

 The discovery of a novel potent p53-MDM2 antagonistic small molecule scaffold using a mix of computational, chemistry and biophysical techniques.
The discovery of a novel potent p53-MDM2 antagonistic small molecule scaffold using a mix of computational, chemistry and biophysical techniques.
Abstract
Using the pharmacophore-based virtual screening platform ANCHOR.QUERY, we morphed our recently described Ugi-4CR scaffold towards a β-lactam scaffold with potent p53-MDM2 antagonizing activities. 2D-HSQC and FP measurements confirm potent MDM2 binding. Molecular modeling studies were used to understand the observed SAR in the β-lactam series.
Introduction
Changing the central scaffold of a small molecule while keeping the key receptor interactions in place is called scaffold hopping and is a very important technique in medicinal chemistry.1,2 Scaffold hopping can yield improved PKPD properties, affinity and selectivity, or intellectual property. We have recently introduced the pharmacophore-based virtual screening platform ANCHOR.QUERY which allows for the fast and efficient screening of very large chemical libraries based on multicomponent reaction chemistries (MCRs).3,4 It is particularly useful to search for protein–protein interactions (PPI), where a deeply buried amino acid anchor motif comprises an energetic hot spot. Such an anchor motif is the Trp23 in the triad Phe19-Trp23-Leu26 present in p53-MDM2 PPI.5 Of the three amino acids of the triad, the central Trp23 is buried most deeply into the MDM2 receptor. Feeding Trp23 as the anchor motif in ANCHOR.QUERY yielded several scaffolds with nM affinity to MDM2.3,4,6–10 A key feature of the anchor approach is the ease of synthesis of predicted binders, typically by one or two chemical steps employing MCR chemistries.11,12
Recently, using this approach, we have discovered a potent Ugi-4CR-based molecule (1) which opens up an additional pocket in MDM2, beyond the well-known three finger pharmacophore model.7,13 Amongst all described,14 this small molecule is unique as it targets and stabilizes the intrinsically disordered N-terminus of MDM2. To explore this binding mode even more, we applied ANCHOR.QUERY as a scaffold hopping tool to discover a novel class of p53-MDM2 antagonists based on a β-lactam scaffold.
Results and discussion
Virtual screening (VS)-based β-lactam scaffold discovery
Scaffolds are often defined by topological computational approaches, e.g. Bemis and Murcko (BM scaffolds).15 Here however, we will use a chemistry-based definition which allows an intuitive immediate application, our recently introduced scaffold definition, “as the smallest atomic denominator and its connectivity resulting from a reaction or reaction sequence using starting materials with common functional groups.”16
The recently found Ugi-4CR scaffold with an experimentally solved cocrystal structure between MDM2 and a potent derivative 1 (PDB ID ; 4MDN) was the starting point of the discovery and development of the current inhibitors.7 β-aminoacylamide 1 (Fig. 1B) binds with a Ki of 600 nM to the MDM2 receptor. Fig. 1A shows the main characteristics of this model. 6-chloroindole-2-carboxylic acid was used as an anchor-building block in order to mimic the Trp23 amino acid. Three additional binding sites Phe19, Leu26 and the induced Leu26 subpocket, enlarged by the Tyr100 ‘open’ position, were filled by tert-butyl, benzyl and p-chlorobenzyl substituents, respectively. Using our open access pharmacophore-based virtual screening web-platform ANCHOR.QUERY (; http://anchorquery.csb.pitt.edu), the aforementioned model was analyzed (Fig. 1A).4 A 4-point pharmacophore point model is proposed by ANCHOR.QUERY (Fig. 1D). The importance of the different parts of the molecule for binding, especially the p-chlorobenzyl groups, was confirmed by the small-network analysis program SCORPION (Fig. 1C).17 Thus, we used the pharmacophore model, including the TRP anchor, two aromatases and a hydrophobe, to query a >30 million virtual compound library based on different MCR scaffolds. An interesting feature of ANCHOR.QUERY is the analysis of the results based on the occurrence of a particular scaffold in the hit list (enrichment factors).
