

Abstract
The development of efficient metal‐promoted bioorthogonal ligations remains as a major scientific challenge. Demonstrated herein is that azides undergo efficient and regioselective room‐temperature annulations with thioalkynes in aqueous milieu when treated with catalytic amounts of a suitable ruthenium complex. The reaction is compatible with different biomolecules, and can be carried out in complex aqueous mixtures such as phosphate buffered saline, cell lysates, fetal bovine serum, and even living bacteria (E. coli). Importantly, the reaction is mutually compatible with the classical CuAAC.
Keywords: alkynes, azides, chemical ligations, click chemistry, ruthenium
The copper‐catalyzed azide–alkyne cycloaddition (CuAAC), paradigm of “click” chemistry,1 can be considered among the most relevant chemical transformations discovered in the last decades, with countless applications in many areas of science.2 The biological relevance of this reaction stems from its robustness and compatibility with aqueous media, as well as from its good bioorthogonality.3 However, the transformation still presents important limitations. Thus, in addition to being fairly incompatible with thiols, the reaction is essentially restricted to terminal alkynes, a consequence of a mechanism which requires the formation of copper acetylide intermediates (Scheme 1 a). An important additional drawback has to do with the side reactivity and toxicity of copper ions in biological contexts.4 Furthermore, to reach efficient conversions in typically diluted biological settings, the reactive copper(I) species need to be generated in situ using excess amounts of a copper(II) source and sodium ascorbate, a reductant which is not innocent in biological contexts.5 These issues have been partially addressed by using copper‐stabilizing ligands which enhance the biocompatibility and kinetic of the reactions.6, 7 Copper‐free, strain‐promoted annulations have been shown to be an efficient alternative,8 however, these reactions also present limitations associated to the side‐reactivity of the reactants. Therefore, the development of new bioorthogonal and biocompatible reactions which address some of the above limitations remains as a major challenge.9 In particular, the discovery of robust and aqueous‐compatible metal‐catalyzed annulations, as alternatives to the CuAAC, represents a highly appealing goal.10
Scheme 1.
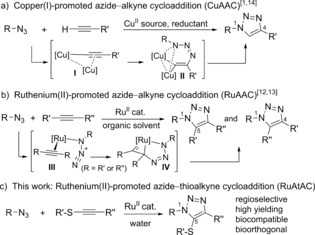
Key mechanistic features of CuAAC and RuAAC.
Several azide–alkyne cycloadditions using metals other than copper have been described in recent years,11 but only the ruthenium variant (RuAAC)12 has shown a meaningful scope (Scheme 1 b).13 In contrast to the CuAAC, which encompasses dinuclear copper intermediates such as I and II,14 the ruthenium‐promoted reaction involves intermediate species like III, which evolve into IV by oxidative cyclometalation, and eventually to the triazole products.15 In keeping with this scenario, the RuAAC, essentially developed in organic solvents, tolerates disubstituted alkynes but can produce mixtures of regioisomers. Probably, the notion that it is not compatible with water and air atmospheres has precluded more biofocused investigations.13, 16 Recent data suggest that some ruthenium complexes can promote the process in water, but the reactions require thermal activation and present a limited scope.17
Herein, we demonstrate that certain ruthenium(II) complexes can indeed catalyze the cycloaddition between azides and alkynes in water, and at room temperature. Importantly, the reaction is especially efficient when thioalkynes are used as reaction partners (Scheme 1 c). Moreover, the process is mutually compatible with the CuAAC, tolerant to different types of biomolecules, including thiols, and can be carried out in either phosphate buffered saline, cell lysates, or cell culture media, even in presence of living bacteria (E. coli).
