

Abstract
Myocardial infarction (MI) is a life-threatening disease. The expression of Smad proteins in the ischemic myocardium changes significantly following myocardial infarction, suggesting a close relationship between Smad proteins and heart remodeling. Moreover, it is known that the expression of Smads is regulated by transforming growth factor-β (TGF-β) and bone morphogenetic proteins (BMP). Based on these findings, regulating the expression of Smad proteins by targeting TGF-β and BMP in the ischemic myocardium may be considered to be a possible therapeutic strategy for the treatment of myocardial infarction.
Keywords: myocardial infarction, Smad, TGF-β, BMP
1. Introduction
Myocardial infarction (MI) is one of the most serious heart diseases in the world and has high rates of morbidity and mortality. When the blood supply to the myocardium is suddenly stopped, cardiomyocyte necrosis occurs within minutes due to the lack of oxygen and nutrition. Cardiomyocyte loss is subsequently accompanied by post-infarction remodeling. Therefore, promising therapies for MI would lead to increased cardiomyocyte survival and proliferation and should potentially prevent or postpone the fibrosis process.
Smads are intracellular proteins that transduce extracellular signals to the nucleus where they activate downstream gene transcription. Smad, small mothers against decapentaplegic, is a portmanteau of the Drosophila protein mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein SMA. The molecular mass of Smads ranges from 42–60Kda, and there are two regions of homology at the amino and carboxy termini named Mad-homology domains MH1 and MH2, respectively (Massaous and Hata, 1997). The Smad MH1 domains interact with the MH2 domains via a proline-rich linker sequence that is in an inactive configuration (Heldin et al, 1997). Once Smads are activated and the interaction is unlocked, Smad proteins form a complex and translocate to the nucleus to regulate target gene transcription.
Smads act as transcription factors to regulate the expression of many genes and participate in serine/threonine kinase signal transduction pathways (Massaous and Hata, 1997) (Patterson and Padgett, 2000). The Smad signaling pathway is a key regulator related to tissue fibrosis in the liver (Xu et al, 2016), kidney (Isono et al, 2002) (Xiao et al, 2014) (Meng et al, 2013), and lungs (Lucarini et al, 2016). Increasing evidence suggest that Smad signals are involved in processes that underlie many heart diseases (Hao et al, 2000) (Schneiders et al., 2005) (Dixon et al., 2000) (Zhao et al., 2015) (Bugyei-Twum et al., 2014). Pressure overload induced by transaortic constriction (TAC) results in excess cardiac fibrosis and myocardial hypertrophy, which are related to increased TGF-β signaling, and activates Smad2/3 and ERK1/2 within endothelial cells in cardiac blood vessels (Wei et al., 2012).
In this mini review, we elaborate on the diverse roles of Smad proteins in myocardial ischemia and examine some promising therapeutic strategies for MI that use pro- or anti-Smad proteins.
2. Smad signaling pathway and its regulation
Smad family proteins can be divided into the following three groups: Group 1, receptor-regulated Smads (R-Smads), which include Smad1, Smad2, Smad3, Smad5, and Smad8 (also known as Smad9); Group 2, common Smad (Co-Smad), Smad4, that acts as a co-factor and combines with activated R-Smads to form a complex that can translocate to the nucleus to regulate target genes; Group 3, inhibitory Smads (I-Smads), which include Smad6 and Smad7 (Moustakas et al., 2001). The intracellular distribution of Smad isoforms is changeable based on whether they are activated or not. Under non-activated conditions, most R-Smads are located in the cytoplasm, I-Smads are located in the nucleus, and Co-Smad is located both in the cytoplasm and the nucleus. Smads are activated once their C-terminal region is phosphorylated by a specific receptor by upstream regulators. Once Smads are phosphorylated and activated, R-Smads combine with Co-Smad to form a complex that translocates into the nucleus (Liu et al., 1997).
Classical activators of R-Smad signaling include TGF-β1 and members of the BMP family, both of which belong to the broader cytokine family. There is counter-regulation that modulates the biological activities between the TGF-β1 and BMP activating signaling pathways (Ebelt et al., 2013). In mammals, TGF-β induces Smad2 and Smad3 phosphorylation, which form heteromeric complexes with Smad4, while BMP activates the R-Smad1/5/8 complex to convey intracellular signals (Heldin et al., 1997). Smad1 is then phosphorylated after BMP-2 (Kretzschmar et al., 1997) or BMP-4 (Liu et al., 1996) activation. Smad5 and Smad8 are regulated by BMP-4 (Suzuki et al., 1997). In contrast, overexpression of Smad8 could inhibit the activation of BMP signaling (Tsukamoto et al., 2014). The effects of R-Smads can be blocked by I-Smads, which results in inhibition of TGF-β superfamily signaling (Nakao et al., 1997) (Imamura et al., 1997).
2.1. TGF-β/Smad signaling pathway
Smads transduce extracellular signals from TGF-β ligands to the nucleus where they activate downstream gene transcription. Three isoforms of TGF-β have been identified, TGF-β1, TGF-β2, and TGF-β3, each of which is encoded by a distinct gene. Although these three isoforms have similar cellular targets, TGF-β1 is the prevalent isoform and is quite ubiquitous, while the other two isoforms are expressed in a limited number and type of cells and tissues (Letterio and Roberts, 1998). TGF-β1 has been reported to be involved in ventricular remodeling by promoting myocardial fibrosis (Zhao et al., 2015) (Edgley et al., 2012), (Khan et al., 2016) and mediating cardiomyocyte apoptosis (Schneiders et al., 2005) or cardiac hypertrophy (Huntgeburth et al., 2011). TGF-β combines with its receptors, which have specific type I and type II serine/threonine kinases (Shi and Massague, 2003). TGF-β first binds to its specific receptor type II (TβIIR), which is located on the cell membrane in an oligomeric form with activated kinase, to form the TGF-β-TβIIR complex. The latter complex recruits and binds to type I receptors (TβIR), which are also known as activin receptor-like kinase (ALK4), resulting in phosphorylation of its GS domain (a region rich in glycine, serine and threonine residues) and new complex activation. The serine/threonine-kinase activity of this receptor complex phosphorylates and activates downstream R-Smad (Euler, 2015). Thus, activation of Smad signaling requires both TβIIR and TβIR. Smad7 negatively regulates the activation of this signaling pathway by binding to TβIR or by counteracting the effects of R-Smads to prevent the recruitment and phosphorylation of R-Smad proteins (Xu et al., 2016).