Fig. 1. Process of scaffold hopping using ANCHOR.QUERY. A: Cocrystal structure (PDB ID 4MDN) of 1 (cyan sticks) in MDM2 aligned with the p53-MDM2 structure (PDB ID ; 1YCR) and showing the only p53 hot spot FYL as pink sticks. B: 2D structure of 1. C: Scores by the small-network analysis program SCORPION. D: Pharmacophore model of 1 in ANCHOR.QUERY (anchor: yellow, aromatase: pink, hydrophobe: green). E: Top β-lactam result of the pharmacophore search based on 1 with the 2D structure inserted.

Using this particular pharmacophore model, the observed order of enrichment of scaffolds is shown in Fig. 2. According to occurrence, the top scaffold is the Ugi-β-lactam, followed by the Ugi-tetrazole-3CR scaffold, Ugi-hydantoin, U-4CR,18 Castagnolli-3CR,19,20 Doebner-3CR21 and van Leusen,22 which are the highest scoring amongst the 27 MCR scaffold space populated with >30 million individual compounds. Several potent MDM2 binders based on these scaffolds and cocrystallized with the MDM2 receptor have been described by us and others in the past.3,6,7,23 A high-ranking β-lactam compound after energy minimization is shown in Fig. 1E. Based on the promising results of the pharmacophore search, we embarked to synthesize several proposed compounds and derivatives based on the unprecedented Ugi-β-lactam 3CR and investigated their binding behavior to the MDM2/X receptor.
Fig. 2. ANCHOR.QUERY derived scaffold enrichment factors using 1 (Fig. 1D) as a query. The top scoring scaffold is the yellow boxed Ugi-β-lactam 3CR. For the last two scaffolds, several cocrystal structures in MDM2 and MDMX have been published in the past.

Chemistry
The designed scaffold 2 could be easily obtained by a union of two MCRs: α-aminoalkylation to produce the required substituted β-amino acids and Ugi-β-lactam reaction (Scheme 1).24 The Ugi-β-lactam reaction incorporates the anchoring 3-indolecarboxaldehyde 4, aliphatic isocyanides 5 and suitably substituted β-aminoacids 6. Aldehyde 4 was synthesized from the 6-chloro-indole derivative using the Vilsmeier–Haack formylation reaction.3,6 For the preliminary SAR analysis of the Leu26 pocket and the induced pockets, we synthesized three different substituted β-aminoacids 10a–c by simply refluxing a mixture of substituted benzaldehydes 7a–c, malonic acid and ammonium acetate (Scheme 2).25 The choice of the starting materials was based on our previous results on designing inhibitors of the p53-MDM2/X interaction.26,27 Concerning the Phe19 pocket, we utilized bulky aliphatic isocyanides, such as the tert-butyl (5a) and the 1-adamantyl isocyanide (5b), which were prepared by the classic dehydration of the corresponding formamide using POCl3.
Scheme 1. Retrosynthesis of the targeted β-lactams 2 based on union of two MCRs: 3-component α-aminoalkylation and Ugi-β-lactam reaction.

Scheme 2. Synthesis of the substituted β-aminoacids by refluxing the substituted benzaldehydes 7a–c, malonic acid and ammonium acetate.

Next, we proceeded with the Ugi-β-lactam reaction and the subsequent hydrolysis in total yields of 41–64%. Thus, a mixture of the substituted aminoacids 10a–c, the 6-chloro-indole carboxaldehyde 4 and the isocyanides 5a and b in trifluoroethanol was irradiated in a microwave oven at 130 °C for 90 min. As expected, we obtained both diastereomers of the desired adducts 3a–e in a d.r. of ∼1 : 1.