At the outset, we were inspired by a report of Jia, Sun, and co‐workers on an iridium‐promoted azide–thioalkyne cycloaddition.18 Although the method was developed in anhydrous CH2Cl2, an isolated example in water using benzyl azide caught our attention. Unfortunately, when we tested the reaction of the thioalkyne 2 a with the fluorogenic anthracenyl‐azide probe 1 a,19 the yield of the corresponding adducts (3 aa/3 aa′) was modest (Table 1, entry 1).20 Remarkably, when using Cp*Ru(cod)Cl as a catalyst,15a we observed, after 24 hours, a substantial formation of the desired cycloadducts with excellent regioselectivity (3 aa/3 aa′=19:1, 58 % combined yield, entry 2). This good result, together with the previously demonstrated biocompatibility of this type of ruthenium complex,21 prompted us to further explore the process. Doubling the equivalents of 2 a led to an excellent yield of 99 % of the desired triazoles after 9 hours of stirring at room temperature. Monitoring the reaction at different times confirmed the formation of the products in 78 % yield after just 30 minutes, with the rate being then gradually reduced (entry 4).22 The performance of [Ir(cod)Cl]2 could not be improved by using 2 equivalents of thioalkyne, (entry 5). And other ruthenium(II) catalysts, such as Cp*Ru(PPh3)2Cl,12 RuH2(CO)(PPh3)3, 17b were not efficient (entries 6 and 7). In contrast, the tetramer [Cp*RuCl]4 23 was quite effective (entry 8). The reaction between 1 a and 2 a, could also be carried out in CH2Cl2. However, obtaining good yields required the use of anhydrous solvent and inert atmospheres (entries 9 vs. 10), which is not necessary in water. It looks like the aqueous solvent is somewhat precluding the ruthenium species from being rapidly deactivated.24
Table 1.
Identification of reaction conditions in water.[a]

| Entry | Cat. (X mol %) | 1 a/2 | Solv. | t [h] | Conv [%][b] | 3/3′ [b] | Yield [%][b,c] |
|---|---|---|---|---|---|---|---|
| 1 | [Ir(cod)Cl]2 (2.5) | 1:1 | H2O | 24 | 42 | 1:0 | 29 |
| 2 | Cp*Ru(cod)Cl (5) | 1:1 | H2O | 24 | 65 | 19:1 | 58 |
| 3 | Cp*Ru(cod)Cl (5) | 1:2 | H2O | 9 | 99 | 19:1 | 99 |
| 4 | Cp*Ru(cod)Cl (5) | 1:2 | H2O | 0.5 | 80 | 19:1 | 78 |
| 5 | [Ir(cod)Cl]2 (2.5) | 1:2 | H2O | 24 | 36 | 1:0 | 20 |
| 6 | Cp*Ru(PPh3)2Cl (5) | 1:2 | H2O | 24 | 47 | 23:1 | 17 |
| 7 | RuH2(CO)(PPh3)3 (5) | 1:2 | H2O | 24 | 0 | – | 0 |
| 8 | [Cp*RuCl]4 (1.25) | 1:2 | H2O | 24 | 99 | 14:1 | 99 |
| 9[d] | Cp*Ru(cod)Cl (5) | 1:2 | CH2Cl2 | 2 | 99 | 17:1 | 99 |
| 10 | Cp*Ru(cod)Cl (5) | 1:2 | CH2Cl2 | 2 | 44 | 18:1 | 37 |
| 11[e] | Cp*Ru(cod)Cl (5) | 1:2 | H2O | 9 | 99 | 5:1 | 95[e] |
| 12[f] | Cp*Ru(cod)Cl (5) | 1:2 | H2O | 24 | 10 | – | <5[f] |
| 13[g] | Cp*Ru(cod)Cl (5) | 1:4 | H2O | 4 | 99 | 19:1[h] | 98[h] |
[a] Unless otherwise noted, 2 a (1–2 equiv), water, and 1 a (1 equiv, 75 mm) were sequentially added under air to a vial containing the catalyst (that had been kept under N2), and the mixture was stirred at RT. [b] Determined by 1H NMR spectroscopy of the crude reaction mixture with an internal standard. [c] Combined yield of 3/3′. [d] Carried out under an inert atmosphere in anhydrous solvent. [e] Carried out with 2 b. Products: 3 ab/3 ab′. [f] Carried out with 2 c. Products: 3 ac/3 ac′. [g] Carried out using both 2 a and 2 b (2 equiv each). [h] Products: 3 aa and 3 aa′. cod=1,5‐cyclooctadiene. ![]()