2.2. BMP/Smad signaling pathway
More than 12 BMP-related proteins have been identified. These proteins can be divided into the following groups based on their respective structures: the BMP-2/-4 group, BMP-5/-6/-7(OP-1)/-8 group, BMP-9/-10 group, and BMP-12/-13/-14 group (Katagiri and Watabe, 2016). BMP signaling is mediated by the activation of combinations of type I (BMP-RI) and type II (BMP-RII) serine/threonine kinase receptors (Miyazono et al., 2005). Activation of the BMP pathway is confirmed by increased phosphorylation of the “canonical” BMP effectors Smad1/5/8 (Sui et al., 2009). Different members of the BMP family have particular spatiotemporal expression profiles (Miyazono et al., 2005). As inhibitory Smad proteins, both Smad6 and Smad7 specifically block the activation of the BMP/Smad signaling pathway (Ishisaki et al., 1999) (Imamura et al., 1997).
2.3. Angiotensin and Smad proteins
Angiotensin II (Ang II) has a positive effect on the Smad protein pathway. Ang II regulates rapid phospho-Smad2 (P-Smad2) nuclear translocation in isolated fibroblasts (Hao et al., 2000). In vascular smooth muscle cells, Smad2 and Smad3 can be activated when TGF-β is blocked and Ang II has a greater effect on activating Smad3 than Smad2 (Rodriguez-Vita et al., 2005). Ang II activates the Smad pathway, resulting in an increase in myocardial collagen expression along with Smad2 and Smad3 phosphorylation (Pokharel et al., 2004) (Ikeda et al., 2005). Ang II stimulates cardiac fibrosis in several different models of heart failure (Schieffer et al., 1994, Gu et al., 2012). Additionally, Ang II infusion stimulates cardiac fibroblast production of IL-6, which activates TGF β/Smad and results in cardiac fibrosis (Ma et al., 2012).(Crabos et al., 1994).
2.4. Activation of Smad signaling
The activation of Smad signaling through R-Smad, Co-Smad and I-Smad have been reviewed previously (Flanders, 2004). Briefly, Smad activation is initiated by the binding of R-Smad to membrane receptors, including TGF-β and BMP receptors, which results in R-Smad phosphorylation. Then, phosphorylated R-Smad is released and binds to Co-Smad (Smad4) to form a heteromeric complex that translocates to the nucleus, where it regulates various transcription factors and induces several transcriptional responses. I-Smads (Smad6 and Smad7 for the BMP pathway and Smad7 for the TGF- β/activin pathway) play an important role in Smad binding to Type I receptors and in preventing R-Smad recruitment and phosphorylation. I-Smads can also block the effects of R-Smad by promoting the E3 ubiquitin ligases Smad ubiquitination regulatory factors 1 and 2 (Smurf 1 and 2) to target, ubiquitinate and degrade Type I receptors.
3. Smad signaling in myocardial infarction
3.1. Smad signaling in infarcted myocardium
The expression of Smad proteins in infarcted or fibrotic myocardium is different from that in the normal myocardium. No matter whether it is assessed in early or late stage cardiomyopathy, the expression of Smad proteins is correlated with cardiac fibrosis and elevated collagen synthesis levels (Dixon et al., 2000) (Hao et al., 1999).
Changes in TGF-β signaling have been reported in a variety of heart diseases, including cardiac hypertrophy (Rosenkranz et al., 2002), transaortic constriction (TAC) induced cardiac fibrosis and myocardial hypertrophy (Wei et al., 2012), and myocardial infarction (Hao et al., 1999) (Vilahur et al., 2011) (Li et al., 2012). TGF-β upregulation results in Smad signaling pathway activation in the heart under pathophysiological conditions (Hao et al., 1999) (Yang et al., 2015). The expression of Smad2, 3 and 4 is upregulated at the infarct scar as well as in the peri-ischemic border zone (Hao et al., 1999) and is closely correlated to increased collagen type I expression (Hao et al., 2000). In contrast, expression of Smad7 and TβIR are down-regulated in both the scar tissue and border area; this lasts for approximately eight weeks post-MI (Wang et al., 2002). It has been demonstrated that excessive TGF-β signaling alters cardiac and muscle performance through the intracellular Smad pathway. For example, a heterozygous mutation in Smad4 (S4) is introduced into Sgcg mice (sgcg/S4 mice) to reduce but not ablate Smad4. Sgcg/S4 mice displayed improved cardiac function (Goldstein et al., 2014). Smad7, an inhibitory Smad in the TGF-β pathway, is required for arch artery remodeling (Papangeli and Scambler, 2013). However, BMP-2 is upregulated in the ischemic myocardium accompanying acute MI (Chang et al., 2008). Coronary artery bypass grafting (CABG) and dilated cardiomyopathy (CMP) patients also displayed increased expression of BMP4 mRNA (Wu et al., 2014). (Banach et al., 2016) (Sanders et al., 2016)
Cross-talk exists between BMP and TGF-β in their regulation of different signaling pathways. For example, BMP-2 induces Smad1/5/8 phosphorylation through TGF-β1-receptor types (ALK1, 2 or 3) (Ebelt et al., 2013). BMP receptor-ligand (e.g., BMP-7) binding can activate Smad1/5/8 signaling and induce the expression of inhibitors of differentiation 2 and 3 (ID2 and ID3). ID2 and ID3 can prevent Smad2/3 phosphorylation and thus counteract TGF-β/Smad signaling (Weiskirchen and Meurer, 2013). It has been indicated that excessive TGF-β1 signaling and reduced BMP signaling have been implicated in pulmonary arterial hypertension (PAH) disease pathogenesis. TGF-β also represses BMP-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3 (Upton et al., 2013).
3.2. Smad signaling associated with cardiac pathophysiology
Activation of most R-Smads may be related to cardiac dysfunction, cardiomyocyte apoptosis and cardiac fibrosis. In a pressure overload model caused by aortic banding, pharmacological inhibition of Smad2 signaling using the small molecule inhibitor SM16 attenuated cardiomyocyte hypertrophy and preserved cardiac function (Bjornstad et al., 2012). Schneiders et al. (Schneiders et al., 2005) have reported that Smad proteins are involved in cardiomyocyte apoptosis. TGF-β1 treatment of cardiomyocytes enhances the number of apoptotic cells expressed, increases caspase 3/7 activity and decreases Bcl-2 expression and the mitochondrial membrane potential with Smad7 upregulation (Heger et al, 2012). More importantly, Smad proteins are associated with cardiac fibrosis (Hao et al., 2000) (Araujo-Jorge et al., 2002) (Dixon et al., 2000) (Lucarini et al., 2016) (Wang et al., 2005). Activation of the TGF-β/Smad pathway results in increased transcription of genes related to the production of extracellular matrix components, such as fibronectin, type 1 collagen, and connective tissue growth factor (CTGF), which leads to fibrosis development (Gabriel, 2009). The function and morphology of Smad3-null hearts under normal, non-ischemic conditions is similar to that of wild-type hearts. Following myocardial infarction, the inflammation levels in the hearts of Smad 3-null mice were not significantly different from those of the hearts of wild-type mice. However, fibrotic remodeling and diastolic dysfunction were attenuated and ventricular dilatation was reduced in association with reduced collagen deposition in the hearts of Smad3 knockout mice (Bujak et al., 2007). These results reveal that TGF-β/Smad3 have a strong influence on fibrosis post-MI. The reduced contraction functions in Smad3-deficient hearts following MI may be related to the TGF-β anti-proliferative effects on isolated fibroblasts through the Smad3 signaling pathway (Dobaczewski et al., 2010).
