

Abstract
Reported herein is a light‐triggered organocatalytic strategy for the desymmetrization of achiral 2‐fluoro‐substituted cyclopentane‐1,3‐diketones. The chemistry is based on an intermolecular aldol reaction of photochemically generated hydroxy‐o‐quinodimethanes and simultaneously forges two adjacent fully substituted carbon stereocenters, with one bearing a stereogenic carbon–fluorine unit. The method uses readily available substrates, a simple chiral organocatalyst, and mild reaction conditions to afford an array of highly functionalized chiral 2‐fluoro‐3‐hydroxycyclopentanones.
Keywords: desymmetrization, fluorine, organocatalysis, photochemistry, synthetic methods
The unique properties of the carbon–fluorine (C−F) bond explain why organofluorine compounds play a central role in agrochemicals, pharmaceuticals, and materials science.1 For example, the selective incorporation of fluorine atoms into drug candidates has produced some best‐selling pharmaceuticals.2 Unsurprisingly, the synthetic community has worked hard to develop catalytic methods for the enantioselective synthesis of stereodefined fluorinated organic molecules.3 There has been great progress in the last decade, mainly by using either electrophilic4 or nucleophilic5 fluorinating agents in combination with chiral metal‐based or organic catalysts. An alternative approach uses racemic molecules, bearing a labile C−F stereogenic unit, which undergo a stereocontrolled carbon–carbon bond‐forming process while setting a configurationally stable fluorine‐containing stereocenter.6
We report herein a different catalytic strategy for the stereoselective construction of valuable fluorine‐containing quaternary stereocenters, which is based on the desymmetrization of achiral 2‐substituted‐2‐fluorocyclopentane‐1,3‐diketones (1), where a prochiral fluorine‐containing carbon center is preinstalled (Figure 1 a). The chemistry exploits the ability of a chiral organic catalyst to choose between enantiotopic carbonyl groups within 1 while facilitating a desymmetric intermolecular aldol process with a suitable nucleophile. The resulting symmetry‐breaking process generates two stereocenters simultaneously, and forges a C−F stereogenic unit far from the reaction site. The desymmetrization of centrosymmetric or meso compounds by chiral catalysts is a powerful method for making chiral molecules,7 and has been used extensively to synthesize natural compounds and biologically relevant molecules.7a However, this strategy has never yet been used to install quaternary fluorine‐containing stereocenters.8
Figure 1.
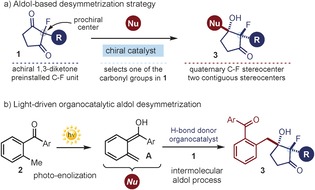
a) The proposed strategy for forging fluorine‐containing quaternary stereocenters by a desymmetrizing aldol reaction of achiral 2‐fluoro‐substituted cyclopentane‐1,3‐diketones (1). b) The photoenolization mechanism of 2‐methylbenzophenones (2) leading to the hydroxy‐o‐quinodimethanes A, which can serve as nucleophiles in the intermolecular aldol desymmetrization process. Nu=nucleophile.