The cycloaddition is also feasible using a typical internal alkyne such as 2 b, albeit somewhat slower (10 % less conversion after 30 min), and it led to a 5:1 mixture of regioisomers (Table 1, entry 11). Other alkynes such as 2 c were unreactive under identical reaction conditions (entry 12). The higher reactivity of the thioalkyne partner was clearly visible in a cross‐competition experiment: when the azide 1 a was reacted with a 1:1 mixture of 2 a and 2 b (2 equiv each), the triazole 3 aa, arising from the cycloaddition with the thioalkyne was exclusively observed in 98 % yield (entry 13). Interestingly, NMR analysis of the interaction between Cp*Ru(cod)Cl and the alkynes (in CD2Cl2) demonstrated that while 2 a displaces the cod ligand at room temperature, 2 b does not induce any change (see Pages S6–S9 of the Supporting Information). Analogous experiments using [Cp*RuCl]4 and 2 a, revealed rapid formation of a new complex identified as [Cp*Ru(2 a)Cl], whereas with alkyne 2 b no new ruthenium species could be detected, even after 3 hours.25 Thus, the good performance of thioalkynes might be in part related to their ability to strongly coordinate the Cp*RuCl moiety at room temperature. Additionally, the presence of the sulfur atom should also favor the formation of the required ruthenacyclic intermediate of type IV (Scheme 1 b).
With these reaction conditions in hand, we analyzed the scope of the method (RuAtAC). Despite the relatively poor water solubility of many of the azides and thioalkynes, the reactions proved to be general at room temperature, and the corresponding triazoles were obtained in good yields and with excellent regioselectivities (Table 2). Thus, aryl and aliphatic substituents either attached to the sulfur atom or to the terminal position of the alkyne were tolerated (e.g. 3 aa–af). Terminal or trimethylsilyl‐substituted thioalkynes (2 g, 2 h) provided the corresponding adducts 3 ag and 3 ah in good yields. Importantly, not only the anthracenyl and benzyl azide (1 a, 1 b) participated in the process, but aliphatic azides such as (2‐azidoethyl)benzene (1 c) or 2‐azidoethan‐1‐ol (1 d) also reacted cleanly to provide the corresponding triazoles (3 ca, 3 ce, 3 de). p‐Tolyl azide reacted with 2 a to provide 3 ea with a moderate 40 % yield, a value that could be improved up to 61 % by using [Cp*RuCl]4 as a catalyst. Interestingly, different types of fluorophore‐equipped trisubstituted triazoles could be generated by either using a dansyl‐based azide (such as in 3 fa) or by incorporating a coumarin moiety, either as part of the thioalkyne (e.g. 3 aj) or of the organic azide (3 ga and 3 ha). Additionally, following work developed by Waser and co‐workers,26 2‐thioglucose and a cysteine‐containing dipeptide were selectively thio‐alkynylated with EBX reagents. Gratifyingly, although the low water solubility of the thioalkynylated protected dipeptide 2 k demanded the use of small amounts of a cosolvent (i.e CH2Cl2, 5–10 % vol.), its reaction with 1 a and 1 f proceeded efficiently, thus affording the desired products 3 ak and 3 fk in good yields. The reaction between an alkynylated thioglucose and 1 a took place smoothly in water to give the triazole 3 al in 64 % yield.
Table 2.
Scope of the RuAtAC in water at room temperature.[a]

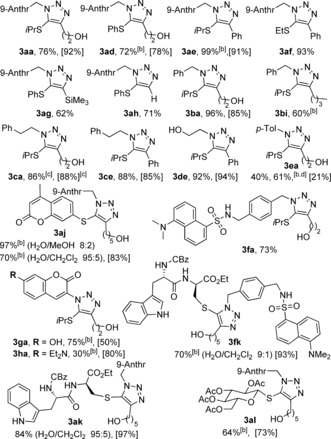
|
[a] Reaction conditions: 2 (2 equiv), water, and 1 a (1 equiv, 75 mm) were sequentially added under air to a vial containing Cp*Ru(cod)Cl (5 mol %) which had been kept under N2. The vial was closed, and the mixture stirred at RT for 15–24 h. Regioselectivities (3/3′) were >18:1 unless otherwise noted (determined by 1H NMR analysis of the crude reaction mixtures with an internal standard). Yield of isolated pure 3, unless otherwise noted. Yields of the reactions carried out in anhydrous CH2Cl2 under an inert atmosphere are shown within brackets. [b] Yield of 3 determined by 1H NMR analysis of the crude reaction mixture with an internal standard. [c] Corresponds to a 18:1 mixture of 3 ca/3 ca′. [d] Carried out with [Cp*RuCl]4 (1.25 mol %).