Smad4 is a central mediator of TGF-β/BMP signaling that controls numerous developmental processes as well as homeostasis in adults. Specific deletion of Smad4 in smooth muscle cells results in vascular defects that lead to embryonic lethality in mice, which may be attributed to decreased VSMC differentiation, proliferation, and migration as well as cell attachment and spreading (Mao et al., 2012).
Inhibitory Smads also display the beneficial effects of reducing apoptosis and fibrosis. Smad6 signaling is involved in cardiomyocyte apoptosis. Smad6 knockdown in mice induces many cardiovascular disorders (Galvin et al., 2000). Smad7 can block myocardial fibrosis (Wang et al., 2005) by reducing the expression of type1 collagen and alpha smooth muscle actin, which prevents excessive scar formation (Kopp et al., 2005) (Wang et al., 2002). Cardiac Smad7 is largely reduced in hypertensive hearts and induced by subcutaneous Ang II infusion. Overexpression of cardiac Smad7 protected against the decrease in the left ventricular (LV) ejection fraction (EF), increase in LV mass, and cardiac inflammation and fibrosis. Treatment with Smad7 halts the progression of cardiac injury by blunting the decrease in EF, increasing LV mass, and blocking TGF-β/Smad3-mediated cardiac fibrosis (Wei et al., 2013). Moreover, Smad3 has been reported to have a beneficial effect on inhibiting hypertrophic growth; for example, Smad3−/− fibroblasts exhibited impaired collagen lattice contraction compared with wild-type cells (Graf and Schaefer-Graf, 2010).
BMP-2 is able to decrease the infarct size and reduce apoptotic cell death following MI in mice; it also enhances the beating frequency and contractile performance of isolated cardiomyocytes in in vitro studies (Ebelt et al., 2013). Masaki et al. (Masaki et al., 2005) showed that BMP2 and Smad1 significantly increased survival and diminished apoptotic death in rat neonatal cardiomyocytes during hypoxia-reoxygenation conditions. BMP-9 has been shown to be involved in angiogenesis (Ricard et al., 2012) (Long et al., 2015). At high concentrations, blocking BMP-9 promotes endothelial cell proliferation (Scharpfenecker et al., 2007), while at low concentrations, increasing BMP-9 enhances endothelial cell proliferation and angiogenesis in Matrigel™ plug assays (Scharpfenecker et al., 2007).
BMP-Smad signaling may also be involved in cardiomyocyte differentiation and proliferation. The differentiation of the BMP antagonist noggin overexpressing P19CL6 (P19CL6noggin) in cardiomyocytes is very low; however, co-overexpression of Smad1 and Smad4 restores the ability of P19CL6noggin to differentiate into cardiomyocytes (Monzen et al., 2001). Transcription factor Tbx20 (T-box) induces adult cardiomyocyte proliferation by activating the BMP/Smad1/5/8, PI3K/Akt, and Hippo/YAP signaling pathways (Xiang et al., 2016).
3.3. Potential therapeutic effects by regulating Smad signaling
BMPs have shown some beneficial effects on cardiac development and anti-apoptosis by acting on Smad proteins. Exogenous BMP-7 reduces myofibroblast generation, inflammatory reactions and expression of MCP-1, TGF-β, and collagen I alpha2 chain in burned corneal tissue (Saika et al, 2005) by inhibiting Smad2/3 signaling and activating Smad 1/5/8 signaling. BMP-7 gene delivery reduces wound healing in rabbit cornea and prevents fibrosis in vivo (Tandon et al, 2013). BMP-2 may have a considerable therapeutic potential for curing chronic myocardial ischemia by improving the contractility of cardiomyocytes and preventing cardiomyocyte cell death (Ebelt et al, 2013). Injection of BMP-2 reduces the infarct size in mice in a left anterior descending artery ligation model. Mice treated with BMP-2 are characterized by reduced cardiomyocyte apoptosis rates. In vitro, BMP-2 increases the frequency of spontaneously beating neonatal cardiomyocytes and the contractile performance, preserves cellular adenosine triphosphate stores, and decreases the rate of apoptosis. BMP-2 specifically induces the phosphorylation of Smad1/5/8 proteins without activating the TGF-β pathway (Ebelt et al, 2013). BMP-4 and BMP-9 can promote endothelial cell differentiation and regulate angiogenesis (Jumabay et al, 2012, Cagavi et al, 2014). Human recombinant BMP-4 promoted survival after H2O2 injury in HL-1 cells, and also protected cardiomyocytes against hypoxia-reoxygenation injury (Wu et al, 2014). Inhibition of BMP1-3 has been suggested as a therapeutic method for heart tissue fibrosis following acute MI (Cvjeticanin et al, 2014).
Low-intensity pulsed ultrasound (LIPUS) might be a useful tool for heart cell therapy because it can activate the BMP-2/Smad1/5/8 pathway and promote cardiomyocyte differentiation (Bernal et al, 2015). It has been reported that chromodomain-helicase-DNA-binding protein 7 (CHD7), an ATP-dependent nucleosome remodeling factor, is a novel interaction partner of the canonical BMP signaling pathway nuclear mediators Smad1/5/8 in the embryonic heart. Moreover, CHD7 associates with the enhancers of the critical cardiac transcription factor Nk×2.5 that contain functional Smad1-binding elements in a BMP-dependent manner. CHD7 is recruited by Smad1/5/8 to the enhancers of BMP-targeted cardiogenic genes to epigenetically regulate their expression (Liu et al., 2014).
PARM-1, prostatic androgen repressed message-1, plays an important role in the cardiomyogenic differentiation of stem cells. PARM-1 overexpression induces BMP-2 mRNA expression in undifferentiated P19CL6 cells and enhances both BMP-2 and BMP-4 mRNA expression in the early phase of cardiomyogenesis. PARM-1 overexpression also enhances Smad1/5/8 phosphorylation (Nakanishi et al., 2012).
However, other opinions exist on the cardioprotective effects of the BMP/Smad signaling pathway. ATP-binding cassette (ABC) transporters are a large family of membrane efflux transporters that transport a variety of substrates including clinically relevant pharmaceutical agents. Recent studies have identified Abcc6, an ABC transporter, as a novel modulator of cardiomyocyte survival after I/R. The cardioprotective effects of Abcc6 may be related to inhibition of the BMP/Smad signaling pathway (Mungrue et al., 2011). Gremlin 2, another BMP antagonist, inhibits BMP-2 pro-inflammation and improves cardiac function post-MI (Sanders et al., 2016). Furthermore, secreted Frizzled-related protein 2 (sFRP2) significantly enhances stem cell engraftment in vivo through the inhibition of both the Wnt and BMP/Smad1/5/8 signaling pathways (Alfaro et al., 2010).
