

Abstract
Background
Vitelliform dystrophies are a group of macular degenerative diseases characterized by round yellow lesions in the macula. While often idiopathic, vitelliform dystrophies include inherited maculopathies such as Best disease and some cases of pattern dystrophy. The prevalence of vitelliform dystrophies in the United States has not been reported. This study examined the prevalence of vitelliform dystrophies in Olmsted County, Minnesota.
Materials and Methods
The Rochester Epidemiology Project database was used to identify all cases of vitelliform or pattern dystrophy in Olmsted County from 1/1/2000–12/31/2014.
Results
27 patients had true vitelliform lesions, indicating a prevalence of 1 in 5500. Of these, 2 had genetically confirmed Best disease, and an additional 5–7 carried a diagnosis of Best disease, which chart reviews confirmed as probable cases; 18–20 patients had adult-onset vitelliform macular dystrophy. The prevalence of Best disease was 1 in 16,500 to 1 in 21,000. Adult-onset vitelliform macular dystrophy was found in 1 in 7400 to 1 in 8200.
Conclusions
Vitelliform dystrophies affect 1 in 5500 individuals in Olmsted County. While the values in this study provide good estimates for the prevalence of Best disease versus adult-onset vitelliform macular dystrophy, the results are limited by dependence on diagnoses made by other ophthalmologists and underutilization of genetic testing. Thus, these diseases should be thought of as at least as prevalent as reported here. As therapies for Best disease and other macular degenerative diseases are quickly becoming a reality, genetic testing should be employed as the gold standard for diagnosis of these diseases.
Keywords: Best disease, epidemiology, vitelliform dystrophy
INTRODUCTION
Vitelliform dystrophies are a group of macular degenerative disorders characterized by the accumulation of yellow subretinal material that, at some stages of disease, clinically resembles an egg yolk.1 Vitelliform lesions are most often acquired, but this group of dystrophies also includes inherited macular degenerations, such as Best vitelliform macular dystrophy (BVMD), adult-onset vitelliform macular dystrophy (AVMD), and some cases of pattern dystrophy.1–4 The prevalence of vitelliform dystrophies, and particularly the prevalence of Best disease, has not previously been reported in the United States.
BVMD is an autosomal dominantly inherited macular dystrophy caused by mutations in the gene BEST1 that results in vision loss as a result of accumulation of lipofuscin in the retinal pigment epithelium.2,5,6 While symptoms can occur during childhood, age of onset is variable, and 7% to 9% of carriers of BVMD-causing mutations may be asymptomatic, consistent with incomplete penetrance of this autosomal-dominant disease.7 The disease has classically been divided into five stages including: 1) subtle retinal pigment epithelium changes in the pre-vitelliform stage, 2) the classic vitelliform egg-yolk lesion, 3) pseudohypopyon, 4) the vitelliruptive scrambled egg stage, and 5) geographic atrophy.8,9 However, not all patients progress predictably through these stages, and some patients also develop choroidal neovascularization.9,10 Patients with BVMD characteristically have a normal electroretinogram (ERG) and an abnormal electrooculogram (EOG) with an Arden ratio <1.55, though cases with normal EOGs have been reported.8,9,11 The prevalence of BVMD in the United States is unknown; and worldwide, only two previous studies have attempted to estimate this value. A study in Sweden estimated the prevalence of BVMD to be 2 in 10,000, while a study in Demark estimated this value at 1.5 in 100,000.12,13
AVMD, also known as adult-onset foveomacular vitelliform dystrophy, is a form of pattern dystrophy. AVMD typically manifests between the ages of 30 and 50 years with symmetric, solitary, round or oval vitelliform lesions of 1/3 to 1 disc diameter in size.1,8 Some patients develop blurry vision and metamorphopsia, while others remain asymptomatic.1,8 Clinically, AVMD is distinguished from BVMD by age of onset, smaller lesion size, slower disease progression, and a normal EOG.1,3,14,15 However, age of onset should arguably not be a criterion as late onset for BVMD is not uncommon.16 Onset of clinical symptoms in BVMD is highly variable, and 7% to 9% of carriers of BVMD-causing mutations never present with clinical disease.7,17 In some cases, autosomal dominant inheritance of AVMD has been demonstrated, and mutations in peripherin/RDS have been implicated in addition to mutations in BEST1.18–21 Whether AVMD, due to BEST1 mutation, should be considered BVMD is a question that is open to debate.7,22,23 We are not aware of any previous reports regarding the incidence of AVMD.