In order to better understand the affinity of the final compounds, separation of the diastereomers was necessary. Thus, after careful chromatographic separation, we were able to isolate both diastereomers which were hydrolyzed to the corresponding acids 2a–e. The hydrolysis of the corresponding esters is necessary based on previous experience with other scaffolds. Esters show an always worse affinity to MDM2 compared to the free carboxylic acid by a factor of 5 or more.3,7,28–30 To investigate if the chloroindole anchor can be replaced by a p-halo phenyl group and following the same synthetic pathway, we also replaced the 6-chloro-indole with a 4-chloro-phenyl group, yielding compounds 2f and g (Scheme 3).
Scheme 3. General reaction scheme and the synthesized library of β-lactams along with the final yields (Ugi-β-lactam and hydrolysis reactions).

Biophysical screening and structure–activity relationship studies
Two complementary assays based on independent physicochemical principles, fluorescence polarization (FP) and HMQC NMR, were used to exclude false positive hits. Fluorescence polarization (FP) assay was employed to determine the inhibitory affinities (Ki) of tetrazole derivatives against MDM2 and MDMX, as previously described.31,32 The results are presented in Table 1. To our delight, our designed and synthesized lactams were active against both MDM2 and MDMX, and in some cases, even below 1 μM (entries 3–5). As expected, in most of the cases, one diastereomer showed better affinity than the other one and this was, in all cases, diastereomer B. The configuration of the two different diastereomers could be assigned based on the two protons on the chiral atoms by 1H NMR. Thus, in the case of diastereomer A, the proton on the lactam ring adjacent to nitrogen (H-19) appears at approximately δ 4.40–4.42, compared with diastereomer B in which the H-19 appears higher at δ 4.68–4.83. Moreover, the proton on the chiral carbon adjacent to the amide (H-11) shows resonances at δ 6.20–6.24 and 6.45–6.48 for diastereomers A and B, respectively. Para-Substituted phenyl lactams gave slightly better results compared with the meta-substituted ones and bulkier isocyanides seem to also contribute to better affinities. Compounds 2c–2e showed similar activity, from which one diastereomer of lactam 2e demonstrated an affinity of 200 nM. Compounds 2a, 2c and 2d showed notable activity against MDMX as well, an interesting feature that most of the current inhibitors fail to exhibit.5,26 Compounds 2f and 2g with different anchors did not show any activity.
Table 1. Activities of the synthesized library of β-lactam-based inhibitors of p53-MDM2/X interaction.
| Entry | Compound | Structure |
K
i [μM] |
|||
| MDM2 |
MDMX |
|||||
| Diastereomer A | Diastereomer B | Diastereomer A | Diastereomer B | |||
| 1 | 2a |
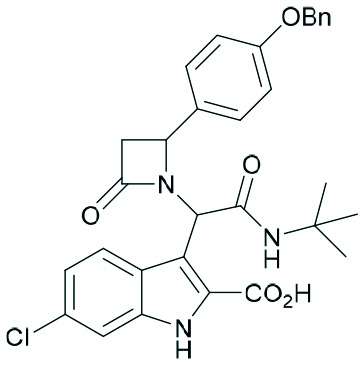
|
3.2 | 1.2 | 12.8 | 2.1 |
| 2 | 2b |
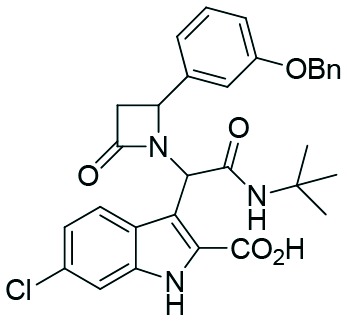
|
2.1 | 1.9 | 2.6 | 6.7 |
| 3 | 2c |
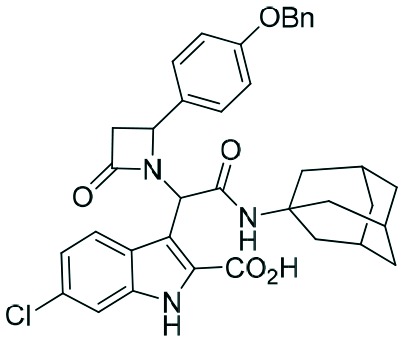
|
1.9 | 0.4 | 4.7 | 1.4 |
| 4 | 2d |
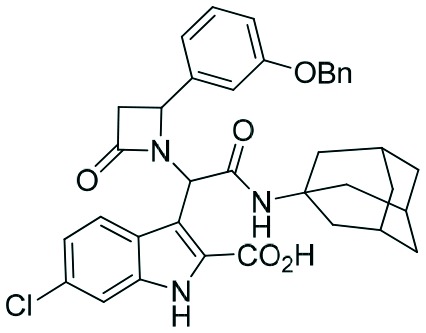
|
0.9 | 0.6 | 2.2 | 2.6 |
| 5 | 2e |
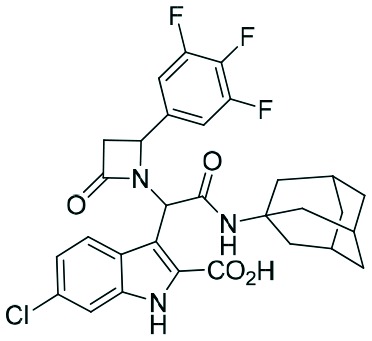
|
2.0 | 0.2 | — | 4.2 |
| 6 | 2f |
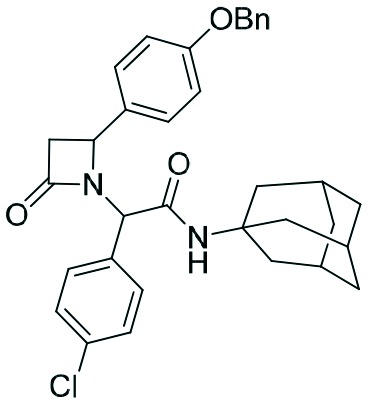
|
n.a. | n.a. | n.a. | n.a. |
| 7 | 2g |
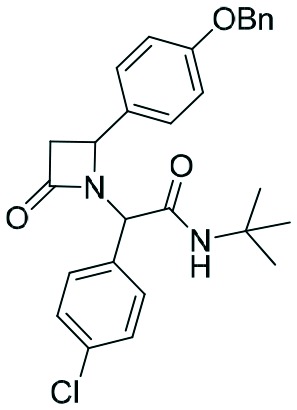
|
n.a. | n.a. | n.a. | n.a. |
FP-based screening of protein–protein interactions often gives a high fraction of false positives especially with hydrophobic molecules, and therefore it is highly advisable to run a second orthogonal biophysical assay. As a second orthogonal screening system, we performed 1H-15N Heteronuclear Multiple Quantum Correlation (HMQC) NMR experiments.33,34
This method is based on the monitoring of chemical shift changes in protein amide backbone resonances upon protein interaction with a small molecule. It allows not only qualitative evaluation of evidence of interaction but also semi-quantitative estimation of the binding affinity. For this NMR experiment, the uniformly 15N-labeled MDM2 was titrated with increasing concentration of the evaluated compound and 1H-15N HMQC spectra were recorded after each new portion of the compound has been added. In the course of the titration, shifts of the cross-peaks assigned to the amino acid affected by the binding of the small molecule, e.g. Gly58 of MDM2, are observed. Moreover, the strong binding of the compound to the target protein results in NMR signal doubling, indicating Kd values of less than 1 μM (and a slow chemical exchange). Such results were observed in the spectra of 15N-labeled MDM2 titrated with compound 2e (Fig. 3). In the titration step where the molar ratio of protein : ligand was 5 : 1, the resonance peak doubling was visible (the first signal from the doubled peak remained in the position of the reference peak, while the second – shifted to another position; inset in Fig. 3). This observation is a confirmation of the tight binding of this inhibitor to MDM2 (Kd < 1 μM). When the protein : ligand ratio reached 1 : 1, the peak shifted to the position of the second split signal. Assignment of the amide groups of MDM2 was obtained from Stoll et al.35
Fig. 3. 1H-15N HMQC titration of MDM2 with 2e. Blue: reference apo-MDM2 alone; green: the molar ratio of protein/ligand is 5 : 1; red: the molar ratio of protein/ligand is 1 : 1. The enlarged fragment shows resonance peak doubling.

Modeling
Despite extensive trials, we were not able to grow crystals of more affine compounds 2c, d, and e with MDM2. For that reason, we tried to rationalize the affinity findings using computational modeling using MOLOC36 and SCORPION.17 The most active compound of the β-lactam series is 2e which incorporates a simple trifluoro-substituted benzene moiety. It thus resembles the previously described α-aminoacylamide 11.6 Therefore, we used the corresponding cocrystal structure to model 2e using MOLOC (Fig. 4). To rationalize qualitatively the binding interactions of compounds 2e and 11 with MDM2, we used the small network analysis software SCORPION (Fig. 4). Compounds 2e and 11 show very similar interactions with the receptor, including hydrophobic, hydrogen bonding and electrostatic interactions. The bulkier adamantyl residue of 2e compared to 11 is making more hydrophobic contacts to Met62, Ile61 and Tyr67; for example, 2e can also make contacts to Val93 whereas 11 is too small to make the corresponding contacts. Notably, the C-3 of the scaffolding β-lactam is also undergoing a short contact to His96. Overall both the binding affinities of 2e and 11 and the Scorpion scores are very similar.
Fig. 4. Comparison of the binding modes of 11 (PDB ID ; 3TU1) and modeled 2e. Above: 11 and 2e in the MDM2 receptor where high scoring atoms are colored red to indicate tight binding. Below: 2D structures of 11 and 2e, Scorpion scores and binding affinities by FP.

Compounds 2c and 2d, with a benzyl elongated ligand moiety, are predicted to bind in a different mode, similar to 1 in our recently described cocrystal structure (Fig. 1A and B).7 Here again we docked 2c and 2d in the crystal structure of 1 and evaluated the ligand receptor complexes using SCORPION (Fig. 5).
Fig. 5. Comparison of the predicted binding modes of 2c (PDB ID ; 3TU1) and 2d modeled in PDB ID ; 4MDN. Above: 2c and 2d in the MDM2 receptor where high scoring atoms are colored red to indicate tight binding. Below: 2D structures of 2c and 2d, Scorpion scores and binding affinities by FP.

The key difference between 2c and 2d is their different aromatic substitution patterns, which are para and meta, respectively. Docking of all four possible stereoisomers make the stereoisomer shown in Fig. 5 most likely, based on predicted interactions using SCORPION analysis. Whereas meta substituted 2d exhibits more hydrophobic contacts in the adamantyl region and also a β-lactam His96 interaction, the interactions in the Leu pocket and the adjacent induced pocket are less compared to 2c. The bulkier adamantyl moiety has superior contribution to the MDM2 affinity compared to the tert-butyl moiety of 1. However, the adamantyl moiety does not seem to be perfectly shaped complementary to the hydrophobic pocket formed by Val93, Val75, His75, Gln72, Tyr67, Il61 and Met62. Therefore, changing the hydrophobic isocyanide building blocks in this position seems to be an attractive possibility to improve the affinity of similar molecules. The second aromatic group including the two linker atoms of 2c can make more and stronger hydrophobic contacts in the induced pocket. For example, the two meta-C of the second aromatic group exhibit multiple van der Waals interactions with Ile19 and Tyr100. The predicted binding mode is confirmed by the 2D NMR data.
Conclusions
Using our pharmacophore platform ANCHOR.QUERY, we morphed an α-aminoacylamide into a β-lactam scaffold, both of which are potent antagonists of the protein–protein interaction in p53-MDM2. Several predicted compounds were resynthesized using a union of two multicomponent reactions: α-aminoalkylation of the synthesized β-amino acids and Ugi-4CR. Their binding behavior was examined by the use of two orthogonal screening methods, namely, FP and 2D-HSQC. The most potent racemic compound 2e binds with 200 nM affinity to MDM2. Surprisingly, the compounds show dual action activity against MDM2 and MDMX, with a factor of only 2–4. Overall, this small library of β-lactams gives an initial idea of the activity of this new scaffold and serves as an excellent starting point for further and more extensive SARs. Modeling studies were used to understand the observed SAR. We believe the herein employed process will be useful in scaffold morphing for other PPI targets as well. Studies are ongoing to investigate the MDM2 protein dynamics in the presence of different β-lactams to learn more about the intrinsically disordered MDM2 terminus and what the rules of stabilization are.
Experimental
General procedures, results of fluorescence polarization binding and NMR studies, characterization data, and exemplary copies of NMR spectra are given in the ESI.‡
Supplementary Material
Acknowledgments
S. Shaabani acknowledges a scholarship from the Ministry of Science, Research and Technology, Tehran, Iran. The work was financially supported by the NIH (2R01GM097082-05) and by the European Union's Horizon 2020 research and innovation programme under MSC ITN “Accelerated Early staGe drug dIScovery” (AEGIS), grant agreement No. 675555.
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available: General procedures, results of fluorescence polarization binding and NMR studies, characterization data, and exemplary copies of NMR spectra. See DOI: 10.1039/c7md00058h
References
- Hu Y., Stumpfe D., Bajorath J. J. Med. Chem. 2017;60(4):1238–1246. doi: 10.1021/acs.jmedchem.6b01437. [DOI] [PubMed] [Google Scholar]
- Schneider G., Neidhart W., Giller T., Schmid G. Angew. Chem., Int. Ed. 1999;38:2894–2896. [PubMed] [Google Scholar]
- Czarna A., Beck B., Srivastava S., Popowicz G. M., Wolf S., Huang Y., Bista M., Holak T. A., Dömling A. Angew. Chem., Int. Ed. 2010;49:5352–5356. doi: 10.1002/anie.201001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D., Khoury K., Huang Y., Wang W., Bista M., Popowicz G. M., Wolf S., Holak T. A., Dömling A., Camacho C. J. PLoS One. 2012;7:e32839. doi: 10.1371/journal.pone.0032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popowicz G. M., Dömling A., Holak T. A. Angew. Chem., Int. Ed. 2011;50:2680–2688. doi: 10.1002/anie.201003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Wolf S., Koes D., Popowicz G. M., Camacho C. J., Holak T. A., Dömling A. ChemMedChem. 2012;7:49–52. doi: 10.1002/cmdc.201100428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista M., Wolf S., Khoury K., Kowalska K., Huang Y., Wrona E., Arciniega M., Popowicz G. M., Holak T. A., Dömling A. Structure. 2013;21:2143–2151. doi: 10.1016/j.str.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Wolf S., Beck B., Köhler L.-M., Khoury K., Popowicz G. M., Goda S. K., Subklewe M., Twarda A., Holak T. A., Dömling A. ACS Chem. Biol. 2014;9:802–811. doi: 10.1021/cb400728e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmiak E., Twarda-Clapa A., Zak K. M., Musielak B., Tomala M. D., Kubica K., Grudnik P., Madej M., Jablonski M., Potempa J., Kalinowska-Tluscik J., Dömling A., Dubin G., Holak T. A. ACS Chem. Biol. 2016;11:3310–3318. doi: 10.1021/acschembio.6b00596. [DOI] [PubMed] [Google Scholar]
- Wang W., Cao H., Wolf S., Camacho-Horvitz M. S., Holak T. A., Dömling A. Bioorg. Med. Chem. 2013;21:3982–3995. doi: 10.1016/j.bmc.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömling A., Wang W., Wang K. Chem. Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömling A. Chem. Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- Dömling A. Curr. Opin. Chem. Biol. 2008;12:281–291. doi: 10.1016/j.cbpa.2008.04.603. [DOI] [PubMed] [Google Scholar]
- Ding K., Lu Y., Nikolovska-Coleska Z., Wang G., Qiu S., Shangary S., Gao W., Qin D., Stuckey J., Krajewski K., Roller P. P., Wang S. J. Med. Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- Bemis G. W., Murcko M. A. J. Med. Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Dömling A., Huang Y. Synthesis. 2010:2859–2883. [Google Scholar]
- Kuhn B., Fuchs J. E., Reutlinger M., Stahl M., Taylor N. R. J. Chem. Inf. Model. 2011;51:3180–3198. doi: 10.1021/ci200319e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugi I., Steinbrückner C. Angew. Chem. 1960;72:267–268. [Google Scholar]
- González-López M., Shaw J. T. Chem. Rev. 2009;109:164–189. doi: 10.1021/cr8002714. [DOI] [PubMed] [Google Scholar]
- Castagnoli N. J. Org. Chem. 1969;34:3187–3189. doi: 10.1021/jo01262a081. [DOI] [PubMed] [Google Scholar]
- Doebner O. Ber. Dtsch. Chem. Ges. 1887;20:277–281. [Google Scholar]
- Van Leusen A. M., Wildeman J., Oldenziel O. H. J. Org. Chem. 1977;42:1153–1159. [Google Scholar]
- Furet P., Chène P., De Pover A., Valat T. S., Lisztwan J. H., Kallen J., Masuya K. Bioorg. Med. Chem. Lett. 2012;22:3498–3502. doi: 10.1016/j.bmcl.2012.03.083. [DOI] [PubMed] [Google Scholar]
- Zarganes-Tzitzikas T., Chandgude A. L., Dömling A. Chem. Rec. 2015;15:981–996. doi: 10.1002/tcr.201500201. [DOI] [PubMed] [Google Scholar]
- Cardillo G., Gentilucci L., Tolomelli A., Tomasini C. J. Org. Chem. 1998;63:2351–2353. [Google Scholar]
- Estrada-Ortiz N., Neochoritis C. G., Dömling A. ChemMedChem. 2016;11:757–772. doi: 10.1002/cmdc.201500487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury K., Popowicz G. M., Holak T. A., Dömling A. MedChemComm. 2011;2:246–260. doi: 10.1039/C0MD00248H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmiak E., Neochoritis C. G., Musielak B., Twarda-Clapa A., Kurpiewska K., Dubin G., Camacho C., Holak T. A., Dömling A. Eur. J. Med. Chem. 2017;126:384–407. doi: 10.1016/j.ejmech.2016.11.029. [DOI] [PubMed] [Google Scholar]
- García-Echeverría C., Chène P., Blommers M. J. J., Furet P. J. Med. Chem. 2000;43:3205–3208. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- Bauer M. R., Boeckler F. M. Structure. 2013;21:2095–2097. doi: 10.1016/j.str.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Huang X. J. Biomol. Screening. 2003;8:34–38. doi: 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
- Czarna A., Popowicz G. M., Pecak A., Wolf S., Dubin G., Holak T. A. Cell Cycle. 2009;8:1176–1184. doi: 10.4161/cc.8.8.8185. [DOI] [PubMed] [Google Scholar]
- Fielding L. Prog. Nucl. Magn. Reson. Spectrosc. 2007;51:219–242. [Google Scholar]
- Barile E., Pellecchia M. Chem. Rev. 2014;114:4749–4763. doi: 10.1021/cr500043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll R., Renner C., Hansen S., Palme S., Klein C., Belling A., Zeslawski W., Kamionka M., Rehm T., Mühlhahn P., Schumacher R., Hesse F., Kaluza B., Voelter W., Engh R. A., Holak T. A. Biochemistry. 2001;40:336–344. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- Gerber P. R., Müller K. J. Comput.-Aided Mol. Des. 1995;9:251–268. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.