In contrast to BMP, the activation of the TGF-β/Smad signaling pathway plays a critical role in cardiomyocyte death and myocardial fibrosis. Chen et al. (Chen et al., 2007) reported that the endothelial nitric oxide synthase (eNOS)/nitric oxide system can protect cardiomyocytes from apoptosis by suppressing TGF-β/Smad2 signaling and blocking Smad2 phosphorylation. Pitavastanin, a HMG-CoA reductase inhibitor, can suppress cardiovascular remodeling by reversing the upregulated TGF-β1-Smad2, 3 signaling pathway (Yagi et al., 2008). βIIV5-3, protein kinase cβII inhibitor, alleviates myocardial damage by inactivating TGF-β1 and inhibiting Smad2/3 phosphorylation (Palaniyandi et al., 2011). Transient receptor potential vanilloid type 1 (TRPV1), a factor that inactivates the TGF-β/Smad2 signaling pathway, can significantly reduce post-MI fibrosis and preserve cardiac function (Huang et al., 2010). High-mobility group box 1 (HMGB1), a pro-inflammatory cytokine, has been reported to attenuate ventricular remodeling by decreasing TGF-β1 and p-Smad2, upregulating Smad7, and suppressing collagen I and III expression (He et al., 2013). Angiotensin-(1–7) could reduce vascular remodeling in a rabbit abdominal aorta injury model via inhibiting both neo-intimal formation and collagen secretion by down-regulating TGF-β1 levels and inhibiting the Smad2 pathway (Zeng et al., 2009). Post-conditioning effectively reduces collagen synthesis and improves cardiac repair, which may be related to attenuating TGF-β1 and p-Smad2/3 expression and up-regulating Smad7 expression (Wang et al., 2013).
Furthermore, many synthetic medicines are considered to improve myocardial infarction through inhibiting the TGF-β/Smad signaling pathway. Statins have been reported to ameliorate ventricular remodeling via the TGF-β1/Smad signaling pathway in rats (Xiao et al., 2016). Fluvastatin, a synthetic HMG-CoA reductase inhibitor, has been found to significantly alleviate myocardial fibrosis by regulating TGF-β1/Smad7 expression post-MI (Zhai et al., 2008). Hydrochlorothiazide and spironolactone, which are used to treat hypertension, effectively reduce cardiac remodeling by alleviating pro-inflammatory cytokine levels and blocking the TGF-β signaling pathway in a congestive heart failure model (Luo et al., 2011). Telmisartan, an Ang II receptor antagonist, can reduce myocardial local Ang II levels and inhibit myocardial fibrosis in hypertensive left ventricular hypertrophy (LVH) by affecting the TGF-β1/Smad signaling pathway (Zhang et al., 2014). It is well-known that doxorubicin increases cardiomyocyte apoptosis, which is secondary to increasing TGF-β/Smad pathway gene expression. Desferrioxamine attenuates doxorubicin-induced acute cardiotoxicity through the TFG-β/Smad p53 pathway (Al-Shabanah et al., 2012).
Sesamin can attenuate hypertensive myocardial fibrosis by suppressing the TGF-β1/Smad signaling pathway. Sesamin markedly reduces TGF-β1 and Smad3 phosphorylation in cardiac tissues and increases Smad7 protein expression. Protein expression of type I and III collagen is also significantly down-regulated (Zhao et al., 2015). Sildenafil, an inhibitor of cyclic guanosine monophosphate-specific phosphodiesterase 5 (PDE5), prevents cardiac fibrosis by inhibiting TGFβ-induced Smad signaling (Gong et al., 2014). In isolated cardiac fibroblasts, sildenafil blocked TGF-β1-induced cardiac fibroblast transformation, proliferation and collagen synthesis (Gong et al., 2014). Oxymatrine (OMT), a component extracted from a traditional Chinese herb named Sophora japonica (Sophora flavescens Ait), significantly reduces left ventricle weight/body weight, prevents myocardial fibrosis in an acute MI model, and downregulates TβR1 and Smad3 expression (Shen et al., 2011). OMT can also promote Smad7 expression and inhibit Smad3 expression (Wu et al., 2008).
It has also been suggested that transplantation of cardiospheres enhances angiogenesis and reduces fibrosis in chronically infarcted myocardium, leading to a partial reversal in cardiac dysfunction. The underlying mechanism may involve the inhibition of TGF-β1/Smad signaling by cardiosphere-secreted soluble endoglins (Tseliou et al., 2014).
4. Conclusions
Smad proteins are involved in many pathological processes in myocardial infarction. Various isoforms play different roles in ischemic myocardium remodeling after being stimulated by either TGF-β or BMP. Smad protein regulation by biomolecules, synthesized medicines and products extracted from some herbal medicines (plant sources) may be considered to be promising therapeutic strategies for treating myocardial infarction (Figure 1).
Figure 1.
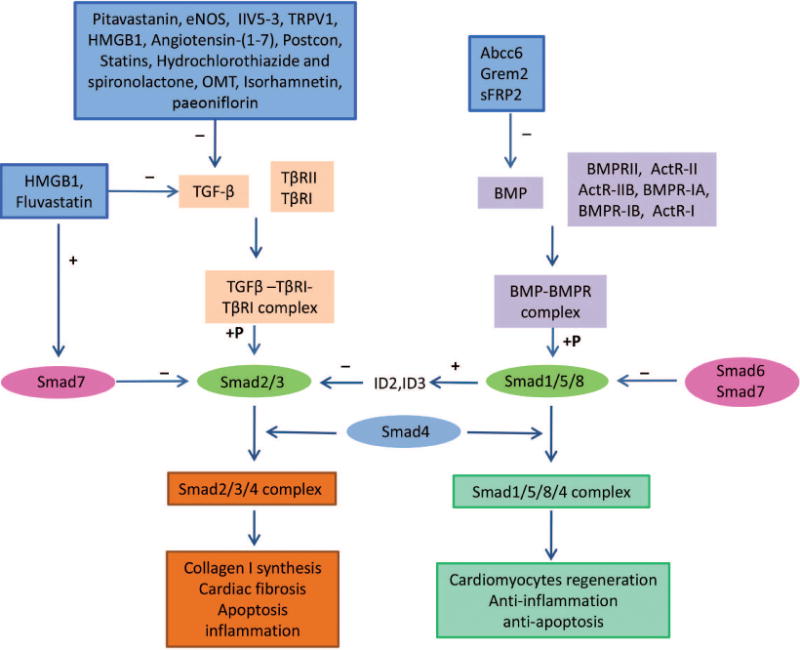
Schematic representation of the role of the TGF-β-Smad and BMP-Smad signaling pathways in myocardial infarction and the mechanisms of some associated potential therapies. Activation of the TGF-β-Smad signaling pathway may be associated with collagen synthesis, cardiac fibrosis, apoptosis, and inflammation. In contrast, activation of the BMP-Smad signaling pathway may be associated with cardiomyocyte regeneration, anti-inflammation, and anti-apoptosis.