Previously, there has been no cure for these diseases, and currently-available therapies are directed toward treating macular edema and neovascularization when it develops.24–26 However, as research in the areas of stem cells and gene therapy appears to be advancing the prospect of therapies for vitelliform dystrophies, knowledge on the prevalence of these diseases has become more relevant.27–30 Thus, this study aims to provide an estimate of the prevalence of vitelliform dystrophies in the United States by utilizing the Rochester Epidemiology Project (REP) database to determine the prevalence of these diseases in Olmsted County, MN.
MATERIALS AND METHODS
This study utilized the REP medical records linkage system. The database consists only of residents of Olmsted County, MN, and queries yield only subjects who have provided informed consent for research. Medical care of this population is provided primarily by Mayo Clinic and Olmsted Medical Center, but additional small independent clinics are included in order to capture nearly all Olmsted County residents.31,32 There are approximately 148,201 unique patient records available for review at the time of this report.
We searched the REP database for ICD-9 codes 362.70–362.77 in order to capture all patients with ophthalmologist-diagnosed vitelliform dystrophy, including BVMD, AVMD, and pattern dystrophy. We found 244 patients carrying one of these diagnosis codes between 1/1/2000 and 12/31/2014. These 244 patients were reviewed by a single investigator to eliminate unrelated diagnoses, such as retinitis pigmentosa, Stargardt’s disease, Leber’s congenital amaurosis, and rod-cone and cone-rod dystrophy. The records of the 40 remaining patients, including any available images, ERGs, EOGs, and genetic testing, were reviewed by all three investigators. This study is in compliance with the Health Insurance Portability and Accountability Act (HIPAA), and the Mayo Institutional Review Board has approved this study.
RESULTS
Of the 40 patients whose records were reviewed in detail, 7 were eliminated due to insufficient data, namely normal or unavailable EOGs in the absence of available imaging studies or genetic testing. An additional six patients were eliminated after record review revealed alternative diagnoses, including age-related macular degeneration, occult macular dystrophy, and central serous chorioretinopathy. No patients were found to have pattern dystrophy. The remaining patients, 27 had true vitelliform lesions. Using 148,201 as the number of unique patients in the REP database, which closely approximates the population of Olmsted County, the prevalence of vitelliform dystrophy in Olmsted County was 27 in 148,201 (~1 in 5500). These patients were further categorized as genetically confirmed BVMD, possible Best disease, and AVMD (Figure 1). Patients were categorized based on available genetic testing, fundus photos, optical coherence tomography (OCT), fluorescein angiography, autofluorescence, ERGs, EOGs, and reported family history of BVMD. The Gass definition for AVMD was used, meaning lesions of approximately 1/3 disc diameter (500 μm) in size.8 Table 1 describes the available data for patients categorized as confirmed or possible BVMD. Of the patients with AVMD, two had genetic testing information, which was unremarkable; three had available EOGs, which showed normal Arden ratios; all patients had available fundus photos; and all except for two patients had available OCTs. Examples of a representative patient with AVMD and a patient with possible BVMD can be seen in Figure 2.
Figure 1.