The idea for selecting a nucleophilic partner suitable for the proposed aldol desymmetrization strategy came from our recent studies9 on the reactivity of hydroxy‐o‐quinodimethanes (A; Figure 1 b). These highly reactive intermediates are easily generated upon light irradiation of 2‐alkylbenzophenones (2).10 Our studies demonstrated that they can participate in a stereoselective processes promoted by chiral organic catalysts.11 Notably, the reactivity of A is not restricted to traditional cycloaddition mechanisms.9a They can also engage in intermolecular addition pathways.9b,9c Building on these precedents, we wondered whether the photoenolization strategy, combined with the action of a chiral organocatalyst, could be applied to the desymmetrization of 2‐fluoro‐2‐alkylsubstituted cyclopentane‐1,3‐diones (1). The resulting desymmetrizing aldol process affords chiral 2‐fluoro‐3‐hydroxycyclopentanones (3) with two adjacent fully substituted carbon stereocenters, with one bearing a stereogenic C−F unit (Figure 1 b).12 To the best of our knowledge, the chemistry provides the first example of an intermolecular‐aldol‐based desymmetrization of achiral cyclic 1,3‐diketones. Since the pioneering work of Hajos and Parrish,13 symmetric 1,3‐diketones of type 1 have been used extensively to design new desymmetric aldol processes. However, these have proceeded through intramolecular manifolds exclusively.7a, 13
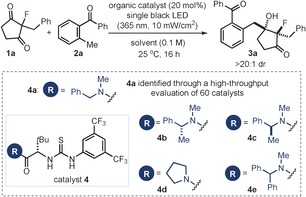
Our exploratory studies focused on the intermolecular aldol reaction between the commercially available 2‐methylbenzophenone (2 a) and 2‐benzyl‐2‐fluorocyclopentane‐1,3‐dione (1 a; see Table 1). The aldol acceptor is easily prepared from cyclopentane‐1,3‐dione following a Knoevenagel condensation/reduction/electrophilic fluorination (using Selectfluor) sequence. The catalytic experiments were conducted in toluene over 16 hours and under irradiation by a single black‐light‐emitting diode (black LED, λ max=365 nm). We initially evaluated a large set of different chiral organic catalysts with a known propensity to activate substrates by hydrogen‐bonding or ion‐pairing interactions.14 Specifically, we used a screening platform to automate the parallel execution of small‐scale reactions to determine the efficiency of a library of 60 novel and known organocatalysts in this desymmetric aldol reaction (see Figure S1 in the Supporting Information for selected results). This evaluation quickly identified the commercially available amido‐thiourea 4 a 15 as the most promising catalyst.
Table 1.
Optimization of the model reaction.[a]
| Entry | Catalyst | Solvent | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|
| 1 | 4 a | toluene | 50 | 76 |
| 2 | 4 b | toluene | 31 | 83 |
| 3 | 4 c | toluene | 41 | 87 |
| 4 | 4 d | toluene | 45 | 89 |
| 5 | 4 e | toluene | 40 | 90 |
| 6 | 4 e | o‐Cl2C6H4 | 69 | 91 |
| 7[d] | 4 e | o‐Cl2C6H4 | 89 | 90 |
| 8[e] | 4 e | o‐Cl2C6H4 | 0 | – |
| 9 | none | o‐Cl2C6H4 | 15 | 0 |
[a] Reactions performed over 16 h on a 0.1 mmol scale using 3 equiv of 2 a under illumination by black LED (λ max=365 nm) with an irradiance of 10±1 mW cm−2. [b] Yield of the isolated 3 a. [c] Enantiomeric excess of 3 a determined by UPC2 analysis on a chiral stationary phase. In all cases a single diastereoisomer was observed by 19F NMR analysis of the crude reaction mixture. [d] Irradiance of 15±1 mW cm−2 and 30 hours of reaction time. [e] In the dark.
The optimization studies were then continued on a meaningful experimental scale (0.1 mmol). We first confirmed that the catalyst 4 a (20 mol %) afforded the product 3 a with good chemical yield and stereoselectivity (Table 1, entry 1; single diastereoisomer). Modification of the benzyl amide moiety in 4 revealed that incorporating an additional stereogenic center imparted a slightly higher stereoinduction, but with a minimal matched/mismatched effect (entries 2 and 3). These results suggested that the steric profile of the tertiary benzyl amide component could play a more prominent role than the presence of a second stereocontrol element in dictating the reaction's stereoselectivity. In consonance with this reasoning, the best results were achieved with the catalysts 4 d and 4 e, both containing a more encumbered amide moiety but a single stereogenic center (entries 4 and 5).
The N‐benzhydryl‐substituted amido‐thiourea catalyst 4 e 15b was selected for further optimization studies. A solvent screening identified o‐dichlorobenzene as the best reaction medium (Table 1, entry 6). Since our illumination system consisted of a black LED connected to an external power supply, we could finely tune and control the intensity of light emission (see Figure S2 for details of the illumination set‐up). Increasing the irradiance from 10±1 (used in the initial experiments) to 15±1 mW cm−2, while extending the reaction time to 30 hours provided 3 a in 89 % yield, as a single diastereoisomer, and with 90 % ee (entry 7). Then, control experiments were conducted to glean insights into the mechanism of the photochemical organocatalytic desymmetrization aldol process. The absence of light irradiation completely suppressed the reaction (entry 8), while the racemic adduct 3 a was isolated in 15 % yield in the absence of 4 (entry 9). The last result, along with the high stereocontrol achieved with the chiral amido‐thiourea 4 e, implies that the rate acceleration facilitated by 4 e is large enough to overcome a racemic background process.