Acknowledgments
This work was supported by National Institutes of Health grants HL105176 and HL114654 (Meifeng Xu), National Natural Science Foundation of China (Grant no. 81270214, Wei Zhu).
References
- Al-Shabanah OA, Aleisa AM, Hafez MM, Al-Rejaie SS, Al-Yahya AA, Bakheet SA, Al-Harbi MM, Sayed-Ahmed MM. Desferrioxamine attenuates doxorubicin-induced acute cardiotoxicity through tfg-beta/smad p53 pathway in rat model. Oxid Med Cell Longev. 2012;2012:619185. doi: 10.1155/2012/619185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro MP, Vincent A, Saraswati S, Thorne CA, Hong CC, Lee E, Young PP. Sfrp2 suppression of bone morphogenic protein (bmp) and wnt signaling mediates mesenchymal stem cell (msc) self-renewal promoting engraftment and myocardial repair. J Biol Chem. 2010;285:35645–35653. doi: 10.1074/jbc.M110.135335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo-Jorge TC, Waghabi MC, Hasslocher-Moreno AM, Xavier SS, Higuchi Mde L, Keramidas M, Bailly S, Feige JJ. Implication of transforming growth factor-beta1 in chagas disease myocardiopathy. J Infect Dis. 2002;186:1823–1828. doi: 10.1086/345882. [DOI] [PubMed] [Google Scholar]
- Banach J, Gilewski W, Slomka A, Buszko K, Blazejewski J, Karasek D, Rogowicz D, Zekanowska E, Sinkiewicz W. Bone morphogenetic protein 6 - a possible new player in pathophysiology of heart failure. Clin Exp Pharmacol Physiol. 2016 doi: 10.1111/1440-1681.12665. [DOI] [PubMed] [Google Scholar]
- Bernal A, Perez LM, De Lucas B, Martin NS, Kadow-Romacker A, Plaza G, Raum K, Galvez BG. Low-intensity pulsed ultrasound improves the functional properties of cardiac mesoangioblasts. Stem Cell Rev. 2015;11:852–865. doi: 10.1007/s12015-015-9608-6. [DOI] [PubMed] [Google Scholar]
- Bjornstad JL, Skrbic B, Marstein HS, Hasic A, Sjaastad I, Louch WE, Florholmen G, Christensen G, Tonnessen T. Inhibition of smad2 phosphorylation preserves cardiac function during pressure overload. Cardiovasc Res. 2012;93:100–110. doi: 10.1093/cvr/cvr294. [DOI] [PubMed] [Google Scholar]
- Bugyei-Twum A, Advani A, Advani SL, Zhang Y, Thai K, Kelly DJ, Connelly KA. High glucose induces smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol. 2014;13:89. doi: 10.1186/1475-2840-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- Cagavi E, Bartulos O, Suh CY, Sun B, Yue Z, Jiang Z, Yue L, Qyang Y. Functional cardiomyocytes derived from isl1 cardiac progenitors via bmp4 stimulation. PloS one. 2014;9:e110752. doi: 10.1371/journal.pone.0110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SA, Lee EJ, Kang HJ, Zhang SY, Kim JH, Li L, Youn SW, Lee CS, Kim KH, Won JY, Sohn JW, Park KW, Cho HJ, Yang SE, Oh WI, Yang YS, Ho WK, Park YB, Kim HS. Impact of myocardial infarct proteins and oscillating pressure on the differentiation of mesenchymal stem cells: Effect of acute myocardial infarction on stem cell differentiation. Stem Cells. 2008;26:1901–1912. doi: 10.1634/stemcells.2007-0708. [DOI] [PubMed] [Google Scholar]
- Chen LL, Yin H, Huang J. Inhibition of tgf-beta1 signaling by enos gene transfer improves ventricular remodeling after myocardial infarction through angiogenesis and reduction of apoptosis. Cardiovasc Pathol. 2007;16:221–230. doi: 10.1016/j.carpath.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Crabos M, Roth M, Hahn AW, Erne P. Characterization of angiotensin ii receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J Clin Invest. 1994;93:2372–2378. doi: 10.1172/JCI117243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvjeticanin B, Prutki M, Dumic-Cule I, Veir Z, Grgurevic L, Vukicevic S. Possible target for preventing fibrotic scar formation following acute myocardial infarction. Med Hypotheses. 2014;83:656–658. doi: 10.1016/j.mehy.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Dixon IM, Hao J, Reid NL, Roth JC. Effect of chronic at(1) receptor blockade on cardiac smad overexpression in hereditary cardiomyopathic hamsters. Cardiovasc Res. 2000;46:286–297. doi: 10.1016/s0008-6363(00)00035-3. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebelt H, Hillebrand I, Arlt S, Zhang Y, Kostin S, Neuhaus H, Muller-Werdan U, Schwarz E, Werdan K, Braun T. Treatment with bone morphogenetic protein 2 limits infarct size after myocardial infarction in mice. Shock. 2013;39:353–360. doi: 10.1097/SHK.0b013e318289728a. [DOI] [PubMed] [Google Scholar]
- Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: A role for transforming growth factor-beta. Cardiovasc Ther. 2012;30:e30–40. doi: 10.1111/j.1755-5922.2010.00228.x. [DOI] [PubMed] [Google Scholar]
- Euler G. Good and bad sides of tgfbeta-signaling in myocardial infarction. Front Physiol. 2015;6:66. doi: 10.3389/fphys.2015.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel VA. Transforming growth factor-beta and angiotensin in fibrosis and burn injuries. J Burn Care Res. 2009;30:471–481. doi: 10.1097/BCR.0b013e3181a28ddb. [DOI] [PubMed] [Google Scholar]
- Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, Jr, Falb D, Huszar D. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–174. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- Goldstein JA, Bogdanovich S, Beiriger A, Wren LM, Rossi AE, Gao QQ, Gardner BB, Earley JU, Molkentin JD, McNally EM. Excess smad signaling contributes to heart and muscle dysfunction in muscular dystrophy. Hum Mol Genet. 2014;23:6722–6731. doi: 10.1093/hmg/ddu390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Yan M, Chen J, Chaugai S, Chen C, Wang D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor beta-induced smad signaling. Front Med. 2014;8:445–455. doi: 10.1007/s11684-014-0378-3. [DOI] [PubMed] [Google Scholar]
- Graf K, Schaefer-Graf UM. Is smad3 the key to inflammation and fibrosis in hypertensive heart disease? Hypertension. 2010;55:1088–1089. doi: 10.1161/HYPERTENSIONAHA.110.150466. [DOI] [PubMed] [Google Scholar]
- Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang WF, Zhou L. Angiotensin ii increases ctgf expression via mapks/tgf-beta1/traf6 pathway in atrial fibroblasts. Exp Cell Res. 2012;318:2105–2115. doi: 10.1016/j.yexcr.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Hao J, Wang B, Jones SC, Jassal DS, Dixon IM. Interaction between angiotensin ii and smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol Heart Circ Physiol. 2000;279:H3020–3030. doi: 10.1152/ajpheart.2000.279.6.H3020. [DOI] [PubMed] [Google Scholar]
- Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IM. Elevation of expression of smads 2, 3, and 4, decorin and tgf-beta in the chronic phase of myocardial infarct scar healing. J Mol and Cell Cardiol. 1999;31:667–678. doi: 10.1006/jmcc.1998.0902. [DOI] [PubMed] [Google Scholar]
- He Y, Zhou X, Zheng X, Jiang X. Exogenous high-mobility group box 1 protein prevents postinfarction adverse myocardial remodeling through tgf-beta/smad signaling pathway. J Cell Biochem. 2013;114:1634–1641. doi: 10.1002/jcb.24505. [DOI] [PubMed] [Google Scholar]
- Heger J, Abdallah Y, Shahzad T, Klumpe I, Piper HM, Schultheiss HP, Schluter KD, Schulz R, Euler G, Dorner A. Transgenic overexpression of the adenine nucleotide translocase 1 protects cardiomyocytes against tgfbeta1-induced apoptosis by stabilization of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2012;53:73–81. doi: 10.1016/j.yjmcc.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. Tgf-beta signalling from cell membrane to nucleus through smad proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Huang W, Rubinstein J, Prieto AR, Wang DH. Enhanced postmyocardial infarction fibrosis via stimulation of the transforming growth factor-beta-smad2 signaling pathway: Role of transient receptor potential vanilloid type 1 channels. J Hypertens. 2010;28:367–376. doi: 10.1097/HJH.0b013e328333af48. [DOI] [PubMed] [Google Scholar]
- Huntgeburth M, Tiemann K, Shahverdyan R, Schluter KD, Schreckenberg R, Gross ML, Modersheim S, Caglayan E, Muller-Ehmsen J, Ghanem A, Vantler M, Zimmermann WH, Bohm M, Rosenkranz S. Transforming growth factor beta(1) oppositely regulates the hypertrophic and contractile response to beta-adrenergic stimulation in the heart. PloS one. 2011;6:e26628. doi: 10.1371/journal.pone.0026628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Aihara K, Sato T, Akaike M, Yoshizumi M, Suzaki Y, Izawa Y, Fujimura M, Hashizume S, Kato M, Yagi S, Tamaki T, Kawano H, Matsumoto T, Azuma H, Kato S, Matsumoto T. Androgen receptor gene knockout male mice exhibit impaired cardiac growth and exacerbation of angiotensin ii-induced cardiac fibrosis. J Biol Chem. 2005;280:29661–29666. doi: 10.1074/jbc.M411694200. [DOI] [PubMed] [Google Scholar]
- Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the tgf-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- Ishisaki A, Yamato K, Hashimoto S, Nakao A, Tamaki K, Nonaka K, ten Dijke P, Sugino H, Nishihara T. Differential inhibition of smad6 and smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in b cells. J Biol Chem. 1999;274:13637–13642. doi: 10.1074/jbc.274.19.13637. [DOI] [PubMed] [Google Scholar]
- Isono M, Chen S, Hong SW, Iglesias-de la Cruz MC, Ziyadeh FN. Smad pathway is activated in the diabetic mouse kidney and smad3 mediates tgf-beta-induced fibronectin in mesangial cells. Biochem Biophys Res Commun. 2002;296:1356–1365. doi: 10.1016/s0006-291x(02)02084-3. [DOI] [PubMed] [Google Scholar]
- Jumabay M, Abdmaulen R, Urs S, Heydarkhan-Hagvall S, Chazenbalk GD, Jordan MC, Roos KP, Yao Y, Bostrom KI. Endothelial differentiation in multipotent cells derived from mouse and human white mature adipocytes. J Mol Cellu Cardiol. 2012;53:790–800. doi: 10.1016/j.yjmcc.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T, Watabe T. Bone morphogenetic proteins. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Dong H, Joyce J, Sasaki T, Chu ML, Tsuda T. Fibulin-2 is essential for angiotensin ii-induced myocardial fibrosis mediated by transforming growth factor (tgf)-beta. Lab Invest. 2016;96:773–783. doi: 10.1038/labinvest.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J, Preis E, Said H, Hafemann B, Wickert L, Gressner AM, Pallua N, Dooley S. Abrogation of transforming growth factor-beta signaling by smad7 inhibits collagen gel contraction of human dermal fibroblasts. J Biol Chem. 2005;280:21570–21576. doi: 10.1074/jbc.M502071200. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massague J. The tgf-beta family mediator smad1 is phosphorylated directly and activated functionally by the bmp receptor kinase. Genes and Dev. 1997;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by tgf-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Li Q, Xu Y, Li X, Guo Y, Liu G. Inhibition of rho-kinase ameliorates myocardial remodeling and fibrosis in pressure overload and myocardial infarction: Role of tgf-beta1-tak1. Toxicol Lett. 2012;211:91–97. doi: 10.1016/j.toxlet.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Liu F, Pouponnot C, Massague J. Dual role of the smad4/dpc4 tumor suppressor in tgfbeta-inducible transcriptional complexes. Genes and Dev. 1997;11:3157–3167. doi: 10.1101/gad.11.23.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massague J. A human mad protein acting as a bmp-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Harmelink C, Peng Y, Chen Y, Wang Q, Jiao K. Chd7 interacts with bmp r-smads to epigenetically regulate cardiogenesis in mice. Hum Mol Genet. 2014;23:2145–2156. doi: 10.1093/hmg/ddt610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial bmpr-ii with bmp9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777–785. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucarini L, Durante M, Lanzi C, Pini A, Boccalini G, Calosi L, Moroni F, Masini E, Mannaioni G. Hydamtiq, a selective parp-1 inhibitor, improves bleomycin-induced lung fibrosis by dampening the tgf-beta/smad signalling pathway. J Cell Mol Med. 2016 doi: 10.1111/jcmm.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Gao X, Peng L, Sun H, Dai G. Effects of hydrochlorothiazide on cardiac remodeling in a rat model of myocardial infarction-induced congestive heart failure. Eur J Pharmacol. 2011;667:314–321. doi: 10.1016/j.ejphar.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J. Macrophage-stimulated cardiac fibroblast production of il-6 is essential for tgf beta/smad activation and cardiac fibrosis induced by angiotensin ii. PloS one. 2012;7:e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Debenedittis P, Sun Y, Chen J, Yuan K, Jiao K, Chen Y. Vascular smooth muscle cell smad4 gene is important for mouse vascular development. Arterioscler Thromb Vasc Biol. 2012;32:2171–2177. doi: 10.1161/ATVBAHA.112.253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki M, Izumi M, Oshima Y, Nakaoka Y, Kuroda T, Kimura R, Sugiyama S, Terai K, Kitakaze M, Yamauchi-Takihara K, Kawase I, Hirota H. Smad1 protects cardiomyocytes from ischemia-reperfusion injury. Circulation. 2005;111:2752–2759. doi: 10.1161/CIRCULATIONAHA.104.490946. [DOI] [PubMed] [Google Scholar]