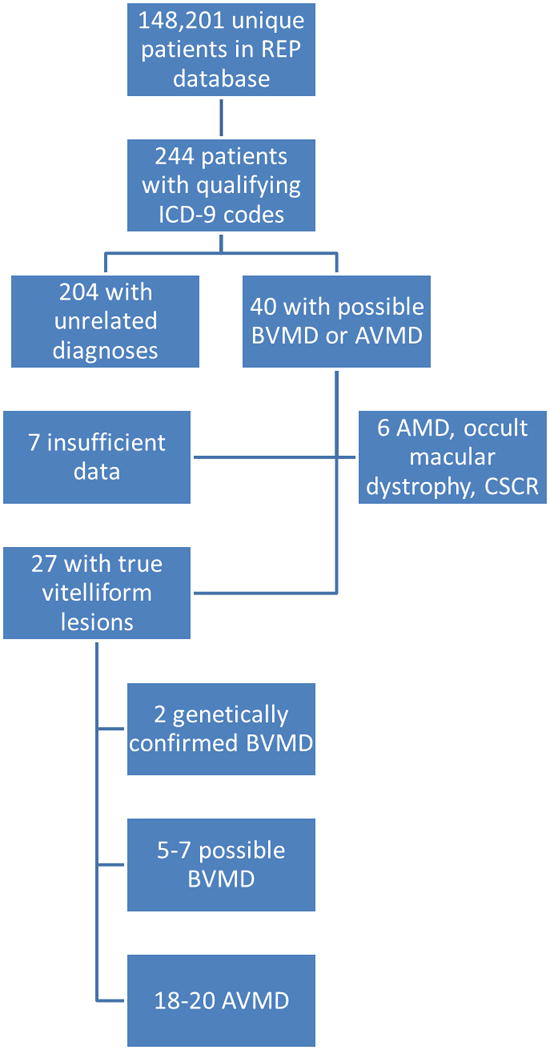
Patients in the REP database with vitelliform dystrophy. Eight unique ICD-9 codes were searched to capture all patients with vitelliform lesions. Patients with unrelated diagnoses, absence of vitelliform lesions, or insufficient available data were eliminated, leaving 27 patients with confirmed vitelliform lesions. Only two patients had genetically confirmed BVMD. There were 5 to 7 patients with possible BVMD and 18 to 20 patients with AVMD.
Table 1.
Available information for patients with confirmed and possible BVMD.
| Patient | Diagnosis | Sex | Age at Diagnosis | Arden Ratio | Review Images | Relatives with Macular Dystrophy |
|---|---|---|---|---|---|---|
| 1 | Genetically-confirmed Best disease | Female | 74 | Right eye 1.28; Left eye 1.16 | Fundus photos and OCT | Mother |
| 2 | Genetically-confirmed Best disease | Female | 33 | No available EOG | Fundus photos and OCT | Brothers and nephew |
| 3 | Possible autosomal recessive bestrophinopathy | Male | 26 | No available EOG | Fundus photos and OCT | Brother |
| 4 | Possible Best disease | Male | 27 | No available EOG | Fundus photos only | Sister |
| 5 | Possible Best disease | Male | 11 | No available EOG | Fundus photos only | None known |
| 6 | Possible Best disease | Female | 10 | No available EOG | Fundus photos and OCT | Father |
| 7 | Possible Best disease | Male | 17 | No available EOG | No imaging available | Son |
| 8 | Possible Best disease | Female | 67 | No available EOG | Fundus photos and OCT | Father |
| 9 | Possible Best disease | Female | 85 | No available EOG | Fundus photos and OCT | None known |
Figure 2.
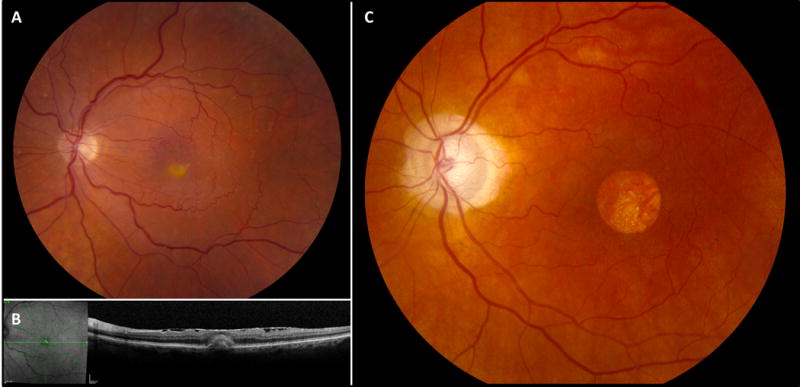
Clinical imaging studies for patients with vitelliform lesions. Color fundus photo of the left eye (A) in a patient with AVMD demonstrates a characteristic vitelliform lesion. The corresponding optical coherence tomography for the patient in A is shown in B. Color fundus photo of the left eye (C) in a patient with possible Best disease demonstrates geographic atrophy.