We then evaluated the generality of the light‐driven organocatalytic aldol desymmetrization process using the optimized reaction conditions described in Table 1, entry 7. We studied the reactivity of a variety of achiral 2‐substituted‐2‐fluorocyclopentane‐1,3‐diketones (1). As displayed in Figure 2, different substituents at the aromatic ring of the benzyl moiety in 1 could be used, regardless of either their electronic and steric properties or position on the phenyl ring (3 a–h). To probe the synthetic utility of the method, we demonstrated that good efficiency was maintained when running the reaction on a 1 mmol scale (3 a). A product bearing a heteroaryl framework could be synthesized, as shown for the furyl‐substituted adduct 3 i. The desymmetric aldol reaction is also effective for 2‐alkyl‐2‐fluoro cyclopentane‐1,3‐diones, thus affording the corresponding adducts 3 j,k with high enantioselectivities. For this substrate class, we observed a correlation between the steric profile of the 2‐alkyl substituent and the relative stereocontrol of the desymmetrization process, since the diastereoselectivity increases with the length of the alkyl chain (d.r. from 5.5:1 to 19:1 when moving from a methyl to a butyl substituent; 3 j and 3 k). X‐ray crystallographic analysis16 performed on crystals from the adduct 3 h allowed the assignment of the absolute configuration for the newly formed fluorine‐containing quaternary stereogenic center while establishing the stereochemical outcome of the aldol‐based desymmetrization. The photochemical organocatalytic system also shows some limitations. The presence of a 2‐phenyl substituent in 1 greatly affected the reactivity of the process, although the stereoselectivity was preserved (3 l). The six‐membered‐ring product 3 m could be obtained only in poor yield and enantiomeric excess.
Figure 2.
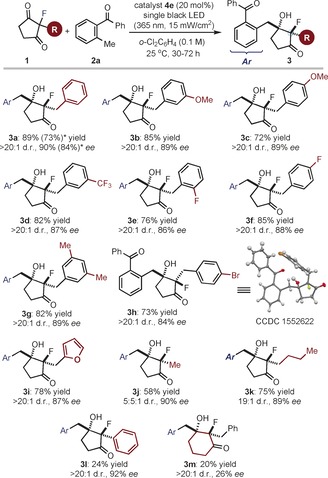
Survey of the 2‐fluoro‐substituted 1,3‐diketones 1 which can participate in the photochemical organocatalytic desymmetrization aldol process. Reactions performed on a 0.1 mmol scale using 3 equiv of 2 a. The d.r. value is inferred by 19F NMR analysis of the crude reaction mixture. Yields and enantiomeric excesses of the isolated products 3 are indicated below each entry (average of two runs). *Performed on a 1 mmol scale.
We then probed the scope with respect to the 2‐alkylbenzophenones 2, which can participate in the reaction by acting as donors upon photochemical generation of the reactive photoenol intermediates. As detailed in Figure 3, a wide range of substituents at both the non‐enolizable (3 n–q) and the enolizable (3 r–u) aromatic ring of 2 are well tolerated, thus granting access to chiral 2‐fluoro‐3‐hydroxycyclopentanones 3 n–u.
Figure 3.
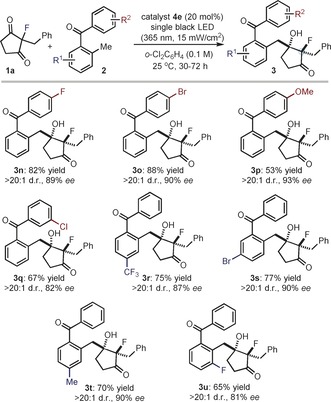
Survey of the 2‐alkylbenzophenones 2 which can participate in the photochemical organocatalytic desymmetrization aldol process. Reactions performed on a 0.1 mmol scale using 3 equiv of 2. The d.r. value is inferred by 19F NMR analysis of the crude reaction mixture. Yields and enantiomeric excesses of the isolated products 3 are indicated below each entry (average of two runs).