- Massaous J, Hata A. Tgf-beta signalling through the smad pathway. Trends Cell Biol. 1997;7:187–192. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- Meng XM, Chung AC, Lan HY. Role of the tgf-beta/bmp-7/smad pathways in renal diseases. Clin Sci. 2013;124:243–254. doi: 10.1042/CS20120252. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Maeda S, Imamura T. Bmp receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16:251–263. doi: 10.1016/j.cytogfr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Monzen K, Hiroi Y, Kudoh S, Akazawa H, Oka T, Takimoto E, Hayashi D, Hosoda T, Kawabata M, Miyazono K, Ishii S, Yazaki Y, Nagai R, Komuro I. Smads, tak1, and their common target atf-2 play a critical role in cardiomyocyte differentiation. J Cell Biol. 2001;153:687–698. doi: 10.1083/jcb.153.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in tgf-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Mungrue IN, Zhao P, Yao Y, Meng H, Rau C, Havel JV, Gorgels TG, Bergen AA, MacLellan WR, Drake TA, Bostrom KI, Lusis AJ. Abcc6 deficiency causes increased infarct size and apoptosis in a mouse cardiac ischemia-reperfusion model. Arterioscler Thromb Vasc Biol. 2011;31:2806–2812. doi: 10.1161/ATVBAHA.111.237420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi N, Takahashi T, Ogata T, Adachi A, Imoto-Tsubakimoto H, Ueyama T, Matsubara H. Parm-1 promotes cardiomyogenic differentiation through regulating the bmp/smad signaling pathway. Biochem Biophys Res Commun. 2012;428:500–505. doi: 10.1016/j.bbrc.2012.10.078. [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of smad7, a tgfbeta-inducible antagonist of tgf-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Palaniyandi SS, Ferreira JC, Brum PC, Mochly-Rosen D. Pkcbetaii inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses. J Cell Mol Med. 2011;15:1769–1777. doi: 10.1111/j.1582-4934.2010.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papangeli I, Scambler PJ. Tbx1 genetically interacts with the transforming growth factor-beta/bone morphogenetic protein inhibitor smad7 during great vessel remodeling. Circ Res. 2013;112:90–102. doi: 10.1161/CIRCRESAHA.112.270223. [DOI] [PubMed] [Google Scholar]
- Patterson GI, Padgett RW. Tgf beta-related pathways. Roles in caenorhabditis elegans development. Trends Genet TIG. 2000;16:27–33. doi: 10.1016/s0168-9525(99)01916-2. [DOI] [PubMed] [Google Scholar]
- Pokharel S, van Geel PP, Sharma UC, Cleutjens JP, Bohnemeier H, Tian XL, Schunkert H, Crijns HJ, Paul M, Pinto YM. Increased myocardial collagen content in transgenic rats overexpressing cardiac angiotensin-converting enzyme is related to enhanced breakdown of n-acetyl-ser-asp-lys-pro and increased phosphorylation of smad2/3. Circulation. 2004;110:3129–3135. doi: 10.1161/01.CIR.0000147180.87553.79. [DOI] [PubMed] [Google Scholar]
- Ricard N, Ciais D, Levet S, Subileau M, Mallet C, Zimmers TA, Lee SJ, Bidart M, Feige JJ, Bailly S. Bmp9 and bmp10 are critical for postnatal retinal vascular remodeling. Blood. 2012;119:6162–6171. doi: 10.1182/blood-2012-01-407593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin ii activates the smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation. 2005;111:2509–2517. doi: 10.1161/01.CIR.0000165133.84978.E2. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schluter KD, Bohm M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing tgf-beta(1) Am J Physiol Heart Circ Physiol. 2002;283:H1253–1262. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- Saika S, Ikeda K, Yamanaka O, Flanders KC, Nakajima Y, Miyamoto T, Ohnishi Y, Kao WW, Muragaki Y, Ooshima A. Therapeutic effects of adenoviral gene transfer of bone morphogenic protein-7 on a corneal alkali injury model in mice. Lab Invest. 2005;85:474–486. doi: 10.1038/labinvest.3700247. [DOI] [PubMed] [Google Scholar]
- Sanders LN, Schoenhard JA, Saleh MA, Mukherjee A, Ryzhov S, McMaster WG, Jr, Nolan K, Gumina RJ, Thompson TB, Magnuson MA, Harrison DG, Hatzopoulos AK. Bmp antagonist gremlin 2 limits inflammation after myocardial infarction. Circulation Res. 2016;119:434–449. doi: 10.1161/CIRCRESAHA.116.308700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, ten Dijke P. Bmp-9 signals via alk1 and inhibits bfgf-induced endothelial cell proliferation and vegf-stimulated angiogenesis. J Cell Sci. 2007;120:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN, Drexler H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin ii type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation. 1994;89:2273–2282. doi: 10.1161/01.cir.89.5.2273. [DOI] [PubMed] [Google Scholar]
- Schneiders D, Heger J, Best P, Michael Piper H, Taimor G. Smad proteins are involved in apoptosis induction in ventricular cardiomyocytes. Cardiovasc Res. 2005;67:87–96. doi: 10.1016/j.cardiores.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Shen XC, Yang YP, Xiao TT, Peng J, Liu XD. Protective effect of oxymatrine on myocardial fibrosis induced by acute myocardial infarction in rats involved in tgf-beta(1)-smads signal pathway. J Asian Nat Prod Res. 2011;13:215–224. doi: 10.1080/10286020.2010.550883. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Sui X, Li D, Qiu H, Gaussin V, Depre C. Activation of the bone morphogenetic protein receptor by h11kinase/hsp22 promotes cardiac cell growth and survival. Circ Res. 2009;104:887–895. doi: 10.1161/CIRCRESAHA.108.192328. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Chang C, Yingling JM, Wang XF, Hemmati-Brivanlou A. Smad5 induces ventral fates in xenopus embryo. Dev Biol. 1997;184:402–405. doi: 10.1006/dbio.1997.8548. [DOI] [PubMed] [Google Scholar]
- Tandon A, Sharma A, Rodier JT, Klibanov AM, Rieger FG, Mohan RR. Bmp7 gene transfer via gold nanoparticles into stroma inhibits corneal fibrosis in vivo. PloS one. 2013;8:e66434. doi: 10.1371/journal.pone.0066434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseliou E, Reich H, de Couto G, Terrovitis J, Sun B, Liu W, Marban E. Cardiospheres reverse adverse remodeling in chronic rat myocardial infarction: Roles of soluble endoglin and tgf-beta signaling. Basic Res Cardiol. 2014;109:443. doi: 10.1007/s00395-014-0443-8. [DOI] [PubMed] [Google Scholar]
- Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, Yoneyama K, Murata E, Machiya A, Jimi E, Kokabu S, Katagiri T. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep. 2014;4:7596. doi: 10.1038/srep07596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton PD, Davies RJ, Tajsic T, Morrell NW. Transforming growth factor-beta(1) represses bone morphogenetic protein-mediated smad signaling in pulmonary artery smooth muscle cells via smad3. Am J Respir Cell Mol Biol. 2013;49:1135–1145. doi: 10.1165/rcmb.2012-0470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilahur G, Juan-Babot O, Pena E, Onate B, Casani L, Badimon L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J Mol Cell Cardiol. 2011;50:522–533. doi: 10.1016/j.yjmcc.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Wang B, Hao J, Jones SC, Yee MS, Roth JC, Dixon IM. Decreased smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol Heart Circ Physiol. 2002;282:H1685–1696. doi: 10.1152/ajpheart.00266.2001. [DOI] [PubMed] [Google Scholar]
- Wang LF, Zhang L, Zhang RY, Li SJ. relationship between expression of smad and ventricular remodeling after myocardial infarction in rats. Zhonghua Xin Xue Guan Bing Za Zhi. 2005;33:932–935. [PubMed] [Google Scholar]
- Wang ZF, Wang NP, Harmouche S, Philip T, Pang XF, Bai F, Zhao ZQ. Postconditioning promotes the cardiac repair through balancing collagen degradation and synthesis after myocardial infarction in rats. Basic Res Cardiol. 2013;108:318. doi: 10.1007/s00395-012-0318-9. [DOI] [PubMed] [Google Scholar]
- Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, Dietz HC, Gabrielson KL, Kass DA, Semenza GL. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of tgf-beta signaling. Proc Natl Acad Sci USA. 2012;109:E841–850. doi: 10.1073/pnas.1202081109. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY, Yan BP, Yu CM, Lan HY. Smad7 inhibits angiotensin ii-induced hypertensive cardiac remodelling. Cardiovasc Res. 2013;99:665–673. doi: 10.1093/cvr/cvt151. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, Meurer SK. Bmp-7 counteracting tgf-beta1 activities in organ fibrosis. Front Biosci. 2013;18:1407–1434. doi: 10.2741/4189. [DOI] [PubMed] [Google Scholar]
- Wu X, Sagave J, Rutkovskiy A, Haugen F, Baysa A, Nygard S, Czibik G, Dahl CP, Gullestad L, Vaage J, Valen G. Expression of bone morphogenetic protein 4 and its receptors in the remodeling heart. Life Sci. 2014;97:145–154. doi: 10.1016/j.lfs.2013.12.030. [DOI] [PubMed] [Google Scholar]
- Wu XL, Zeng WZ, Jiang MD, Qin JP, Xu H. Effect of oxymatrine on the tgfbeta-smad signaling pathway in rats with ccl4-induced hepatic fibrosis. World J Gastroenterol. 2008;14:2100–2105. doi: 10.3748/wjg.14.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang FL, Guo M, Yutzey KE. Overexpression of tbx20 in adult cardiomyocytes promotes proliferation and improves cardiac function after myocardial infarction. Circulation. 2016;133:1081–1092. doi: 10.1161/CIRCULATIONAHA.115.019357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Chang G, Liu J, Sun G, Liu L, Qin S, Zhang D. Simvastatin ameliorates ventricular remodeling via the tgfbeta1 signaling pathway in rats following myocardial infarction. Mol Med Rep. 2016;13:5093–5101. doi: 10.3892/mmr.2016.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zhang J, Peng X, Dong Y, Jia L, Li H, Du J. The notch gamma-secretase inhibitor ameliorates kidney fibrosis via inhibition of tgf-beta/smad2/3 signaling pathway activation. Int J Biochem Cell Biol. 2014;55:65–71. doi: 10.1016/j.biocel.2014.08.009. [DOI] [PubMed] [Google Scholar]
- Xu F, Liu C, Zhou D, Zhang L. Tgf-beta/smad pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64:157–167. doi: 10.1369/0022155415627681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi S, Aihara K, Ikeda Y, Sumitomo Y, Yoshida S, Ise T, Iwase T, Ishikawa K, Azuma H, Akaike M, Matsumoto T. Pitavastatin, an hmg-coa reductase inhibitor, exerts enos-independent protective actions against angiotensin ii induced cardiovascular remodeling and renal insufficiency. Circ Res. 2008;102:68–76. doi: 10.1161/CIRCRESAHA.107.163493. [DOI] [PubMed] [Google Scholar]
- Yang YZ, Fan TT, Gao F, Fu J, Liu Q. Exogenous cytochrome c inhibits the expression of transforming growth factor-beta1 in a mouse model of sepsis-induced myocardial dysfunction via the smad1/5/8 signaling pathway. Mol Med Rep. 2015;12:2189–2196. doi: 10.3892/mmr.2015.3629. [DOI] [PubMed] [Google Scholar]
- Zeng W, Chen W, Leng X, He JG, Ma H. Chronic angiotensin-(1–7) administration improves vascular remodeling after angioplasty through the regulation of the tgf-beta/smad signaling pathway in rabbits. Biochem Biophys Res Commun. 2009;389:138–144. doi: 10.1016/j.bbrc.2009.08.112. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Gao X, Wu Q, Peng L, Lin J, Zuo Z. Fluvastatin decreases cardiac fibrosis possibly through regulation of tgf-beta(1)/smad 7 expression in the spontaneously hypertensive rats. Eur J Pharmacol. 2008;587:196–203. doi: 10.1016/j.ejphar.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Shao L, Ma A, Guan G, Wang J, Wang Y, Tian G. Telmisartan delays myocardial fibrosis in rats with hypertensive left ventricular hypertrophy by tgf-beta1/smad signal pathway. Hypertens Res. 2014;37:43–49. doi: 10.1038/hr.2013.119. [DOI] [PubMed] [Google Scholar]
- Zhao M, Zheng S, Yang J, Wu Y, Ren Y, Kong X, Li W, Xuan J. Suppression of tgf-beta1/smad signaling pathway by sesamin contributes to the attenuation of myocardial fibrosis in spontaneously hypertensive rats. PloS one. 2015;10:e0121312. doi: 10.1371/journal.pone.0121312. [DOI] [PMC free article] [PubMed] [Google Scholar]