Disappointingly, only two patients, both female, had BVMD confirmed by genetic testing. One (Patient 1) underwent genetic testing via a research protocol and was found to have a novel mutation R13C.33,34 The other (Patient 2) underwent clinical genetic testing and had a heterozygous AAG>AGG nucleotide substitution, resulting in an amino acid change of Lys30Arg in the coding sequence of BEST1, which is known to be disease-causing. An additional five patients (four male, one female) were categorized as possible Best disease, which was consistent with the original ophthalmology diagnosis. None of the additional five had undergone genetic testing. One of the five, a male whose parents were cousins, had multifocal vitelliform lesions consistent with a diagnosis of autosomal-recessive bestrophinopathy (Figure 3). Of these seven patients, six had a family history positive for Best disease. The remaining 20 (11 male, 9 female) patients were categorized as AVMD. However, at least two of these patients (Patients 8 and 9 in Table 1), both female, could possibly have BVMD, and insufficient data were available to categorize these patients with a higher degree of certainty. Thus, we report the prevalence for these diseases as a range. The prevalence of BVMD was 7 to 9 in 148,201 (~1 in 21,000 to 1 in 16,500), and the prevalence of AVMD was 18 to 20 in 148,201 (~1 in 8200 to 1 in 7400).
Figure 3.
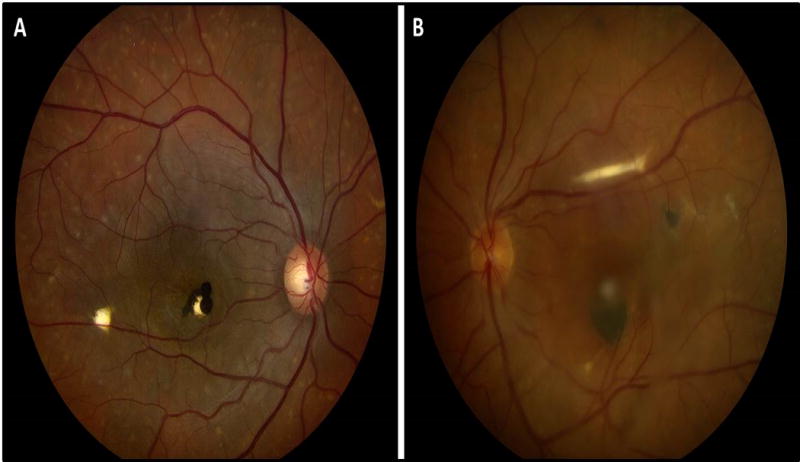
Color fundus photographs of the right (A) and left (B) eyes of a patient with presumed autosomal-recessive bestrophinopathy (ARB). Both eyes demonstrate a darkly pigmented subretinal mass in the fovea with multifocal vitelliform lesions throughout the fundus, more clearly seen in the right eye. The presence of multifocal vitelliform lesions, rather than a classic egg yolk lesion in the setting of consanguinity, is consistent with ARB.
DISCUSSION
The prevalence of vitelliform dystrophy in this study was approximately 1 in 5500. While this provides a good estimation of the prevalence of these diseases, a notable shortcoming of this study is that we had to rely upon the diagnoses made by other ophthalmologists. It is possible that additional patients with vitelliform dystrophy were misdiagnosed such that they were not found in our initial REP search. Thus, we find it necessary to state that the prevalence in Olmsted County is at least as high as reported in this study. It could possibly be higher.
The prevalence of BVMD in this study falls between the previously reported values of 2 in 10,000 and 1.5 in 100,000 in the Swedish and Denmark studies, respectively.12,13 Thus, we believe this to be a good approximation of the prevalence of BVMD in whites in the United States. Of note, the population in Olmsted County is primarily white, so this prevalence may be less representative of a more racially diverse population.
It is also important to note that our study methods differed significantly compared to those in the Swedish and Denmark studies. The author of the Swedish paper maintained continuous personal contact with the departments of pediatrics and ophthalmology at the regional University Hospital, which serves as a referral center for the country.13 Over the course of many years, seven probands came to the author’s attention. Additionally, chart review was undertaken on all patients born from the 1st–8th of each month from 1966 through 1971, which resulted in the discovery of six new probands. Results of an unpublished patient survey regarding hereditary macular degeneration led to the discovery of one additional proband, and review of membership records for the Association for the Blind and Weaksighted, as well as a social record for the Adviser for weaksighted people in the county of Västerbotten, resulted in four new probands. Finally, chart review was performed for all patients with a retinal disorder who received an eye exam at the University Hospital in 1971, and three additional probands were found. Patients with uncertain diagnoses were interviewed and further investigated when possible. Available fundus photographs were reviewed; and in many cases, relatives of patients with hereditary macular degeneration were examined at the University Hospital. Of the 95 subjects confirmed to have hereditary macular degeneration in the study, 75 were from a single family. This differs significantly from our study, given the multitude of sources that were searched to identify new probands. Our search was limited to a single computer database. This method was chosen, however, to avoid referral bias, which would undoubtedly lead to an inaccurate estimation of prevalence if a single academic hospital’s records were reviewed for all patients regardless of place of residence. Unfortunately, however, these methods did not include patient contact and prevented the discovery of accurate pedigree information. With the information in the available records, none of the patients with confirmed or possible BVMD in our study appeared to be related. While many of them did have a positive family history reported, it is likely that their affected relatives did not reside in Olmsted County, leading to exclusion from our study.
The Denmark study identified probands from files of the National Low Vision Eye Clinic, which is a national referral center.12 Retrospective chart review was undertaken for all patients examined between 1980 and 2006, with additional patients being identified from a national family archive for genetic eye diseases. Patients with a recently established genetic diagnosis were offered a clinical follow-up exam. Again, this study allowed for patient contact, and family members were able to be identified through a national family archive, which was not possible in our study. The search for probands in this study did not span as many different methods at the Swedish study, nor were efforts made to examine patient family members. This may somewhat explain the lower prevalence that was found. However, it is still curious that our study results aligned more with those of the Swedish study, as our study did not permit patient contact. This could possibly be due to patient ancestry, but this remains unclear.
The prevalence of AVMD has not previously been reported. However, given the frequency with which vitelliform lesions are encountered in the clinic and the fact that AVMD encompasses a broad array of inherited and idiopathic diseases, it is not surprising that it is about three times as common as BVMD.
Unfortunately, unlike the Swedish and Denmark studies, details regarding pedigree structure could not be sufficiently determined in this study, and all but three of the patients with AVMD lacked both EOG and genetic testing. Thus, it is possible that more of the patients categorized as AVMD truly have BVMD, which could not be determined by examination of available clinical records. Furthermore, even of the patients categorized as BVMD in this study, only two had available genetic testing. We found this to be disappointing, as it significantly hampered our ability to definitively confirm the diagnoses that these patients carried.
We perceive two significant barriers to genetic testing in this patient population. The first is lack of insurance coverage for the tests, which makes genetic testing cost-prohibitive for many patients. The second is that, in the past, there has been no cure for vitelliform dystrophies. Thus, it was widely accepted that genetic testing would not change the management of these patients. However, as treatments are available for complications of these diseases, such as macular edema and choroidal neovascularization and new therapies for these disorders are on the horizon, this is no longer an appropriate assumption. Patients with genetically-confirmed bestrophinopathies now have the opportunity to enroll in research trials that may yield effective treatments for these diseases.33 While these trials are still in the data-gathering and laboratory phases, future therapeutic clinical trials may come as a result of this work, possibly involving gene therapy or stem cells.28,29 These therapies will likely be somewhat individualized, depending on the specific causative mutation(s) found in a given patient, which will undoubtedly make genetic testing a critical aspect of patient care. Moreover, the knowledge of the underlying genetics of these inherited diseases has important implications for family members who may not currently have any symptoms but are at risk of developing vision loss in the future. With further research advances, retinal damage may be prevented before it occurs if certain genetic mutations are known to be present.