We then applied the photochemical organocatalytic desymmetrization strategy to the straightforward one‐pot synthesis of the highly functionalized Hajos–Parrish‐type ketone 5 (Scheme 1). This type of scaffold belongs to a class of valuable chiral intermediates which are useful for synthesizing biologically relevant compounds and natural products.7a, 13, 17 The organocatalytic desymmetrization of 2‐fluoro‐2‐(3‐oxobutyl) cyclopentane 1,3‐dione (1 v), which transiently affords the enantioenriched fluorinated 3‐hydroxyketone 3 v, was followed by an enamine‐mediated cyclization upon addition of pyrrolidine (50 mol %) to the same reaction flask.
Scheme 1.
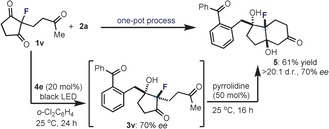
One‐pot organocatalytic enantioselective synthesis of the fluorinated Hajos–Parrish‐type ketone 5 proceeding through an intermolecular desymmetrizing aldol reaction of 1 v with subsequent enamine‐mediated intramolecular aldolization.
In summary, we have documented a novel catalytic strategy to stereoselectively construct valuable fluorine‐containing quaternary stereocenters, thus demonstrating that the desymmetrization of centrosymmetric compounds can be used for this purpose. Specifically, we used the ability of a readily available amido‐thiourea catalyst to selectively activate one of the enantiotopic carbonyl groups within achiral 2‐substituted‐2‐fluorocyclopentane‐1,3‐diketones (1). The resulting desymmetric intermolecular aldol process with photochemically generated, highly reactive hydroxy‐o‐quinodimethanes generates two stereocenters simultaneously, and forges a stereogenic C−F unit far from the reaction site. This methodology could be used to develop other asymmetric catalytic entries into useful stereogenic carbon‐fluorine synthons.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the Generalitat de Catalunya (CERCA Program), MINECO (CTQ2016‐75520‐P and Severo Ochoa Excellence Accreditation 2014–2018, SEV‐2013‐0319), and the European Research Council (ERC 681840—CATA‐LUX). S.C. is grateful to the MECD for a FPU fellowship (ref. FPU14/06541). L.D. thanks the Marie Curie COFUND action (2014‐1‐ICIQ‐IPMP) for a postdoctoral fellowship. We also thank the CELLEX Foundation through the CELLEX‐ICIQ high throughput experimentation platform
S. Cuadros, L. Dell'Amico, P. Melchiorre, Angew. Chem. Int. Ed. 2017, 56, 11875.
Contributor Information
Sara Cuadros, http://www.iciq.org/research/research group/prof‐paolo‐melchiorre/.
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Organofluorine compounds. Chemistry and applications (Ed.: T. Hiyama), Springer, New York, 2000; [Google Scholar]
- 1b. Jeschke P., ChemBioChem 2004, 5, 570–589; [DOI] [PubMed] [Google Scholar]
- 1c. Ametamey S. M., Honer M., Schubiger P. A., Chem. Rev. 2008, 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 2b. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 2c. O'Hagan D., J. Fluorine Chem. 2010, 131, 1071–1081. [Google Scholar]
- 3.For reviews, see:
- 3a. Prakash G. K. S., Beier P., Angew. Chem. Int. Ed. 2006, 45, 2172–2174; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2228–2230; [Google Scholar]
- 3b. Brunet V. A., O'Hagan D., Angew. Chem. Int. Ed. 2008, 47, 1179–1182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1198–1201; [Google Scholar]
- 3c. Ma J.-A., Cahard D., Chem. Rev. 2008, 108, PR1–PR43; [DOI] [PubMed] [Google Scholar]
- 3d. Yang X., Wu T., Phipps R. J., Toste F. D., Chem. Rev. 2015, 115, 826–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For pioneering work, see:
