google Scholar]
- 2.For recent reviews, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew. Chem. Int. Ed. 2014;53:9142. doi: 10.1002/anie.201403873.Dechert-Schmitt A-MR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat. Prod. Rep. 2014;31:504. doi: 10.1039/c3np70076c.Perez F, Oda S, Geary LM, Krische MJ. Top. Curr. Chem. 2016;374:365. doi: 10.1007/s41061-016-0028-0.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ. Science. 2016;354:aah5133. doi: 10.1126/science.aah5133.
- 3.For selected reviews on enantioselective carbonyl allylation, see: Ramachandran PV. Aldrichim. Acta. 2002;35:23.Denmark SE, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu C-M, Youn J, Jung H-K. Bull. Korean Chem. Soc. 2006;27:463.Marek I, Sklute G. Chem. Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.Lachance H, Hall DG. Org. React. 2008;73:1.Yus M, Gonzalez-Gomez JC, Foubelo F. Chem. Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 4.(a) Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]; (b) Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054. doi: 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Han SB, Kim IS, Han H, Krische MJ. J. Am. Chem. Soc. 2009;131:6916. doi: 10.1021/ja902437k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zbieg JR, McInturff EL, Leung JC, Krische MJ. J. Am. Chem. Soc. 2011;133:1141. doi: 10.1021/ja1104156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Moran J, Preetz A, Mesch RA, Krische MJ. Nature Chem. 2011;3:287. doi: 10.1038/nchem.1001. [DOI] [PubMed] [Google Scholar]; (g) Sam B, Montgomery TP, Krische MJ. Org. Lett. 2013;15:3790. doi: 10.1021/ol401771a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sam B, Luong T, Krische MJ. Angew. Chem. Int. Ed. 2015;54:5465. doi: 10.1002/anie.201500238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Oda S, Sam B, Krische MJ. Angew. Chem. Int. Ed. 2015;54:8525. doi: 10.1002/anie.201503250. [DOI] [PubMed] [Google Scholar]; (j) Holmes MT, Nguyen KD, Schwartz LA, Luong T, Krische MJ. J. Am. Chem. Soc. 2017;139:8114. doi: 10.1021/jacs.7b04374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Barrett AGM, Seefeld MA. J. Chem. Soc. Chem. Commun. 1993:339. [Google Scholar]; (b) Barrett AGM, Seefeld MA. Tetrahedron. 1993;49:7857. [Google Scholar]; (c) Barrett AGM, Seefeld MA, Williams DJ. J. Chem. Soc. Chem. Commun. 1994:1053. [Google Scholar]; (d) Barrett AGM, Seefeld MA, White AJP, Williams DJ. J. Org. Chem. 1996;61:2677. doi: 10.1021/jo9522062. [DOI] [PubMed] [Google Scholar]
- 6.(a) Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054. doi: 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR, McInturff EL, Krische MJ. Org. Lett. 2010;12:2514. doi: 10.1021/ol1007235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walia A, Kang S, Silverman RB. J. Org. Chem. 2013;78:10931. doi: 10.1021/jo401778e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For alkyne mediated carbonyl allylation via tandem ruthenium catalysis, see: Park BY, Nguyen KD, Chaulagain MR, Komanduri V, Krische MJ. J. Am. Chem. Soc. 2014;136:11902. doi: 10.1021/ja505962w.Liang T, Nguyen KD, Zhang W, Krische MJ. J. Am. Chem. Soc. 2015;137:3161. doi: 10.1021/jacs.5b00747.Liang T, Zhang W, Chen T-Y, Nguyen KD, Krische MJ. J. Am. Chem. Soc. 2015;137:13066. doi: 10.1021/jacs.5b08019.
- 9.For alkyne mediated carbonyl allylation via tandem iridium catalysis, see: Obora Y, Hatanaka S, Ishii Y. Org. Lett. 2009;11:3510. doi: 10.1021/ol901366q.Obora Y, Sawaguchi T, Tsubakimoto K, Yoshida H, Ogawa S, Hatanaka S. Synthesis. 2013:2115.Liang T, Zhang W, Krische MJ. J. Am. Chem. Soc. 2015;137:16024. doi: 10.1021/jacs.5b12131.
- 10.For a review on the use of alkynes as electrophilic or nucleophilic allylmetal precursors in transition metal catalysis, see: Haydl AM, Breit B, Liang T, Krische MJ. Angew. Chem. Int. Ed. 2017;56 doi: 10.1002/anie.201704248.
- 11.For reviews on hydrogen-auto transfer, see: Guillena G, Ramón DJ, Yus M. Angew. Chem. Int. Ed. 2007;46:2358. doi: 10.1002/anie.200603794.Hamid MHSA, Slatford PA, Williams JMJ. Adv. Synth. Catal. 2007;349:1555.Nixon TD, Whittlesey MK, Williams JMJ. Dalton Trans. 2009:753. doi: 10.1039/b813383b.Guillena G, Ramón DJ, Yus M. Chem. Rev. 2010;110:1611. doi: 10.1021/cr9002159.Quintard A, Rodriguez J. Chem. Comm. 2016:10456. doi: 10.1039/c6cc03486a.Quintard A, Rodriguez J. ChemSusChem. 2016;9:28. doi: 10.1002/cssc.201501460.
- 12.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]
- 13.For a related competition experiment, see reference 4e.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.