Thus, we believe all patients with vitelliform dystrophies should undergo genetic testing, as this is now the gold standard for diagnosis. In fact, in the era of whole exome sequencing, ERGs and EOGs are no longer necessary for the diagnosis of bestrophinopathies. A shift toward genetic testing will save patients the time and discomfort of ERG and EOG testing and will allow for correct diagnoses to be made in practices that do not have the means to perform reliable ERGs and EOGs.
While the data in this paper provide a good estimate of the impact of vitelliform dystrophies, and particularly BVMD, on the population in the United States, this prevalence will be better defined in the future after widespread genetic testing is in place. The difficulty categorizing the patients in this study combined with current research that holds promise for genetically-driven cures for certain types of vitelliform dystrophies highlights the importance of shifting practice patterns toward genetic confirmation of inherited retinopathies.
Acknowledgments
FUNDING
This study was supported by unrestricted grants from Research to Prevent Blindness Inc., New York, NY, the Paul Family, and the Deshong Family.
This study was made possible using the resources of the Rochester Epidemiology Project, which is supported by the National Institute on Aging of the National Institutes of Health under Award Number R01AG034676. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Color versions of one or more figures in the article can be found online at http://www.tandfonline.com/iopg.
DECLARATION OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974;72:139–156. [PMC free article] [PubMed] [Google Scholar]
- 2.Blodi CF, Stone EM. Best’s vitelliform dystrophy. Ophthalmic Paediatr Genet. 1990;11:49–59. [PubMed] [Google Scholar]
- 3.Brecher R, Bird AC. Adult vitelliform macular dystrophy. Eye (Lond) 1990;4(Pt 1):210–215. doi: 10.1038/eye.1990.28. [DOI] [PubMed] [Google Scholar]
- 4.Gutman I, Walsh JB, Henkind P. Vitelliform macular dystrophy and butterfly-shaped epithelial dystrophy: a continuum? Br J Ophthalmol. 1982;66:170–173. doi: 10.1136/bjo.66.3.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marquardt A, Stohr H, Passmore LA, et al. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease) Hum Mol Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 6.Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 7.Marmorstein AD, Cross HE, Peachey NS. Functional roles of bestrophins in ocular epithelia. Prog Retin Eye Res. 2009;28:206–226. doi: 10.1016/j.preteyeres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal A. Gass’ Atlas of Macular Diseases. Philadelphia, PA: Elsevier Saunders; 2012. p. 1378. [Google Scholar]
- 9.Sohn EH, Mullins RF, Stone EM. Macular Dytsrophies. In: Ryan S, editor. Retina. 4. Vol. 2. Elsevier Inc.; 2006. pp. 852–890. [Google Scholar]
- 10.MacDonald IM, Lee T. Best Vitelliform Macular Dystrophy - Synonyms: Best Macular Dystrophy, Vitelliform Macular Dystrophy Type 2.2003 [Updated 2013 Dec 12] In: Pagon Ra, AM, Ardinger Hh., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2016. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1167/ [Google Scholar]
- 11.Testa F, Rossi S, Passerini I, et al. A normal electro-oculography in a family affected by best disease with a novel spontaneous mutation of the BEST1 gene. Br J Ophthalmol. 2008;92:1467–1470. doi: 10.1136/bjo.2008.143776. [DOI] [PubMed] [Google Scholar]
- 12.Bitner H, Schatz P, Mizrahi-Meissonnier L, et al. Frequency, genotype, and clinical spectrum of best vitelliform macular dystrophy: data from a national center in Denmark. Am J Ophthalmol. 2012;154:403–412 e404. doi: 10.1016/j.ajo.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 13.Nordstrom S. Hereditary macular degeneration–a population survey in the country of Vsterbotten, Sweden. Hereditas. 1974;78:41–62. doi: 10.1111/j.1601-5223.1974.tb01427.x. [DOI] [PubMed] [Google Scholar]
- 14.Epstein GA, Rabb MF. Adult vitelliform macular degeneration: diagnosis and natural history. Br J Ophthalmol. 1980;64:733–740. doi: 10.1136/bjo.64.10.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kingham JD, Lochen GP. Vitelliform macular degeneration. Am J Ophthalmol. 1977;84:526–531. doi: 10.1016/0002-9394(77)90446-9. [DOI] [PubMed] [Google Scholar]
- 16.Renner AB, Tillack H, Kraus H, et al. Late onset is common in best macular dystrophy associated with VMD2 gene mutations. Ophthalmology. 2005;112:586–592. doi: 10.1016/j.ophtha.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 17.Nordstrom S, Thorburn W. Dominantly inherited macular degeneration (Best’s disease) in a homozygous father with 11 children. Clin Genet. 1980;18:211–216. doi: 10.1111/j.1399-0004.1980.tb00874.x. [DOI] [PubMed] [Google Scholar]
- 18.Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Kramer F, White K, Pauleikhoff D, et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 20.Vine AK, Schatz H. Adult-onset foveomacular pigment epithelial dystrophy. Am J Ophthalmol. 1980;89:680–691. doi: 10.1016/0002-9394(80)90288-3. [DOI] [PubMed] [Google Scholar]
- 21.Wells J, Wroblewski J, Keen J, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 22.Boon CJ, Klevering BJ, Leroy BP, et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Chowers I, Tiosano L, Audo I, et al. Adult-onset foveomacular vitelliform dystrophy: A fresh perspective. Prog Retin Eye Res. 2015;47:64–85. doi: 10.1016/j.preteyeres.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Celea C, Pop M, Avidis-Zamfiroiu N. Evolution of Choroidal Neovascular Membrane in Best Disease after Single Intravitreal Bevacizumab. Case Report. Maedica (Buchar) 2015;10:61–64. [PMC free article] [PubMed] [Google Scholar]
- 25.Sodi A, Murro V, Caporossi O, et al. Long-Term Results of Photodynamic Therapy for Choroidal Neovascularization in Pediatric Patients with Best Vitelliform Macular Dystrophy. Ophthalmic Genet. 2015;36:168–174. doi: 10.3109/13816810.2015.1009121. [DOI] [PubMed] [Google Scholar]
- 26.Tiosano L, Jaouni T, Averbukh E, et al. Bevacizumab treatment for choroidal neovascularization associated with adult-onset foveomacular vitelliform dystrophy. Eur J Ophthalmol. 2014;24:890–896. doi: 10.5301/ejo.5000486. [DOI] [PubMed] [Google Scholar]
- 27.Johnson AA, Bachman LA, Gilles BJ, et al. Autosomal Recessive Bestrophinopathy Is Not Associated With the Loss of Bestrophin-1 Anion Channel Function in a Patient With a Novel BEST1 Mutation. Invest Ophthalmol Vis Sci. 2015;56:4619–4630. doi: 10.1167/iovs.15-16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R, Shen W, Kuai D, et al. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013;22:593–607. doi: 10.1093/hmg/dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang T, Justus S, Li Y, Tsang SH. BEST1: the Best Target for Gene and Cell Therapies. Mol Ther. 2015 doi: 10.1038/mt.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yvon C, Ramsden CM, Lane A, et al. Using Stem Cells to Model Diseases of the Outer Retina. Comput Struct Biotechnol J. 2015;13:382–389. doi: 10.1016/j.csbj.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurland LT, Molgaard CA. The patient record in epidemiology. Sci Am. 1981;245:54–63. doi: 10.1038/scientificamerican1081-54. [DOI] [PubMed] [Google Scholar]
- 32.Melton LJ., 3rd History of the Rochester Epidemiology Project. Mayo Clin Proc. 1996;71:266–274. doi: 10.4065/71.3.266. [DOI] [PubMed] [Google Scholar]
- 33.Marmorstein AD. ClinicalTrials.gov. Bethesda, MD: National Library of Medicine (US); 2014–2016. Mayo Clinic Stem Cell Models of Best Disease and Other Retinal Degenerative Diseases. [cited 2015 Nov 11] National Institutes of Health - https://clinicaltrials.gov/ct2/show/NCT02162953. https://clinicaltrials.gov/ct2/show/NCT02162953. Last Updated. [Google Scholar]
- 34.Shulz H. Leiden Open Variation Database – Variant Listings for Bestrophin 1 (BEST1) Regensburg, Germany: 2016. http://www-huge.uni-regensburg.de/BEST1_database/home.php?select_db=BEST1. Last Updated (May 27, 2013) [Google Scholar]