We next explored the bioorthogonality of the chemistry by performing the reaction in either the presence of different biomolecular additives or under biologically relevant conditions (Table 3). Gratifyingly, the RuAtAC could be efficiently carried out in the presence of glutathione (20 fold excess respect to the [Ru]; entry 1), different aminoacids (entry 2), and even in the presence of a random miniprotein (entry 3). The desired triazole was obtained in moderate to excellent yields and similar regioselectivities compared to those obtained in pure water (>15:1). The reaction could also be carried out in phosphate‐buffered saline (PBS; entry 4). Additionally, frequently used culture cell media, such as DMEM, fetal bovine serum (FBS), and Hela cell lysates are also excellent reaction media, so the triazole 3 aa was obtained in yields varying from 77 to 91 %, (entries 5–7). Analogue reactions in these media between (2‐azidoethyl)benzene (1 c) and the thioalkyne 2 e gave good yields of 3 ce (see Table S3).
Table 3.
Analysis of the biocompatibility of the method.[a]

| Entry | Conditions[b] | Conv. [%][c] | 3/3′[c] | Yield [%][c,d] |
|---|---|---|---|---|
| 1 | H2O/glutathione | 80 | 19:1 | 60 |
| 2 | H2O/Hist + Fmoc‐ala | 93 | 18:1 | 82 |
| 3 | H2O/Peptide 39 aa (0.5 mm) | 99 | 23:1 | 98 |
| 4 | PBS | 99 | 18:1 | 97 |
| 5 | cell cultured media (DMEM) | 99 | 15:1 | 84 |
| 6 | fetal bovine serum (FBS) | 88 | 17:1 | 77 |
| 7 | cell lysates (Hela) | 99 | 16:1 | 91 |
[a] Reaction conditions: 2 a (2 equiv) was added to a suspension of Cp*Ru(cod)Cl (5 mol %), 1 a (1 equiv, 75 mm), and the additive, in the selected milieu, and the resulting mixture was stirred for 24 h. [b] The additives in entries 1–2 are in 20‐fold excess with respect to the ruthenium catalyst. [c] Determined by 1H NMR analysis of the crude reaction mixture using an internal standard. [d] Combined yield of 3 aa/3 aa′.
At this point, it was of interest to contrast the performances of the RuAtAC and CuAAC in water to establish strengths and weaknesses of each method. Thus, we carried out parallel experiments using (2‐azidoethyl)benzene (1 c; 75 mm), 5 mol % of each catalyst, and either 2 e (for Ru) or phenylacetylene (for Cu). While the ruthenium‐promoted reaction was significantly faster than the copper counterpart, when using CuSO4 and sodium ascorbate for the latter (60 % vs. 22 % yield, after 2 h), the CuAAC became faster by including additives such as BTTAA (75 % yield after 2 h; see Table S4). Importantly, the CuAAC failed with internal alkynes, including thioalkynes like 2 a and, not surprisingly, it is essentially inhibited in the presence of thiols like glutathione (0 % yield after 24 h). In contrast, the RuAtAC works effectively even in the presence of a 20‐fold excess of glutathione (75 % yield), and provides the products with both internal and terminal thioalkynes (as shown in Table 2). On the weak side, the efficiency of the RuAtAC decreases upon dilution (16 % yield at 250 μm), while the ligand‐accelerated CuAAC provided a 57 % yield under similar micromolar conditions (see Pages S11–S13).