- 4a. Hintermann L., Togni A., Angew. Chem. Int. Ed. 2000, 39, 4359–4362; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 4530–4533; For selected examples: [Google Scholar]
- 4b. Hamashima Y., Yagi K., Takano H., Tamás L., Sodeoka M., J. Am. Chem. Soc. 2002, 124, 14530–14531; [DOI] [PubMed] [Google Scholar]
- 4c. Beeson T. D., MacMillan D. W. C., J. Am. Chem. Soc. 2005, 127, 8826–8828; [DOI] [PubMed] [Google Scholar]
- 4d. Marigo M., Fielenbach D., Braunton A., Kjærsgaard A., Jørgensen K. A., Angew. Chem. Int. Ed. 2005, 44, 3703–3706; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3769–3772; [Google Scholar]
- 4e. Steiner D. D., Mase N., C. F. Barbas III , Angew. Chem. Int. Ed. 2005, 44, 3706–3710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3772–3776; [Google Scholar]
- 4f. Ishimaru T., Shibata N., Horikawa T., Yasuda N., Nakamura S., Toru T., Shiro M., Angew. Chem. Int. Ed. 2008, 47, 4157–4161; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4225–4229; [Google Scholar]
- 4g. Lozano O., Blessley G., Martinez del Campo T., Thompson A. L., Giuffredi G. T., Bettati M., Walker M., Borman R., Gouverneur V., Angew. Chem. Int. Ed. 2011, 50, 8105–8109; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8255–8259; [Google Scholar]
- 4h. Kwiatkowski P., Beeson T. D., Conrad J. C., MacMillan D. W. C., J. Am. Chem. Soc. 2011, 133, 1738–1741; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4i. Erb J., Paull D. H., Dudding T., Belding L., Lectka T., J. Am. Chem. Soc. 2011, 133, 7536–7546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4j. Rauniyar V., Lackner A. D., Hamilton G. L., Toste F. D., Science 2011, 334, 1681–1684; [DOI] [PubMed] [Google Scholar]
- 4k. Yang X., Phipps R. J., Toste F. D., J. Am. Chem. Soc. 2014, 136, 5225–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected examples, see:
- 5a. Kalow J. A., Doyle A. G., J. Am. Chem. Soc. 2010, 132, 3268–3269; [DOI] [PubMed] [Google Scholar]
- 5b. Katcher M. H., Doyle A. G., J. Am. Chem. Soc. 2010, 132, 17402–17404; [DOI] [PubMed] [Google Scholar]
- 5c. Katcher M. H., Sha A., Doyle A. G., J. Am. Chem. Soc. 2011, 133, 15902–15905; [DOI] [PubMed] [Google Scholar]
- 5d. Woerly E. M., Banik S. M., Jacobsen E. N., J. Am. Chem. Soc. 2016, 138, 13858–13861; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Banik S. M., Medley J. W., Jacobsen E. N., Science 2016, 353, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected examples, see:
- 6a. Nakamura M., Hajra A., Endo K., Nakamura E., Angew. Chem. Int. Ed. 2005, 44, 7248–7251; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7414–7417; [Google Scholar]
- 6b. Jiang X., Gandelman M., J. Am. Chem. Soc. 2015, 137, 2542–2547; [DOI] [PubMed] [Google Scholar]
- 6c. Saadi J., Wennemers H., Nat. Chem. 2016, 8, 276–280; [DOI] [PubMed] [Google Scholar]
- 6d. Balaraman K., Wolf C., Angew. Chem. Int. Ed. 2017, 56, 1390–1395; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1411–1416. [Google Scholar]
- 7.For recent reviews, see:
- 7a. Zeng K.-P., Cao Z.-Y., Wang Y.-H., Zhou F., Zhou J., Chem. Rev. 2016, 116, 7330–7396; [DOI] [PubMed] [Google Scholar]
- 7b. Borissov A., Davies T. Q., Ellis S. R., Fleming T. A., Richardson M. S. W., Dixon D. J., Chem. Soc. Rev. 2016, 45, 5474–5540; For recent examples of catalytic desymmetrization processes of 2,2-dialkyl substituted cyclopentane-1,3-diones, see: [DOI] [PubMed] [Google Scholar]
- 7c. Partridge B. M., Callingham M., Lewis W., Lam H. W., Angew. Chem. Int. Ed. 2017, 56, 7227–7232; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7333–7338; [Google Scholar]
- 7d. Prévost S., Dupré N., Leutzsch M., Wang Q., Wakchaure V., List B., Angew. Chem. Int. Ed. 2014, 53, 8770–8773; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8915–8918. [Google Scholar]
- 8.Recently, the asymmetric ring opening of meso epoxides with fluoride anions has been reported. This desymmetrization process is based on a C−F bond-forming step and can only forge fluorine-containing tertiary (and not quaternary) stereocenters. See Ref. [5a] and:
- 8a. Kalow J. A., Doyle A. G., J. Am. Chem. Soc. 2011, 133, 16001–16002; [DOI] [PubMed] [Google Scholar]
- 8b. Zhu J., Ysui G. C., Lautens M., Angew. Chem. Int. Ed. 2012, 51, 12353–12356; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12519–12522. [Google Scholar]
- 9.Diels–Alder reaction:
- 9a. Dell'Amico L., Vega-Peñaloza A., Cuadros S., Melchiorre P., Angew. Chem. Int. Ed. 2016, 55, 3313–3317; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3374–3378; Mannich-type reaction: [Google Scholar]
- 9b. Hepburn H. B., Magagnano G., Melchiorre P., Synthesis 2017, 76–86; Conjugate addition: [Google Scholar]
- 9c. Dell'Amico L., Fernández-Alvárez V. M., Maseras F., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 3304–3308; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3352–3356. [Google Scholar]
- 10.
- 10a. Yang N. C., Rivas C., J. Am. Chem. Soc. 1961, 83, 2213–2213; [Google Scholar]
- 10b. Sammes P. G., Tetrahedron 1976, 32, 405–422; [Google Scholar]
- 10c.“Photoenolization and its applications”: Klán P., Wirz J., Gudmundsdottir A. in CRC Handbook of Organic Photochemistry and Photobiology, 3 rd ed. (Ed.: A. Griesbeck), CRC, Boca Raton, 2012, chap. 26, pp. 627–651. [Google Scholar]
- 11.For a review on enantioselective catalytic photochemical processes, see: Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 12.Similar fluorine-containing chiral cyclic scaffolds, prepared in racemic fashion, have been used as intermediates in the synthesis of pyrrolopyridazine JAK3 inhibitors. See: Bristol-Meyers Squibb Company; Stephen T. W., Brown G. D., Doweyko L. M., Duan J., Guo J., Hynes J., Jian B., Kempson J., Lin S., Lu Z., Spergel S. H., Tokarski J. S., Wu H., Yang B. V., Patent WO2012/125886A1, 2012.
- 13.
- 13a.“Asymmetric Synthesis of Optically Active Polycyclic Organic Compounds”: Hajos Z. G., Parrish D. R., German. Patent, DE 2102623, 1971;
- 13b. Hajos Z. G., Parrish D. R., J. Org. Chem. 1974, 39, 1612–1615; [Google Scholar]
- 13c. Eder U., Sauer G., Wiechert R., Angew. Chem. Int. Ed. Engl. 1971, 10, 496–497; [Google Scholar]; Angew. Chem. 1971, 83, 492–493. [Google Scholar]
- 14.
- 14a. Taylor M. S., Jacobsen E. N., Angew. Chem. Int. Ed. 2006, 45, 1520–1543; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1550–1573; [Google Scholar]
- 14b. Brak K., Jacobsen E. N., Angew. Chem. Int. Ed. 2013, 52, 534–561; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 558–588. [Google Scholar]
- 15.
- 15a. Reisman S. E., Doyle A. G., Jacobsen E. N., J. Am. Chem. Soc. 2008, 130, 7198–7199; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Zuend S. J., Coughlin M. P., Lalonde M. P., Jacobsen E. N., Nature 2009, 461, 968–971; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Ford D. D., Lehnherr D., Kennedy C. R., Jacobsen E. N., J. Am. Chem. Soc. 2016, 138, 7860–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.CCDC 1552622 (3 h) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 17. Eagan J. M., Hori M., Wu J., Kanyiva K. S., Snyder S. A., Angew. Chem. Int. Ed. 2015, 54, 7842–7846; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7953–7957. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary