

Abstract
Genomic interactions reveal the spatial organization of genomes and genomic domains, which is known to play key roles in cell function. Physical proximity can be represented as two‐dimensional heat maps or matrices. From these, three‐dimensional (3D) conformations of chromatin can be computed revealing coherent structures that highlight the importance of nonsequential relationships across genomic features. Mainstream genomic browsers have been classically developed to display compact, stacked tracks based on a linear, sequential, per‐chromosome coordinate system. Genome‐wide comparative analysis demands new approaches to data access and new layouts for analysis. The legibility can be compromised when displaying track‐aligned second dimension matrices, which require greater screen space. Moreover, 3D representations of genomes defy vertical alignment in track‐based genome browsers. Furthermore, investigation at previously unattainable levels of detail is revealing multiscale, multistate, time‐dependent complexity. This article outlines how these challenges are currently handled in mainstream browsers as well as how novel techniques in visualization are being explored to address them. A set of requirements for coherent visualization of novel spatial genomic data is defined and the resulting potential for whole genome visualization is described.
Keywords: FAIR principles, genome browsers, multiscale, RICH visualization, three‐dimensional data
Abbreviations
1D, first dimension
2D, second dimension
3C, chromosome conformation capture
3D, three‐dimensional
APIs, application programming interface
BGRA, Browser Genome Release Agreement
EMDB, Electron Microscopy Data Bank
GB, Gigabyte
HPC, high‐performance computing
IDR, image data resource
kb, Kilobase (1000 genomic base pairs)
LOD, level of detail
MB, Megabyte
MD, molecular dynamics
mmTF, macromolecular transmission format
nm, nanometers
OME, open microscopy environment's
TADs, topologically associating domains
The microscope is the emblematic tool for visualizing biological material. From Hooke's Micrographia to super‐resolution imaging, observation resolves biology at progressively higher levels of detail. And at the far atomic scale, physical models of molecules, from DNA to proteins, have pieced together the invisible toolkit of life. Between these two resolution ends (i.e., from 10 to 200 nm), the genome cannot be easily observed 1. However, this is now being bridged by a number of techniques revealing a hierarchical, spatial, and dynamic architecture of the genome 2. Closing this ‘resolution gap’ would bring a convergence of knowledge with the opportunity for integrating multiple data types to produce whole genome models, that is, providing a virtual genomic ‘microscope’.
The genome is encoded in DNA as a first dimension (1D) of instructions, which correspond to genes and their regulatory elements. A second dimension (2D) of elements, generally referred to as epigenetics, influence how genes are regulated. Finally, the spatial organization of chromosomes has been seen as a third dimension (3D) of the genome. For the last decade, and thanks to technological developments in imaging and molecular biology, we now know that these three dimensions of genomes form a complex hierarchy of functional and dynamic relationships that change between cell types and throughout the cell cycle 3. This has cemented the importance of integrating all dimensions to a complete understanding of the regulation of the genome.
Research into this 3D has emerged from breakthroughs in microscopy, molecular genetics, and high‐performance computing. Super‐resolution microscopy is now surpassing light‐diffraction limitations below 200 nm to reveal fine grain structures for in vivo nucleus topology 4, the development of chromatin networks 5, chromatin folding for different epigenetic states 6, and tracking of genomic elements 7. Computational techniques 8 are now used to construct a spatial understanding of genome function, inferring gene regulation from chromosome conformation capture (3C)‐based experiments 9, exposing functional organization through polymer physics 10, and chromatin dynamics by atomistic nucleosome models 11, among others. Data recording these 3D aspects of the genome, principally from sequence‐derived quantitative methods, has started to be aggregated into existing genome browsers but the process presents a number of challenges 12. For example, the data are large and diverse, with multiple, interrelated attributes 13 and requiring clear, coordinated strategies to data stewardship 14 to enable analysis.
Three‐dimensional visualization of complex and large‐scale genomic data has been key to discoveries over the last 50 years 15. Human perception has evolved to quickly and intuitively find and make patterns to build comprehension and, when correctly constructed, to reflect ‘truthfulness’ of data 16. Graphic representations make data accessible, tractable, and digestible, for fast and precise communication. Today's genome browsers were developed in direct response to the task of handling and analyzing a plethora of 1D and 2D data 17 typically displaying linear sequences as horizontal tracks, aligned and stacked below the reference nucleotide sequence. However, beyond the logistics of data management, there are challenges in visualizing 3D data. In general, 3D representation can cause distortion and occlusion of the data due to perspective, lighting, and the complexities of human visual processing 18. Specifically, the novel, dense, and complex nature of genomic 3D data present challenges in representing, interrogating, and interacting with visualizations to facilitate the discovery of novel insights 19.
Previous publications have reviewed the wealth of tools to browse genomic data 20, 21 and recently, visualization tools for genomic 3D data 22. Yet this is a fast maturing field with new discoveries, techniques, and tools that requires ongoing analysis to ensure valid and innovative investigation. Moreover, the nature of the data and the novel demands they place on visualization oblige reconsideration of conventions of existing tools. We describe first the challenges inherent in 3D data: massive, multiple data types and scales, uncertainty and states, dynamic time dependence, accessibility, and integration of the data. Then, we discuss the visualization challenges specific to 3D data: task definition, grammar, abstraction, rendering, inspection, navigation, workflow, responsiveness, and publication. For each, we present existing examples to help describe the challenges and the current state of the art in addressing them. Finally, we discuss the overall situation and indicate further lines of investigation into the visualization of 3D genomic data.
Multifaceted genomics data
Three‐dimensional genomic data encompass a number of very distinct data types. Microscopy technologies scan subcellular structures and can detect specific genomic features via ‘painted’ loci producing layered images that can be used to generate 3D models as well as accurate spatial measurements for calculation and validation of genome architecture 23. 3C‐like experiments can also identify the proximity of chromatin fragments by indicating interactions between regions of resolutions up to 1 kb 24. In particular, Hi‐C experiments, which result in an all‐to‐all genome‐wide interactions map, can be used to infer chromatin compartments, domains (or also called topologically associating domains, TADs), and loops 25. Computational processing of these matrices can derive into 3D models of the most probable overall spatial conformation. Such coarse grain models can also be constructed from molecular dynamics (MD) simulations of chromatin properties in stochastic environments 10. At the smallest resolution, small biomolecule and nucleosome models can be constructed from strings of discrete, atomic crystals with MD also being used to describe their behavior 26. Therefore, a large range of types and file sizes are needed for a complete description of the 3D genome, which generates a consequent burden on browsers. This variety of data (Fig. 1) is further complicated by the nature and extents of the data attributes of multiscale, multistate, time dependence, and uncertainty 13.
Figure 1.
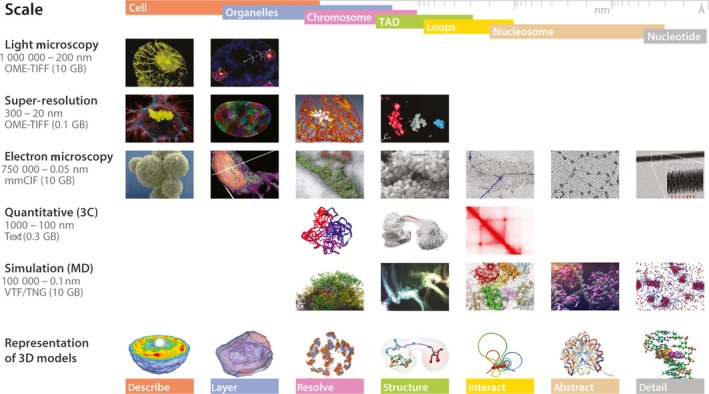
Multifaceted genomic 3D data. The 3D data in genomics are multiscale, multistate, time‐dependent, and contain uncertainties that make their representation challenging. For example, there has been multiple types and forms to visualize genomic data in 3D from the cell to the nucleotide, each of them with specific particularities and specific representations.
Multiscale
We can now record 3D aspects of entire genomes across a large range of physical sizes, from the few microns of the nucleus to the few nanometers of nucleosomes 27. These can be pictured as the levels of magnification of a virtual microscope, each of which may contain several distinct data types, with distinct experimental resolutions, as a consequence of the advantages/limitations of the used experimental methods to study them. Situating each piece of data within this hierarchy helps delineating the scope and resolution of each scale from which the coverage and relationships of the datasets can be determined. In conjunction, these levels not only form a multiscale model of the data but also conceptualize the multiscale nature of the nucleus. The data at each scale can also depict the genome in distinct forms, with stratification by physical delimitation (e.g., chromosome territories), assembled colocation (e.g., compartments or TADs) or derived functionality (e.g., TADs or loops).
Multistate
This multiscale model is not static as the data also likely represent distinct states of the genome. Uppermost, distinct epigenetic states describe diverse genomic functions that may alter the expression of resident genes. It is now known that variable states depend on biochemical mechanisms altering the physical nature of chromatin, and thus altering its physical properties and spatial organization. Chromatin may be folded into the so‐called open or closed states 23, and may be compacted at different densities 28 making genomic regions accessible or inaccessible to processing biomolecules. These two coarse states of the genome, which relate to the original hetero‐ and eu‐chromatin, have been further classified into several other states 24, which can have an impact on how genomes are structured.
Time dependence
Experimentally, data generation may be affected by time in the process, protocols, execution, interruptions, time required for biochemical reactions etc. These time‐dependent aspects of experimentally interrogating the genome architecture, are embedded within a source of variability that is more fundamental. Indeed, experiments interrogating 3D genomes also capture the order of events describing how the genome structure changes over time due to biologically relevant processes. For example, 3C‐based experiments can capture nuclear transformation during cell cycle 29, 30, which show that changes in chromatin interactions over time play a role in controlling expression fundamental to cell function. Finally, polymer physics and MD are being used to reflect time as a fourth dimension for biochemical mechanisms of the nucleus. As such, 3D data extend the dimensionality of the genome into what is nowadays described as the dynamic genome or the 4D genome.
Uncertainty
In addition to reflecting biology, experiments imbue data with uncertainty due to limited coverage, unspecific samples, unusable output, or restricted focus. Furthermore, raw experimental data, which needs to be processed considering the particularities of each experimental protocol, may increase its noise/uncertainty once parsed and analyzed. Each data type therefore contains biases that need to be accounted and robust analytical tools are needed to assess the confidence on the final 3D data to visualize 31.
Data accessibility
As mentioned above, experimental analysis of genome organization requires various data types, which may be produced by others elsewhere. Publically accessible 1D reference genomes have not only ensured efficient use of resources but also provided a coherent baseline against which related data can be assessed and validated. For example, retrieval under the Browser Genome Release Agreement (BGRA, http://www.ensembl.org/info/about/legal/browser_agreement.html) requires consistent sequence and assembly identifiers. These providers manage data streaming to reduce network load and end‐user wait times by creating endpoints and application programming interface (APIs) like Track Hubs for easy access. Unfortunately, most 3D data remain accessible only by direct download from the labs, through nonspecialized databases or as supplementary publication data. While some 3D datasets have been submitted to UCSC and Ensembl as part of the BGRA or other special collections, there are no generally accepted and established specific repositories for 3D genomic data.
The data from super‐resolution microscopy consists of the raw images with metadata and processed binary spot list, which contain coordinate data and image attributes such as frame, intensity, peak shape, etc. These files are unwieldy with raw file sizes of around 10 GB (60 MB when processed). However, these are now being catered for the new image data resource (IDR) initiative for coordinating imaging with genomic data built on the open microscopy environment's (OME) OMERO image data management platform 32. OMERO uses the BioFormats tool that also promotes harmonization of formats via the open OME‐TIFF, which enable rich metadata while capturing multiple states or time frames within a single file 33. At smaller scales, EMPAIR archives electron microscopy raw 2D data and the electron microscopy data bank (EMDB) stores 3DEM models.
Interaction data (i.e., 3C‐based experimental data) lack dedicated repositories but can be made to fit with conventional repositories and track browsers as it records genomic loci pairs and associated scores (interaction counts). For example, such data can be visualized by the WashU Epigenome Browser by reading in tab‐separated text 34 similar to Bedtools BEDPE format 35. The Juicer software can also read and analyze 3C‐based interaction data by converting experimental reads into a tool‐specific Hi‐C format, which can then be visualized using the Juicebox browser 36. Finally, the tadbit software 8 can output complete experimental dataset in standard JSON format, which can then be imported and visualized using the TADkit 3D browser. Importantly, data accessibility and portability in 3D genomics still require some time to develop a widely accepted format for storing and disseminating interaction data and models. The most recently developed longTabix format from UCSC Genome Browser 37 could be used for such task but still lacks ways of representing multiple states or time frame datasets.
Coarse grain restraint‐based models derived from interaction data result in 3D objects represented by sets of Cartesian coordinates 38. A large number of formats can capture such positional data with states and dynamics (e.g., mmCIF with TNG trajectory format 39 or the VMD trajectory format 40). However, and unfortunately, such datasets are usually provided as flat text files or hacked into extended PDB files 41. While the latest facilitates distribution and portability to computational tools, this obliges the results to be adapted on an ad hoc basis, reducing potential for standardized, reproducible data integration, which may induce undocumented errors. Moreover, the classical PDB format must contain ‘< 62 chains and/or 99999 ATOM records’ and so it is unsuited to larger genome structures. The updated format, mmCIF, addresses these limitations but remains atom‐based, which still requires hacking to store large objects such as entire genomes. Finally, this approach is complicated to import into existing molecular tools that expect atomic coordinates as well as pushes the limits of the GL‐based visualizations. Given the size and complexity of the new macromolecular structures, RSCB‐PDB recently launched the innovative macromolecular transmission format (mmTF), which has greater compression for faster transfer while retaining efficient parsing. While the technique is instructive, the format is targeted at macromolecules and does not address the issues of file access in genomic structures. High‐speed Internet connections and chunked data requests may ease data transfer. However, the ideal strategy for both coarse grain and atomistic 3D models of genomes would be use of spatial data structures (e.g., OctTree, BallTree k‐d, etc.) although currently none of the newer formats discussed here take advantage of this.
Data integration
The data detailed above are varied and the forms of access have been uncoordinated. However, these datasets are better studied in coordination by integrative approaches. Bringing together multiple types of data from a variety of sources, many of which are not easily centralized is a challenge. It is essential for confidence and utility of the data that ease of access and explicit identification is assigned to any dataset. This labeling would ideally be done on output from production (i.e., by the software or machine) but including backlog or historic data (i.e., verification and assignation). To implement this, a number of common attributes must be identified and included in all data, for example, experimental technique, domain, organism, chromosome, source, etc. These attributes can be added as file annotation in various ways (i.e., metadata, tags, headers, xml, or in an independent database). And filenames can, as in our own 4DGenome experimental procedures 42, represent experimentally pertinent details: User, Experiment ID, Biological replicate ID, Technical replicate ID, HTS application. This would lead toward the goal of producing whole genome models, storing 3D data, or DOI links to them, within a single file.
There have been a number of attempts at establishing standard formats describing multiscale genomic data. The earliest genome3d generates OctTree spatial data structures with four levels: ‘nuclear’ coarse grain model; ‘fiber’ of random‐walk chromatin; ‘nucleosome’ decorated strands; ‘atomic’ sequence‐derived helices 43. Gmol developed a GSS format which stores models in a six‐level hierarchy of genome organization: ‘genome’, ‘chromosome’, ‘loci’, ‘fiber’, ‘nucleosome’, and ‘nucleotide’ 44. More recently, following recommendations by the wwPDB integrative methods task force 13, the Sali Lab has developed the Integrative Hybrid Model format IHM‐mmCIF to store integrative models based on 3D data including restraints. For standard genome browsers, the mmCIF format import is not currently available and, apart from the interaction data, would need the development of new display modules.
As such, these 3D data are novel, massive, and complex. They present challenges in coordinating and implementing an integrated view of the genome beyond a simple cartographic zoom for transitions across data types, resolutions, forms, and ontologies. Associations need to be created between disparate data on different levels so as to assemble a multiscale/state model. Moreover, it is important to portray variance and time in the models. However, this is not typically displayed in browsers, and the way it can be included is currently neither easy nor standardized. These issues of data stewardship have been widely discussed in the field of molecular biology from which has emerged the FAIR set of principles for scientific data management: Findability, Accessibility, Interoperability and Reuse 14. Against this framework, the flaws inherent to 3D genomic data are: lack of common, consistent, descriptive metadata, storage, and formats. And even if genomic 3D data would match the FAIR principles, its visualization in current genome browsers would represent a frozen moment/scale in the genome.
Data visualization
Visualization is a task‐driven process that assists the user on iterative steps toward the goals of identifying the patterns, finding the connections, and determining the mechanisms behind the data 45. The complex nature of biological processes reflected in the interleaved scales, states, and phases of 3D data, and the multidisciplinary research that this requires, makes it impossible to clearly define all required tasks. Specialized 3D data browsers, such as 3D‐GNOME 46 and Genome3D.eu 47, are task focused, leading the user through data selection, filtering, and mining to produce 3D visualizations aligned to tracks. Of particular note is NCBI's ‘Twelve Elements’ white paper 48, which explicitly defines tasks for their new icn3d browser 49. Their recommendations are in line with other task analyses for 3D protein 19 and biological pathway 50 visualization tools and serve as a comprehensive basis for other classes of 3D data. From this, we define a taxonomy of 3D tasks outlining essential features of 3D genome visualization. These tasks indicate the core challenges in giving the data context and in highlighting key facets of the data 51. Together with the FAIR treatment of the data detailed above, these form the initial steps in visualization 52: acquire large, multifaceted, diverse datasets; parse the data to manage, structure, and annotate them; filter to correct for experimental artifacts, biases, and select the relevant data; and mine for statistical relevance 22. The subsequent visualization steps present further challenges: to represent new objects, abstractions, and variations of the data; to refine by adding definition, clarity, and discoverability to find patterns in the data; to interact by selection, navigation, curating to test hypotheses, form conclusions, and to share the insights.
Data representation
Grammar
A fundamental rule of visualization is that the choice of representation should most correctly portray the data and dataset types 53. 3D genomic data record spatial structures, in particular, the compaction of DNA as chromatin conformations that form compartments and TADs, which emerged from interpretation of the 3D data. Therefore, although the data can be analyzed as is (interaction maps), interpretation and representation of the underlying physical form remains a core task 54. By taking advantage of human shape perception, 3D representation can also assist the exploratory analysis of complex objects. However, there are caveats that need to be addressed: visualization is affected by the limitations of spatial perception and memory, namely comparison of nonplanar elements, judgment of depth, and comparison of animated objects 55. In particular, 3D representations in visualization are a challenge as they can reduce effective visualization due to object occlusion, perspective distortion, and loss of legibility of tilted graphic elements 56. Given the nascent state and the diverse scientific approaches of 3D genomics research, there is yet to emerge a standard form of representation of the spatial nature of genomes. In contrast to proteins, where the field has accepted a ‘cartoon’ representation of their structures 57, genomes have been visualized with in a variety of representations (Fig. 2). However, a limited number of 3D genome structures have been characterized so far, with definitions generally approaching: the nucleus membrane containing a nebulous globule of chromosome territories with a skeleton network and an inner core of conserved structures, condensing at interphase; the chromatin tubules of varying width and densities of compaction 28; amorphous bundles of these chromatin differentiate into loose, active ‘A’ compartments and compact, repressed ‘B’ compartments; smaller fractal bundles of subdivisions from TADs to loops; and at the lowest scale, uncompacted chromatin as helical fibers of DNA; all suspended within a maelstrom of biomolecules. So far, most 3D representations have focused on chromatin strands rather than other higher order structures. Visualization tools such as genome browsers can provide ‘dictionaries’ of visual grammar. Unfortunately, no automated 3D generation of such grammar exist for chromatin data, save for the simple example of Chimera's multiscale models extension which outputs low‐resolution models of molecular structures 58. That said, the library of macromolecules and the rendering software is increasing in capacity and may eventually encompass these larger scale genomic structures for inclusion in whole cell models 59.
Figure 2.
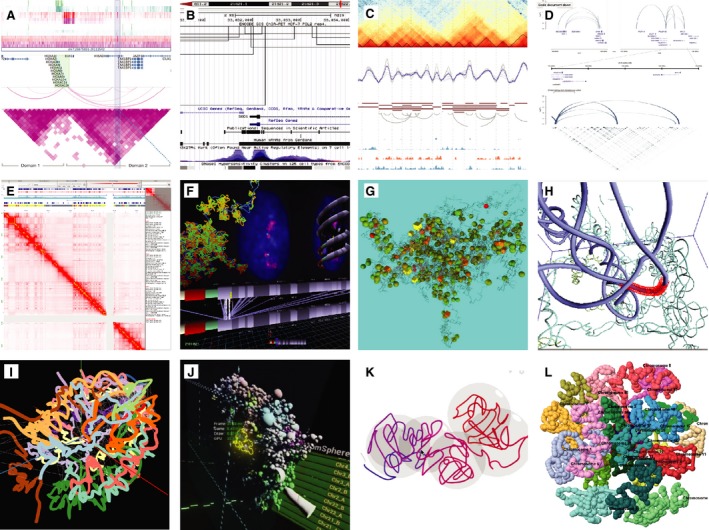
Data representation. Examples of genomic data representation. (A) WashU Browser (1D/2D), expansive matrices and arcs; (B) UCSF Browser (1D/2D), bracketed interactions: (C) HiCExplorer (1D/2D), track annotations; (D) VisPIG (1D/2D), segmented browsing with detail zoom; (E) Jucier (2D), matrix focused browsing; (F) Globe3DV (2D/3D), everything on view; (G) Genome3D (3D), multiscale browser; (H) 3DGB (3D), spatial paradigm; (I) HiC‐3DViewer (3D), multichromosome view; (J) Chrom3D‐VR (3D), interactive manipulation of data; (K) TADkit (1D/2D/3D), TAD visualization; (L) Gmol (3D), multiscale browsing.
Abstraction
Another approach to overcoming the challenges of representing 3D data is to abstract or reduce data dimensions (Fig. 3), a habit innate to humans. The mapped surface can then also become a visualization 2D space, aiding cohesion, and orientation of data, for example, as used in Multeesum 60, Space Syntax 61, and Route‐Zooming 62, among others. The sequence coordinates themselves can be abstracted to adapt to other data, (e.g., by stacking regions 63 or focusing only on transcript scopes 64). ABySS‐Explorer adds granularity to permit visual accentuation of the sequence 65. The sequence can also be arranged nonlinearly represented as a Hilbert graph, a fractal arrangement of the coordinates within a square producing compact, navigable maps of whichever data values are assigned to each coordinate 66. An interesting variation on this is the H‐curve which assigns nucleotides to quadrants and draws the genome along the Z‐axis to reveal features such as translocations, motifs 67. Finally, a more radical approach is to take advantages of other information channels, such as the auditory, to augment the visual field with fine grain information 68.
Figure 3.
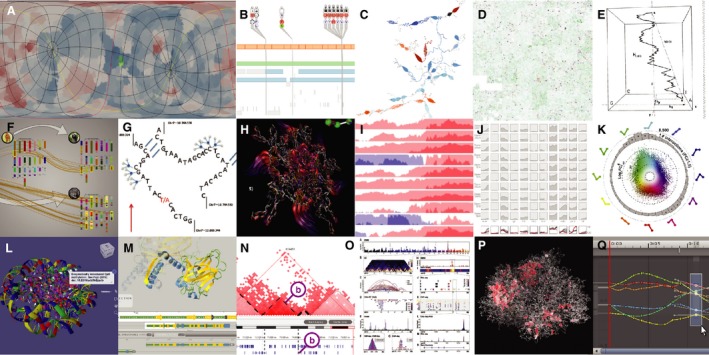
Data abstraction. (A) Flat surface map of green colored docking sites; (B) Transcript focused VariantView; (C) Added granularity in ABySS‐Explorer; (D) Conserved genomic regions in a Hilbert curve; (E) Genomes as 3D H‐curves; (F) Synteny Explorer; (G) Abstraction in Shavit genome browser; (H) Flowlines indicating submolecular motion; (I) Horizon graphs for stacked data; (J) Small multiples as curvemaps; (K) Multistate expression data as rose‐ring; (L) 3D annotation in AutoDesk Molecular viewer; (M) 3D selection from track in Aquaria; (N) Cross‐dimensional section in TADkit; (O) Layout design in Sushi; (P) Chromos VR visualization of chromatin active loops; (Q) Synchronized object and track selection in Unity.
Interaction data are also, by definition, an abstraction of structure captured within the cell nucleus. A whole genome view of interactions is gained by creating a matrix or heatmap, which can be the basis for a browser layout. This has been demonstrated by Juicebox that aligns tracks to matrix edges 36. Alternatively, being symmetrical along the diagonal, half‐matrices have been aligned along this diagonal to data in genome browsers, although this causes excessive track height. The more typical method is to create a visual link between the interacting regions, first shown in the WashU Epigenome Browser 34. The UCSC browser has only just introduced the ability to display interactions albeit styled like architectural dimension lines 37. More importantly, the far genomic distances they span, which is unaddressed by most track browsers, can be addressed by sectioning tracks to display side‐by‐side only the ranges around two nodes being connected, as in washu and vispig 69. Another approach is that of annular graphs, such as the archetypal Circos browser, which tackle this with circular track showing transgenomic connections across the central area 70. In contravening formal reading direction, they reduce legibility but this is counterbalanced by the greater overview of data and exposure of patterns compared to linear tracks as shown in the Rondo genome browser 71. When 3D structures are inferred from the interaction data, their forms need not be simulacrums of the chromatin. This is comparable to ribosome hairpins which lend themselves to a flattening of their conformations 72. While genomic structures are topologically more complex, such layouts may also be applied to the genome as seen in the synteny Explorer 73, by judicious summary of secondary structures as in ligplot+ 74, by characterization of molecular interaction forces 75, or by accepting a degree of overlap achieved by ChromoVis for chromatin fibers (D. Filippova, G. Duggal, R. Patro & C. Kingsford, unpublished data). Surface molecular models can also be abstracted by spherical projection of accessible external surfaces to create a cartographic map 76.
Variation
Visualizing the variability within genomic data is essential for giving a true representation of the attributes, states, uncertainty, and dynamics described by the data, and for setting the biological context. Animation conveys multiple changes in data form, situation, and attributes over time while allowing the viewer to maintain context and easily tracking changes 77. However, static images and orthographic projections have been shown to enable more precise judgments of measurement 78. Conventional browsers have limited ability to toggle or transition between superimpositions or segments of states or phases. Furthermore, 3D data capture collections of change events, and although it can be animated, unlike a cinematic animation, is not structured for narrative clarity. However, techniques such as flowlines 79 or streamlines 80 can annotate models and extend tracking to enhance comparison 60.
Data refining
Classification
Finding patterns within data is a core task of visualization. Ontologies such as those managed by the EBO assist identification of data objects through the Zooma tool 81. While a number of 3D structures have been described as detailed above, classification of genomic 3D data is still in its infancy. Although the representation of 3D genomic form is not yet fully developed, the structure of genomes can be classified by known features, processes, and mechanisms. For example, a region of chromatin could be classified by its density, activity, or accessibility 28. Indeed, the application of validation in browsers for 1D and 2D data already assists, but is far behind for, the classification of 3D data. Aquaria is a successful example of the use of precalculated classifications in the study and analysis of protein structures. Aquaria not only integrates 3D models with sequence data but also assists analysis with careful visual design choices guiding and accentuating relevant correlations across data dimensions 82. More dramatically, the potential brought by machine learning with high‐performance computing (HPC) is already being applied to the rapid exploration of structure and function. However, here the challenge is in finding synchronization between the processing time required and the user interaction within a browser.
Comparison
Pattern discovery/classification is not only useful for creative expression but also facilitates comparison 83. Side‐by‐side comparison is a common didactic resource in infant education and is suited to 3D data slices like bioimages or 2D abstractions as graphs and matrices 84. However, such a comparison places demand on user's internal memory which becomes untenable with large or dispersed datasets. Track‐based browsers rely on vertical visual assessment, which imposes significant distance between the data to compare once more than two tracks are displayed. One solution could be avoiding vertical stacking by clustering or stacking in the z‐axis similar to horizon graphs 85. 3D data, such as interaction matrices and 3D viewports, only serve to increase the problem of layout spread. Moreover, 3D datasets contain large point‐sets that are spatially and temporally distributed, which make comparison especially challenging. Animation permits comparison while maintaining context and easily tracking changes, especially when forward‐reverse controls are to hand, or the animation swings forward‐reversed looping of a short‐animated path. But when there are a large number of changes, change‐blindness outside the focus of attention renders comparison almost impossible 86. Removal of unchanged data can mitigate this effect by Boolean operations on the representation, as seen in VariantView, or by taking advantage of visual acuity in view switching (e.g., creating jump‐cuts for ‘blink’ comparison) 87. Another method that has proved particularly useful for comparison is fragmentation of large sets or sequences into smaller frames or multiples, which when placed side‐by‐side highlight salient features 88 such as pathlines or condensed normalized icons in Multeesum 60 or rose‐like rings 89.
Annotation
Quality visualizations provide fluid experiences, which sometimes are haltered by the process of documenting or annotating. Track browsers already provide tools for doing this although few do so ‘on the fly’ and ‘over the track’. Ideally this should be implemented such that it remains not only within the user session but also as metadata to the project archive and even the data file itself. There are clearly issues in treating 3D annotating concerning object obfuscation and text legibility. However, text lists and genome tracks can serve as guides to annotation in 3D, with map features such as icon clustering providing means to add not only readability but also summarization.
Data interaction
Navigation
An overview of the data is essential for an initial orientation and subsequent navigation. These starting points could be adapted from Ben Fry's Strippy, which, while not a physical representation, extols the state of the art in both genomics and computational graphics of 2002 90. More recently, other abstractions present a textured overview from which navigation can be intuited in, for example, whole matrices 84 or 3D models 91. In current genome browsers, often a karyogram gives overall location and a scale‐bar of the chromosome tracks and coordinates of the main base‐pair track view. This overall view can show different marks or glyphs, which at higher zoom levels aggregate or transform to the next scale depiction. Below the chromosome, genome browsers lack further scales and, unfortunately, lose situational awareness. Studies show limits to mental registration in visualization navigation in 2D and 3D 92. Addressing this challenge would require both new data and spatial overviews at different scales. The ontology of genomics ranges from environment and population through organisms, with 3D data covering the cellular, organelle, molecular, and atomic scales (Fig. 1). These biological scales equate to the levels of details used in interactive visualizations and, in particular, in efficient 3D computer graphics. The levels therefore form the basis from which coherent and effective visualization can be constructed. Each data type can be assigned at each level for clear orientation while navigating. This has been attempted through file formats explicitly as in GSS (genome, chromosome, loci, fiber, nucleosome) and assigned per data type in the case of IHM‐mmCIF. However, these do not enable VRML‐style level of detail (LOD) navigation as in the Genome3D viewer 43. The key to address this challenge is data inclusivity (i.e., all available data across all scales). Unfortunately, in 3D genomics not all data are readily available for all scales. Moreover, data may also be dimensionless, unorientated, or simply reflect unstructured conformations. Where no data are available, the level must be indicated as deficient, again giving clarity of extent and limitations of what is being represented and also leaving room for future clarifications. While navigating, the visualization must supply details on demand. Advances in molecular graphics technologies have made accurate rendering of highly detailed and intricate models possible 26. In particular, the WebGL standards enable interactive rendering from online data. Of course, there are limits given the size and complexity of the whole genome model, but this can still be provided with chunking, on‐demand loading, spatial data structures, integrative formats, and ontology‐based hierarchy of scales. Combined with high‐resolution screens, there is a good opportunity to provide such large‐scale high‐detail visualizations within a genome browser. The challenge is to find a balance trade‐off between adequate resolution and responsive user experience.
Selection
The benefits from refining data by classification facilitate selection. Again, 3D data selection is a challenge because of 3D obfuscation and the complexity of 3D models 48. The classification described above can help address these challenges and advanced selection is now being designed into genome browsers such as icn3d 49 and ChimeraX 93. Conventional tracks can also assist in selection of 3D elements as demonstrated in the Aquaria protein browser 82 or 3D animation software such as Blender or Unity. As such, the track as mere slices of the whole 3D model, remain essential to navigation.
Curation and hosting
Visualization of data is not only an experimental step but can serve as an experimental record or lab book. The path of the data from source through processing to representation and investigation can be recorded as user projects, which will record the goals, the decisions and actions taken, the arrangement and comparisons, and the conclusions drawn. A pair of genomic coordinates is a concise reference, defining simultaneously the data range and the scale of investigation, from homogenized data sources and correspondences visible by vertical alignment. In contrast, 3D data visualization is complex to document. Under the Integrative Hybrid Models, there is a collection of scales and states and times. In addition, given the 3D space represented, the point of view and level of observation is as important as the visualization state. Collating this can be assisted by automated saving of user actions with default naming conventions. This becomes more essential in collaborative working environments such as magi 94. As mentioned above, 3D data do not contain a single linear narrative, and so the visualization tools should be able to combine different types of data into a single interactive workspace on screen—a dashboard or storyboard. The visualization process may appear to be exploratory, but by being task‐based it is a process of constructive story‐telling, relying on a flexibility of layout, and arrangeable components from a library familiar to users of the specific research domain. Such a shared vocabulary has been developed over the years for genome tracks with similar glyphs but there is no standard for the semantics of coding tracks. Reduction of an experiment and the visualization process into a 2D representation, for example, SushiR 95, and in 3D by guided rendering suites like PresentaBALL 96 and Molecular Flipbook 97. But interactivity is fundamental to the process of investigation through visualizations, and although publication files can be easily shared through common hosting services like Zenodo and Figshare, these are mainly shown as static figures and animations. Embedding of 3D models as U3D files in the proprietary PDF format is possible but cumbersome to generate and has not been widely adopted. In contrast, there has been significant recent adoption of web components which give the possibility of shared visualizations, through common syntax such as the D3js‐based Vega‐lite 98 or suites of graphs such as those found in the biojs archive 99, although these currently lack components specific to 3D data.
Conclusions and future perspective
The genome is three dimensional and its investigation is no longer limited to a flat Petri dish or a linear sequence, making it essential to record and depict its spatial nature. The data that can be obtained today present a number of challenges as they document the genome in unprecedented extent and detail and with new forms and relationships. There are distinct types of data coincident at the different levels of biological function, with complex interconnected attributes incompatible with conventional storage and access. Furthermore, the genome is shown to be dynamic, changing in state over time both as cellular machinery and as an evolutionary continuum across cell types and species. A wealth of understanding can emerge from coordination of the data, for example, by establishing ‘golden sets’ of reference structures. To this end, standard formats for integrated, multifaceted data are being created, and computational infrastructure and storage criteria are being established for processing and serving up genomic 3D data. Achieving this can be aided by the application of FAIR principles of data management but the challenges in visualization exceed the capabilities of current genome tools.
The genome browser is an archetypal tool due to its historic role providing unified access 1D and 2D data but is evolving to integrate the new spatial data. By combining techniques from across the sciences, these data are closing the resolution gap around 20 nm, however each discipline has a distinct perspective reflected in the way they represent the data. Cellular biology focuses downward from physiology on sequence tracks, whereas the biochemistry gazes upward from nucleotides at macromolecular models. Neither perceives the whole and there remain structures in the data, between chromosome and chromatin, that are unsatisfactorily represented by either browser or 3D viewer. Bridging this visualization gap to provide a homogenized view of these genomic data presents specific challenges: defining a novel visual grammar; augmenting analysis at scale; arranging sensible viewpoints; and capturing the interactive reasoning. We have described above a number of approaches and tools for conveying of 3D data that inform the design and construction of such a tool. These sections also define a data visualization process that is: Representative of the data and the intent of the user; Interactive for thorough investigation; Curated to map the scientific exploration; and Hosted, extolling FAIR Reuse to enable validation and reproducibility. For a complete and coherent view of the genome the data must be FAIR and the visualization must be RICH.
Visualization relies on having the human ‘in the loop’ to lift it above mere data processing and as such, it must integrate with the need and methods of the user. Today's web‐based technologies permit development of tools that can be created as individual presentations and yet work fluidly in ensembles allowing the novel tool can adapt to the different tasks of each researcher. New forms of interfacing with the data may produce unforeseen insights, as appears to be occurring with the penetration of virtual/augmented reality technologies, which are driving new standards and explorations within genomics and its visualization (e.g., spatial augmented reality haptics 100). But while these are seductive and well suited for expressive communication through projects such as Chromos 101, it is more important to maximize the use of the 3D data being produced by representing its detail rather than increasing immersion. The domain of structural genomics is still developing and new tasks and further challenges will arise in the near future. For now, current directions in visualization of 3D data development are marking the first moves toward the creation of an integral multifaceted view of genomics.
Terminology and abbreviations
3D: The abbreviation ‘3D’ can prove confusing. Popular usage signifies objects of Euclidian geometry, typically described by Cartesian x, y and z coordinates. However, it is commonly used in this field to refer to datasets which describe the 3D structure of the genome, even if the data is ‘2D’, that is, tabular or a matrix. We therefore use the terms ‘3D data’ and ‘3D model’ to differentiate them.
Structure: ‘that which defines and maintains physical relationships within an object or groups of components’, can be used in genomics both for 1D sequence, that is, ‘large‐scale structural genomic variations are insertions, deletions,’ and, as used in this review, for 3D spatial form, that is, ‘something which defines and maintains spatial form’.
Author's contributions
MG surveyed and tested the software. MG and MAM‐R wrote the paper. Both authors read and approved the final manuscript.
Acknowledgements
We thank François Serra, David Castillo, Irene Farabella, Marco Di Stefano, Yannick Spill, François Le Dily, Andrew Yates, Mark McDowall, Jürgen Walther, Sean O'Donoghue, Christian Stolte, Jeffery Heer, Jim Zheng, Philippe Youkharibache, Graham Johnson, Luc Patiny, Diana Roldan, Ian Sillitoe, Alexander Rose, Josep Lluis Gelpi, Adam Hospital, Elaine Meng, Tom Goddard, Daniel Russel, Merry Wang, Ben Webb, Brinda Vallat, Jianlin Chang, Oluwatosin Oluwadare, Victoria Neguembor, Nico Stuurman, and Lucy Natarajan for responding to our queries about their work.
We received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC Synergy grant agreement 609989 (4DGenome) as well as the European Union's Horizon 2020 research and innovation programme (agreement 676556). We acknowledge Centro de Excelencia Severo SEV‐2012‐0208 and CERCA Programme/Generalitat de Catalunya to CRG. The content of this manuscript reflects only the author's views and the Union is not liable for any use that may be made of the information contained therein.
Edited by Wilhelm Just
References
- 1. Marti‐Renom MA and Mirny LA (2011) Bridging the resolution gap in structural modeling of 3D genome organization. PLoS Comput Biol 7, e1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dekker J, Marti‐Renom MA and Mirny LA (2013) Exploring the three‐dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonev B and Cavalli G (2016) Organization and function of the 3D genome. Nat Rev Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- 4. Follain G, Mercier L, Osmani N, Harlepp S and Goetz JG (2017) Seeing is believing ‐ multi‐scale spatio‐temporal imaging towards in vivo cell biology. J Cell Sci 130, 23–38. [DOI] [PubMed] [Google Scholar]
- 5. Le Gros MA, Clowney EJ, Magklara A, Yen A, Markenscoff‐Papadimitriou E, Colquitt B, Myllys M, Kellis M, Lomvardas S and Larabell CA (2016) Soft X‐ray tomography reveals gradual chromatin compaction and reorganization during neurogenesis in vivo. Cell Rep 17, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guan J, Liu H, Shi X, Feng S and Huang B (2017) Tracking multiple genomic elements using correlative CRISPR imaging and sequential DNA FISH. Biophys J 112, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engreitz JM, Pandya‐Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES et al (2013) The Xist lncRNA exploits three‐dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serra F, Bau D, Goodstadt M, Castillo D, Filion G and Marti‐Renom MA (2017) Automatic analysis and 3D‐modelling of Hi‐C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput Biol 13, e1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dekker J, Rippe K, Dekker M and Kleckner N (2002) Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- 10. Di Stefano M, Paulsen J, Lien TG, Hovig E and Micheletti C (2016) Hi‐C‐constrained physical models of human chromosomes recover functionally‐related properties of genome organization. Sci Rep 6, 35985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dans PD, Walther J, Gomez H and Orozco M (2016) Multiscale simulation of DNA. Curr Opin Struct Biol 37, 29–45. [DOI] [PubMed] [Google Scholar]
- 12. Im W, Liang J, Olson A, Zhou HX, Vajda S and Vakser IA (2016) Challenges in structural approaches to cell modeling. J Mol Biol 428, 2943–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sali A, Berman HM, Schwede T, Trewhella J, Kleywegt G, Burley SK, Markley J, Nakamura H, Adams P, Bonvin AM et al (2015) Outcome of the first wwPDB hybrid/integrative methods task force workshop. Structure 23, 1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten JW, da Silva Santos LB, Bourne PE et al (2016) The FAIR guiding principles for scientific data management and stewardship. Sci Data 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richardson JS (1981) The anatomy and taxonomy of protein structure. Adv Protein Chem 34, 167–339. [DOI] [PubMed] [Google Scholar]
- 16. Tufte ER (2001) The Visual Display of Quantitative Information. Graphics Press, Cheshire, Conn. [Google Scholar]
- 17. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM and Haussler D (2002) The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munzner T (2014) Visualization Analysis and Design. CRC Press, Boca Raton, FL. [Google Scholar]
- 19. Heinrich J, Burch M and O'Donoghue SI (2014) On the use of 1D, 2D, and 3D visualisation for molecular graphics In 2014 IEEE VIS International Workshop on 3DVis, 3DVis 2014, pp. 55–60. [Google Scholar]
- 20. Pavlopoulos GA, Malliarakis D, Papanikolaou N, Theodosiou T, Enright AJ and Iliopoulos I (2015) Visualizing genome and systems biology: technologies, tools, implementation techniques and trends, past, present and future. Gigascience 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Kong L, Gao G and Luo J (2013) A brief introduction to web‐based genome browsers. Brief Bioinform 14, 131–143. [DOI] [PubMed] [Google Scholar]
- 22. Yardimci GG and Noble WS (2017) Software tools for visualizing Hi‐C data. Genome Biol 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT and Zhuang X (2016) Super‐resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS and Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozlíková B, Krone M, Falk M, Lindow N, Baaden M, Baum D, Viola I, Parulek J and Hege HC (2016) Visualization of Biomolecular Structures: state of the Art Revisited. Comput Graphics Forum, https://doi.org/10.1111/cgf.13072. [Google Scholar]
- 27. Risca VI and Greenleaf WJ (2015) Unraveling the 3D genome: genomics tools for multiscale exploration. Trends Genet 31, 357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ et al (2010) Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagano T, Lubling Y, Varnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P and Tanay A (2017) Cell‐cycle dynamics of chromosomal organization at single‐cell resolution. Nature 547, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA and Dekker J (2013) Organization of the mitotic chromosome. Science 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trussart M, Serra F, Bau D, Junier I, Serrano L and Marti‐Renom MA (2015) Assessing the limits of restraint‐based 3D modeling of genomes and genomic domains. Nucleic Acids Res 43, 3465–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams E, Moore J, Li SW, Rustici G, Tarkowska A, Chessel A, Leo S, Antal B, Ferguson RK, Sarkans U et al (2017) Image data resource: a bioimage data integration and publication platform. Nat Methods 14, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, Macdonald D et al (2010) Metadata matters: access to image data in the real world. J Cell Biol 189, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou X, Lowdon RF, Li D, Lawson HA, Madden PAFF, Costello JF, Wang T, Costello JF and Wang T (2013) Exploring long‐range genome interaction data using the WashU Epigenome Browser. Nat Methods 10, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quinlan AR and Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES and Aiden EL (2016) Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Syst 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tyner C, Barber GP, Casper J, Clawson H, Diekhans M, Eisenhart C, Fischer CM, Gibson D, Gonzalez JN, Guruvadoo L et al (2017) The UCSC genome browser database: 2017 update. Nucleic Acids Res 45, D626–D634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bau D, Sanyal A, Lajoie BR, Capriotti E, Byron M, Lawrence JB, Dekker J and Marti‐Renom MA (2011) The three‐dimensional folding of the alpha‐globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol 18, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lundborg M, Apostolov R, Spangberg D, Gardenas A, van der Spoel D and Lindahl E (2014) An efficient and extensible format, library, and API for binary trajectory data from molecular simulations. J Comput Chem 35, 260–269. [DOI] [PubMed] [Google Scholar]
- 40. Lenz O (2016) VTF format in https://github.com/olenz/vtfplugin/wiki/VTF-format.
- 41. Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O'Shaughnessy‐Kirwan A et al (2017) 3D structures of individual mammalian genomes studied by single‐cell Hi‐C. Nature 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quilez J, Vidal E, Dily FL, Serra F, Cuartero Y, Filion G, Graf T and Marti‐Renom MA (2017) Managing the analysis of high‐throughput sequencing data. bioRxiv, https://doi.org/10.1101/136358. [Google Scholar]
- 43. Arndt W, Asbury TM, Zheng WJ, Mitman M and Jijun T (2011) Genome3D: a viewer‐model framework for integrating and visualizing multi‐scale epigenomic information within a three‐dimensional genome In 2011 IEEE International Conference on Bioinformatics and Biomedicine Workshops, BIBMW 2011, pp. 936–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nowotny J, Wells A, Oluwadare O, Xu L, Cao R, Trieu T, He C and Cheng J (2016) GMOL: an interactive tool for 3D genome structure visualization. Sci Rep 6, 20802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shneiderman B (1996) The eyes have it: a task by data type taxonomy for information visualizations. Proceedings of the 1996 IEEE Symposium on Visual Languages, 336.
- 46. Szalaj P, Michalski PJ, Wroblewski P, Tang Z, Kadlof M, Mazzocco G, Ruan Y and Plewczynski D (2016) 3D‐GNOME: an integrated web service for structural modeling of the 3D genome. Nucleic Acids Res 44, W288–W293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lewis TE, Sillitoe I, Andreeva A, Blundell TL, Buchan DW, Chothia C, Cozzetto D, Dana JM, Filippis I, Gough J et al (2015) Genome3D: exploiting structure to help users understand their sequences. Nucleic Acids Res 43, D382–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Youkharibache P (2017) Twelve elements of visualization and analysis for tertiary and quaternary structure of biological molecules. bioRxiv, https://doi.org/10.1101/153528. [Google Scholar]
- 49. National Library of Medicine NCBI (2010) iCn3D: web‐based 3D structure viewer in https://www.ncbi.nlm.nih.gov/Structure/icn3d/icn3d.html.
- 50. Murray P, McGee F and Forbes AG (2017) A taxonomy of visualization tasks for the analysis of biological pathway data. BMC Bioinformatics 18, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yau N (2013) Data Points: Visualization That Means Something. John Wiley & Sons Inc, Indianapolis, IN. [Google Scholar]
- 52. Fry B (2008) Visualizing Data. O'Reilly Media Inc, Sebastopol, CA. [Google Scholar]
- 53. Tufte ER (1990) Envisioning Information. Graphics Press, Cheshire, CT. [Google Scholar]
- 54. Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni‐Schmidt O, Williams R, Chait BT et al (2007) Determining the architectures of macromolecular assemblies. Nature 450, 683–694. [DOI] [PubMed] [Google Scholar]
- 55. Ashikhmin M, Gleicher M, Hoffman N, Johnson G, Munzner T, Reinhard E, Sung K, Thompson WB, Willemsen P and Wyvill B (2005) Fundementals of Computer Graphics. A. K. Peters, Ltd. Natick, MA. [Google Scholar]
- 56. Risden K, Czerwinski MP, Munzner T and Cook DB (2000) An initial examination of ease of use for 2D and 3D information visualizations of web content. Int J Hum Comput Stud 53, 695–714. [Google Scholar]
- 57. Richardson JS (2000) Early ribbon drawings of proteins. Nat Struct Biol 7, 624–625. [DOI] [PubMed] [Google Scholar]
- 58. Goddard TD, Huang CC and Ferrin TE (2005) Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure 13, 473–482. [DOI] [PubMed] [Google Scholar]
- 59. Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y and Feig M (2016) Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 5, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meyer M, Munzner T, DePace A and Pfister H (2010) MulteeSum: a tool for comparative spatial and temporal gene expression data. IEEE Trans Vis Comput Graph 16, 908–917. [DOI] [PubMed] [Google Scholar]
- 61. Hillier B (2007) Space is the Machine. Space Syntax, London, UK. [Google Scholar]
- 62. Sun G, Liang R, Qu H and Wu Y (2017) Embedding spatio‐temporal information into maps by route‐zooming. IEEE Trans Vis Comput Graph 23, 1506–1519. [DOI] [PubMed] [Google Scholar]
- 63. Nielsen C and Wong B (2012) Points of view: representing the genome. Nat Methods 9, 423. [DOI] [PubMed] [Google Scholar]
- 64. Ferstay JA, Nielsen CB and Munzner T (2013) Variant view: visualizing sequence variants in their gene context. IEEE Trans Vis Comput Graph 19, 2546–2555. [DOI] [PubMed] [Google Scholar]
- 65. Nielsen CB, Jackman SD, Birol I and Jones SJ (2009) ABySS‐Explorer: visualizing genome sequence assemblies. IEEE Trans Vis Comput Graph 15, 881–888. [DOI] [PubMed] [Google Scholar]
- 66. Pak Chung W, Kwong Kwok W, Foote H and Thomas J (2003) Global visualization and alignments of whole bacterial genomes. IEEE Trans Visual Comput Graphics 9, 361–377. [Google Scholar]
- 67. Hamori E and Ruskin J (1983) H curves, a novel method of representation of nucleotide series especially suited for long DNA sequences. J Biol Chem 258, 1318–1327. [PubMed] [Google Scholar]
- 68. Temple MD (2017) An auditory display tool for DNA sequence analysis. BMC Bioinformatics 18, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Scales M, Jager R, Migliorini G, Houlston RS and Henrion MY (2014) visPIG–a web tool for producing multi‐region, multi‐track, multi‐scale plots of genetic data. PLoS ONE 9, e107497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Connors J, Krzywinski M, Schein J, Gascoyne R, Horsman D, Jones SJ and Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garvan Institute of Medical Research (2016) Rondo – interactive visual exploration of chromosome 3D connectivity maps in, commonwealth scientific and industrial research organisation (CSIRO), http://rondo.ws/.
- 72. Bernier CR, Petrov AS, Waterbury CC, Jett J, Li F, Freil LE, Xiong X, Wang L, Migliozzi BL, Hershkovits E et al (2014) RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss 169, 195–207. [DOI] [PubMed] [Google Scholar]
- 73. Bryan C, Guterman G, Ma KL, Lewin H, Larkin D, Kim J, Ma J and Farre M (2017) Synteny explorer: an interactive visualization application for teaching genome evolution. IEEE Trans Vis Comput Graph 23, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Laskowski RA and Swindells MB (2011) LigPlot+: multiple ligand‐protein interaction diagrams for drug discovery. J Chem Inf Model 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- 75. Hermosilla P, Estrada J, Guallar V, Ropinski T, Vinacua A and Vazquez PP (2017) Physics‐based visual characterization of molecular interaction forces. IEEE Trans Vis Comput Graph 23, 731–740. [DOI] [PubMed] [Google Scholar]
- 76. Krone M, Friess F, Scharnowski K, Reina G, Fademrecht S, Kulschewski T, Pleiss J and Ertl T (2017) Molecular surface maps. IEEE Trans Vis Comput Graph 23, 701–710. [DOI] [PubMed] [Google Scholar]
- 77. Brehmer M, Lee B, Bach B, Henry Riche N and Munzner T (2017) Timelines revisited: a design space and considerations for expressive storytelling IEEE Trans Vis Comput Graph 23, 2151–2164. [DOI] [PubMed] [Google Scholar]
- 78. Tory M, Kirkpatrick AE, Atkins MS and Moller T (2006) Visualization task performance with 2D, 3D, and combination displays. IEEE Trans Vis Comput Graph 12, 2–13. [DOI] [PubMed] [Google Scholar]
- 79. Fowler D and Ware C (1989). Strokes for Representing Univariate Vector Field Maps. In Proceedings of the Graphics Interface ‘89 London, Ontario, Canada: pp. 249–253 [Google Scholar]
- 80. Marchesin S, Chen CK, Ho C and Ma KL (2010) View‐dependent streamlines for 3D vector fields. IEEE Trans Vis Comput Graph 16, 1578–1586. [DOI] [PubMed] [Google Scholar]
- 81. Jupp S, Burdett T, Malone J, Leroy C, Pearce M, McMurry J and Parkinson H (2015). A new ontology lookup service at EMBL‐EBI. In Proceedings of SWAT4LS International Conference 2015, pp. 118–119. [Google Scholar]
- 82. O'Donoghue SI, Sabir KS, Kalemanov M, Stolte C, Wellmann B, Ho V, Roos M, Perdigao N, Buske FA, Heinrich J et al (2015) Aquaria: simplifying discovery and insight from protein structures. Nat Methods 12, 98–99. [DOI] [PubMed] [Google Scholar]
- 83. Shoresh N and Wong B (2012) Points of view: data exploration. Nat Methods 9, 5. [DOI] [PubMed] [Google Scholar]
- 84. Lekschas F, Bach B, Kerpedjiev P, Gehlenborg N and Pfister H (2017) HiPiler: visual exploration of large genome interaction matrices with interactive small multiples. bioRxiv, https://doi.org/10.1101/123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heer J, Kong N and Agrawala M (2009) Sizing the horizon: the effects of chart size and layering on the graphical perception of time series visualizations. Paper presented at the Proceedings of the SIGCHI Conference on Human Factors in Computing Systems.
- 86. Simons DJ, Franconeri SL and Reimer RL (2000) Change blindness in the absence of a visual disruption. Perception 29, 1143–1154. [DOI] [PubMed] [Google Scholar]
- 87. Nowell L, Hetzler E and Tanasse T (2001) Change blindness in information visualization: a case study. 15–22.
- 88. Meyer M, Wong B, Styczynski M, Munzner T and Pfister H (2010) Pathline: a tool for comparative functional genomics. Comput Graph Forum 29, 1043–1052. [Google Scholar]
- 89. Siersbaek R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P et al (2017) Dynamic rewiring of promoter‐anchored chromatin loops during adipocyte differentiation. Mol Cell 66, 420–435.e5. [DOI] [PubMed] [Google Scholar]
- 90. Fry B (2004) Computational Information Design. Massachusetts Institute of Technology, Thesis (Ph. D.)–Massachusetts Institute of Technology, http://hdl.handle.net/1721.1/26913. [Google Scholar]
- 91. Bustos B, Keim DA, Panse C and Schreck T (2004) 2D maps for visual analysis and retrieval in large multi‐feature 3D model databases. In Proceedings of the IEEE Visualization Conference (VIS) Austin, TX, pp. 598–599. [Google Scholar]
- 92. Scarr J, Cockburn A and Gutwin C (2013) Supporting and exploiting spatial memory in user interfaces. Found Trends Hum Comput Interact 6, 1–84. [Google Scholar]
- 93. UCSF Resource for Biocomputing Visualization and Informatics (2017) UCSF ChimeraX advantages in https://www.cgl.ucsf.edu/chimerax/docs/user/advantages.html.
- 94. Leiserson MD, Gramazio CC, Hu J, Wu HT, Laidlaw DH and Raphael BJ (2015) MAGI: visualization and collaborative annotation of genomic aberrations. Nat Methods 12, 483–484. [DOI] [PubMed] [Google Scholar]
- 95. Phanstiel DH, Boyle AP, Araya CL and Snyder MP (2014) Sushi.R: flexible, quantitative and integrative genomic visualizations for publication‐quality multi‐panel figures. Bioinformatics 30, 2808–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nickels S, Stockel D, Mueller SC, Lenhof H‐P, Hildebrandt A and Dehof AK (2013) PresentaBALL: A powerful package for presentations and lessons in structural biology. Paper presented at the BioVis 2013 ‐ IEEE Symposium on Biological Data Visualization 2013, Proceedings.
- 97. Iwasa JH (2015) Bringing macromolecular machinery to life using 3D animation. Curr Opin Struct Biol 31, 84–88. [DOI] [PubMed] [Google Scholar]
- 98. Satyanarayan A, Moritz D, Wongsuphasawat K and Heer J (2017) Vega‐lite: a grammar of interactive graphics. IEEE Trans Vis Comput Graph 23, 341–350. [DOI] [PubMed] [Google Scholar]
- 99. Gomez J, Garcia LJ, Salazar GA, Villaveces J, Gore S, Garcia A, Martin MJ, Launay G, Alcantara R, Del‐Toro N et al (2013) BioJS: an open source JavaScript framework for biological data visualization. Bioinformatics 29, 1103–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Thomas BH, Marner M, Smith RT, Elsayed NAM, Von Itzstein S, Klein K, Adcock M, Eades P, Irlitti A, Zucco J et al (2014) Spatial augmented reality ‐ A tool for 3D data visualization. In 2014 IEEE VIS International Workshop on 3DVis (3DVis), IEEE, Paris: pp. 45–50. [Google Scholar]
- 101. Cooper M, Lomas A, Spivakov M, Fraser P and Varnai C (2017) Chromos EP: Expressing genetic interactions through music in http://maxcooper.net/chromos-ep.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
As mentioned above, experimental analysis of genome organization requires various data types, which may be produced by others elsewhere. Publically accessible 1D reference genomes have not only ensured efficient use of resources but also provided a coherent baseline against which related data can be assessed and validated. For example, retrieval under the Browser Genome Release Agreement (BGRA, http://www.ensembl.org/info/about/legal/browser_agreement.html) requires consistent sequence and assembly identifiers. These providers manage data streaming to reduce network load and end‐user wait times by creating endpoints and application programming interface (APIs) like Track Hubs for easy access. Unfortunately, most 3D data remain accessible only by direct download from the labs, through nonspecialized databases or as supplementary publication data. While some 3D datasets have been submitted to UCSC and Ensembl as part of the BGRA or other special collections, there are no generally accepted and established specific repositories for 3D genomic data.
The data from super‐resolution microscopy consists of the raw images with metadata and processed binary spot list, which contain coordinate data and image attributes such as frame, intensity, peak shape, etc. These files are unwieldy with raw file sizes of around 10 GB (60 MB when processed). However, these are now being catered for the new image data resource (IDR) initiative for coordinating imaging with genomic data built on the open microscopy environment's (OME) OMERO image data management platform 32. OMERO uses the BioFormats tool that also promotes harmonization of formats via the open OME‐TIFF, which enable rich metadata while capturing multiple states or time frames within a single file 33. At smaller scales, EMPAIR archives electron microscopy raw 2D data and the electron microscopy data bank (EMDB) stores 3DEM models.
Interaction data (i.e., 3C‐based experimental data) lack dedicated repositories but can be made to fit with conventional repositories and track browsers as it records genomic loci pairs and associated scores (interaction counts). For example, such data can be visualized by the WashU Epigenome Browser by reading in tab‐separated text 34 similar to Bedtools BEDPE format 35. The Juicer software can also read and analyze 3C‐based interaction data by converting experimental reads into a tool‐specific Hi‐C format, which can then be visualized using the Juicebox browser 36. Finally, the tadbit software 8 can output complete experimental dataset in standard JSON format, which can then be imported and visualized using the TADkit 3D browser. Importantly, data accessibility and portability in 3D genomics still require some time to develop a widely accepted format for storing and disseminating interaction data and models. The most recently developed longTabix format from UCSC Genome Browser 37 could be used for such task but still lacks ways of representing multiple states or time frame datasets.
Coarse grain restraint‐based models derived from interaction data result in 3D objects represented by sets of Cartesian coordinates 38. A large number of formats can capture such positional data with states and dynamics (e.g., mmCIF with TNG trajectory format 39 or the VMD trajectory format 40). However, and unfortunately, such datasets are usually provided as flat text files or hacked into extended PDB files 41. While the latest facilitates distribution and portability to computational tools, this obliges the results to be adapted on an ad hoc basis, reducing potential for standardized, reproducible data integration, which may induce undocumented errors. Moreover, the classical PDB format must contain ‘< 62 chains and/or 99999 ATOM records’ and so it is unsuited to larger genome structures. The updated format, mmCIF, addresses these limitations but remains atom‐based, which still requires hacking to store large objects such as entire genomes. Finally, this approach is complicated to import into existing molecular tools that expect atomic coordinates as well as pushes the limits of the GL‐based visualizations. Given the size and complexity of the new macromolecular structures, RSCB‐PDB recently launched the innovative macromolecular transmission format (mmTF), which has greater compression for faster transfer while retaining efficient parsing. While the technique is instructive, the format is targeted at macromolecules and does not address the issues of file access in genomic structures. High‐speed Internet connections and chunked data requests may ease data transfer. However, the ideal strategy for both coarse grain and atomistic 3D models of genomes would be use of spatial data structures (e.g., OctTree, BallTree k‐d, etc.) although currently none of the newer formats discussed here take advantage of this.