The lack of reactivity of internal thioalkynes in the presence of copper catalysts suggested that the CuAAC and the RuAtAC could be mutually orthogonal.27 Gratifyingly, the engineered diyne 4 was quantitatively converted into the bis(triazole) 5, without cross‐reactivity, by performing a CuAAC with the dansyl azide 1 f, and subsequent in situ addition of the ruthenium catalyst and the anthracenyl azide 1 a (1 equiv with respect to thioalkyne; Scheme 2). Considering the scarcity of mutually compatible bioorthogonal reactions, the possibility of using both annulations in tandem in one pot is certainly promising.27
Scheme 2.
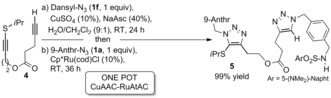
Tandem CuAAC and RuAtAC in water.
Finally, and importantly, we found that the RuAtAC can also be carried in the presence of bacteria (E. coli) without compromising their viability. Therefore, incubation of PBS containing E coli with 1 a (1 mm), 2 a (2 mm), and Cp*Ru(cod)Cl (100 μm) led to a rapid increase in the fluorescence. After 24 hours, centrifugation and analysis in a plate reader of both the extracellular supernatant and the methanol/water (8:2) extracts of the resulting bacteria pellet showed a combined increase in fluorescence of eight times with respect to controls (see Figure S29). Importantly, the fluorescence was mainly concentrated inside the bacteria, and the product 3 aa was also detected by HPLC‐ESI. Analysis of the optical density of the bacterial cultures revealed that neither the catalyst nor the reactants are meaningfully toxic (see Table S5).
In summary, we have discovered a new methodology to achieve catalytic, orthogonal chemical annulations in water, at room temperature. The reaction is promoted by specific ruthenium(II) catalysts, works efficiently with a variety of azides and thioalkynes, and can be carried out in presence of biomolecules (glutathione, aminoacids, peptides). The reaction is also efficient in phosphate buffered saline, and in complex biological media such as cell lysates and fetal bovine serum, and even in presence of living bacteria. Importantly, the reaction is mutually compatible with the classical CuAAC, thus providing the option of tandem biorthogonal processes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work has received financial support from Spanish grants (SAF2016‐76689‐R and SAF2013‐41943‐R), the Xunta de Galicia (2015‐CP082 and Centro Singular de Investigación de Galicia accreditation 2016‐2019 ED431G/09), the European Union (European Regional Development Fund ‐ ERDF), and the ERC (Adv. Grant 340055). We also thank the Orfeo‐Cinqa network CTQ2016‐81797‐REDC, and T. Seedat and V. Fraga for preliminary experiments.
P. Destito, J. R. Couceiro, H. Faustino, F. López, J. L. Mascareñas, Angew. Chem. Int. Ed. 2017, 56, 10766.
Contributor Information
Dr. Fernando López, Email: fernando.lopez@csic.es.
Prof. José L. Mascareñas, Email: joseluis.mascarenas@usc.es.
References
- 1. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056. [Google Scholar]
- 2.For instance, see:
- 2a. Hein J. E., Fokin V. V., Chem. Soc. Rev. 2010, 39, 1302; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Musumeci F., Schenone S., Desogus A., Nieddu E., Deodato D., Botta L., Curr. Med. Chem. 2015, 22, 2022; [DOI] [PubMed] [Google Scholar]
- 2c. Zhang T., Zheng Z., Cheng X., Ding X., Peng Y., Prog. Chem. 2008, 20, 1090; [Google Scholar]
- 2d. Matyašovský J., Perlíková P., Malnuit V., Pohl R., Hocek M., Angew. Chem. Int. Ed. 2016, 55, 15856; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16088. [Google Scholar]
- 3.
- 3a. Li L., Zhang Z., Molecules 2016, 21, 1393; [Google Scholar]
- 3b. Tiwari V. K., Mishra B. B., Mishra K. B., Mishra N., Singh A. S., Chen X., Chem. Rev. 2016, 116, 3086; [DOI] [PubMed] [Google Scholar]
- 3c. Schulz D., Rentmeister A., ChemBioChem 2014, 15, 2342. [DOI] [PubMed] [Google Scholar]
- 4. Kennedy D. C., McKay C. S., Legault M. C. B., Danielson D. C., Blake J. A., Pegoraro A. F., Stolow A., Mester Z., Pezacki J. P., J. Am. Chem. Soc. 2011, 133, 17993. [DOI] [PubMed] [Google Scholar]
- 5. Hong V., Steinmetz N. F., Manchester M., Finn M. G., Bioconjugate Chem. 2010, 21, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected examples, see:
- 6a. Rodionov V. O., Presolski S. I., Díaz Díaz D., Fokin V. V., Finn M. G., J. Am. Chem. Soc. 2007, 129, 12705; [DOI] [PubMed] [Google Scholar]
- 6b. Presolski S. I., Hong V., Cho S.-H., Finn M. G., J. Am. Chem. Soc. 2010, 132, 14570; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Besanceney-Webler C., Jiang H., Zheng T., Feng L., Soriano del Amo D., Wang W., Klivansky L. M., Marlow F. L., Liu Y., Wu P., Angew. Chem. Int. Ed. 2011, 50, 8051; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8201; [Google Scholar]
- 6d. Uttamapinant C., Tangpeerachaikul A., Grecian S., Clarke S., Singh U., Slade P., Gee K. R., Ting A. Y., Angew. Chem. Int. Ed. 2012, 51, 5852; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5954; [Google Scholar]
- 6e. Li S., Wang L., Yu F., Zhu Z., Shobaki D., Chen H., Wang M., Wang J., Qin G., Erasquin U. J., Ren L., Wang Y., Cai C., Chem. Sci. 2017, 8, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For the use of nanoparticle-based catalysts, see:
- 7a. Clavadetscher J., Hoffmann S., Lilienkampf A., Mackay L., Yusop R. M., Rider S. A., Mullins J. J., Bradley M., Angew. Chem. Int. Ed. 2016, 55, 15662; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15891; [Google Scholar]
- 7b. Bai Y., Feng X., Xing H., Xu Y., Kim B. K., Baig N., Zhou T., Gewirth A. A., Lu Y., Oldfield E., Zimmerman S. C., J. Am. Chem. Soc. 2016, 138, 11077. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Jewett J. C., Bertozzi C. R., Chem. Soc. Rev. 2010, 39, 1272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Devaraj N. K., Weissleder R., Acc. Chem. Res. 2011, 44, 816. For photoinducible annulations, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Lim R. K. V., Lin Q., Acc. Chem. Res. 2011, 44, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Vrabel M., Carell T., Cycloadditions in Bioorthogonal Chemistry, Springer, Heidelberg, 2016; [Google Scholar]
- 9b. Shieh P., Bertozzi C. R., Org. Biomol. Chem. 2014, 12, 9307; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. McKay C. S., Finn M. G., Chem. Biol. 2014, 21, 1075; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. King M., Wagner A., Bioconjugate Chem. 2014, 25, 825; [DOI] [PubMed] [Google Scholar]
- 9e. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592; [DOI] [PubMed] [Google Scholar]
- 9f. Ramil C. P., Lin Q., Chem. Commun. 2013, 49, 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Yang M., Yang Y., Chen P. R., Top. Curr. Chem. 2016, 374, 2; [DOI] [PubMed] [Google Scholar]
- 10b. Yang M. Y., Li J., Chen P. R., Chem. Soc. Rev. 2014, 43, 6511. [DOI] [PubMed] [Google Scholar]
- 11. Wang C., Ikhlef D., Kahlal S., Saillard J.-Y., Astruc D., Coord. Chem. Rev. 2016, 316, 1. [Google Scholar]
- 12. Zhang L., Chen X., Xue P., Sun H. H. Y., Williams I. D., Sharpless K. B., Fokin V. V., Jia G., J. Am. Chem. Soc. 2005, 127, 15998. [DOI] [PubMed] [Google Scholar]
- 13. Johansson J. R., Beke-Somfai T., Said Stalsmeden A., Kann N., Chem. Rev. 2016, 116, 14726. [DOI] [PubMed] [Google Scholar]
- 14.For a recent review, see:
- 14a. Zhu L., Brassard C. J., Zhang X., Guha P. M., Clark R. J., Chem. Rec. 2016, 16, 1501. See also: [DOI] [PubMed] [Google Scholar]
- 14b. Worrell B. T., Malik J. A., Fokin V. V., Science 2013, 340, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Boren B. C., Narayan S., Rasmussen L. K., Zhang L., Zhao H., Lin Z., Jia G., Fokin V. V., J. Am. Chem. Soc. 2008, 130, 8923; [DOI] [PubMed] [Google Scholar]
- 15b. Boz E., Tüzün N. Ş., J. Organomet. Chem. 2013, 724, 167. [Google Scholar]
- 16. Oakdale J. S., Fokin V. V., Umezaki S., Fukuyama T., Org. Synth. 2013, 90, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Wang T.-H., Wu F.-L., Chiang G.-R., He S.-T., Lo Y.-H., J. Organomet. Chem. 2014, 774, 57; [Google Scholar]
- 17b. Siyang H. X., Liu H. L., Wu X. Y., Liu P. N., RSC Adv. 2015, 5, 4693; [Google Scholar]
- 17c. Molla R. A., Roy A. S., Ghosh K., Salam N., Iqubal M. A., Tuhina K., Islam S. M., J. Organomet. Chem. 2015, 776, 170. [Google Scholar]
- 18.
- 18a. Ding S., Jia G., Sun J., Angew. Chem. Int. Ed. 2014, 53, 1877; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1908; [Google Scholar]
- 18b. Luo Q., Jia G., Sun J., Lin Z., J. Org. Chem. 2014, 79, 11970. [DOI] [PubMed] [Google Scholar]
- 19. Le Droumaguet C., Wang C., Wang Q., Chem. Soc. Rev. 2010, 39, 1233. [DOI] [PubMed] [Google Scholar]
- 20.In anhydrous CH2Cl2 under argon, 3 aa was obtained in 78 % yield (see Table S1).
- 21.
- 21a. Streu C., Meggers E., Angew. Chem. Int. Ed. 2006, 45, 5645; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5773; [Google Scholar]
- 21b. Völker T., Dempwolff F., Graumann P. L., Meggers E., Angew. Chem. Int. Ed. 2014, 53, 10536; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10705; [Google Scholar]
- 21c. Hsu H.-T., Trantow B. M., Waymouth R. M., Wender P. A., Bioconjugate Chem. 2016, 27, 376; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Sánchez M. I., Penas C., Vázquez M. E., Mascareñas J. L., Chem. Sci. 2014, 5, 1901; [PMC free article] [PubMed] [Google Scholar]
- 21e. Tomás-Gamasa M., Martínez-Calvo M., Couceiro J. R., Mascareñas J. L., Nat. Commun. 2016, 7, 12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Increasing the amount of the catalyst doesn't lead to substantial changes in the rate (see the Supporting Information).
- 23. Rasmussen L. K., Boren B. C., Fokin V. V., Org. Lett. 2007, 9, 5337. [DOI] [PubMed] [Google Scholar]
- 24.This might be associated to the lower solubility of O2 and/or the ruthenium catalysts in water than in the organic solvent. See:
- 24a. Li C.-J., Chen L., Chem. Soc. Rev. 2006, 35, 68; [DOI] [PubMed] [Google Scholar]
- 24b. Chanda A., Fokin V. V., Chem. Rev. 2009, 109, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a.According to Fürstner et al., Cp*Ru(2 a)Cl would be an 18 e− Ru species, with the alkyne acting as a 4 e− donor ligand: Roşca D.-A., Radkowski K., Wolf L. M., Wagh M., Goddard R., Thiel W., Fürstner A., J. Am. Chem. Soc. 2017, 139, 2443. [DOI] [PubMed] [Google Scholar]
- 25b.With 2 b, a fast exchange between the cod ligand and the alkyne cannot be discarded.
- 25c.Importantly, [Cp*Ru(2 a)Cl] is catalytically competent (see Pages S6–S9).
- 26. Frei R., Wodrich M. D., Hari D. P., Bonin P.-A., Chauvier C., Waser J., J. Am. Chem. Soc. 2014, 136, 16563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patterson D. M., Prescher J. A., Curr. Opin. Chem. Biol. 2015, 28, 141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary