

Abstract
Fast pyrolysis bio‐oils possess unfavorable physicochemical properties and poor stability, in large part, owing to the presence of carboxylic acids, which hinders their use as biofuels. Catalytic esterification offers an atom‐ and energy‐efficient route to upgrade pyrolysis bio‐oils. Propyl sulfonic acid (PrSO3H) silicas are active for carboxylic acid esterification but suffer mass‐transport limitations for bulky substrates. The incorporation of macropores (200 nm) enhances the activity of mesoporous SBA‐15 architectures (post‐functionalized by hydrothermal saline‐promoted grafting) for the esterification of linear carboxylic acids, with the magnitude of the turnover frequency (TOF) enhancement increasing with carboxylic acid chain length from 5 % (C3) to 110 % (C12). Macroporous–mesoporous PrSO3H/SBA‐15 also provides a two‐fold TOF enhancement over its mesoporous analogue for the esterification of a real, thermal fast‐pyrolysis bio‐oil derived from woodchips. The total acid number was reduced by 57 %, as determined by GC×GC–time‐of‐flight mass spectrometry (GC×GC–ToFMS), which indicated ester and ether formation accompanying the loss of acid, phenolic, aldehyde, and ketone components.
Keywords: acidity, bio-oil, esterification, mesoporous silica, sulfonic acid
Introduction
Biofuels have an important role to play in mitigating anthropogenic climate change arising from the combustion of fossil fuels.1 In the context of energy, despite significant growth in fossil fuel reserves, great uncertainties remain in the economic and environmental impact of exploitation, and crucially, approximately 65–80 % of such carbon resources cannot be burned without breaching the United Nations framework convention on climate change (UNFCC) target to keep the global temperature rise this century well below 2 °C. Biofuels will prove critical in helping many countries meet their renewable energy commitments, which for the UK are 15 % by 2020, alongside greenhouse gas (GHG) emission reductions of 34 % by 2020 and 80 % by 2050 (compared with 1990 levels). They also represent drop‐in fuels able to utilize existing pipeline and filling station distribution networks.2 Thermochemical processing of waste biomass such as lignocellulosic materials sourced from agriculture or municipal waste offers a promising route to biofuels through pyrolysis.3
Pyrolysis is a widespread approach for bio‐oil4 synthesis, in which biomass is thermally decomposed in an oxygen‐free or oxygen‐limited environment.5 The resulting crude bio‐oil is a complex mixture of acids, alcohols, furans, aldehydes, esters, ketones, sugars, and multifunctional compounds such as hydroxyacetic acid, hydroxyl‐acetaldehyde and hydroxyacetone (derived from cellulose and hemicellulose), together with 3‐hydroxy‐3‐methoxy benzaldehyde, phenols, guaiacols, and syringols derived from the lignin component.1b, 6 Pyrolysis bio‐oils thus require “upgrading” through deoxygenation and neutralization to enhance their energy density, stability, and physical properties.6a, 7 A range of catalytic upgrading methods are known,8 at least at the laboratory scale, including esterification,9 ketonization,10 hydrodeoxygenation,11 and condensation.12
Carboxylic acids comprise 5–10 wt % of pyrolysis bio‐oils,9, 13 and are largely responsible for their poor chemical stability. Hence, esterification (particularly employing bio‐derived alcohols such as methanol, ethanol, or phenols9, 14) offers an energy‐efficient and atom‐economical route to upgrading.8b, 15 Homogeneous mineral acid catalysts are historically employed for esterification, however their process disadvantages and poor (environmental) E‐factors are well‐documented; hence, strong drivers remain for the development of heterogeneous solid acid counterparts.11 Although base catalysts are widely used for the transesterification of vegetable oils (triacylglycerides) to yield biodiesel, they are unsuitable for catalytic esterification owing to neutralization/saponification.1d
Diverse solid acids have been explored for esterification, including zeolites,16 heteropolyacids,17 sulfated metal oxides,18 carbon‐based acid catalysts,19 and functionalized mesoporous silicas.20 Research on the latter indicates that mesoporous SBA‐15,21 KIT‐6,22 and PMO23 sulfonic acids, and a macroporous–mesoporous SBA‐15 (MM‐SBA‐15)20g analogue, are among the most promising owing to their tunable pore architecture strong Brønsted acidity and hydrophobicity.2a, 14, 20g, 23, 24 3‐Propylsulfonic acid (PrSO3H)/SBA‐15 has been reported as an efficient catalyst for acetic acid esterification with methanol2a, 25 and other alcohols in simulated bio‐oils,26 and the most widely used sulfonic acid in solid acid catalyzed esterification.27 Such catalysts exhibit improved water tolerance during esterification when the sulfonated silica surface is co‐functionalized with alkyl chains.2a, 5, 25b We recently reported a post‐modification hydrothermal saline‐promoted grafting (HSPG) route to introduce higher sulfonic acid loadings into mesoporous silicas than those achievable by conventional grafting methods,24a and confer stability towards leaching during the esterification of model acids.24b, 28 Hydrophobicity and catalytic reactivity, can also be enhanced through incorporating organic groups into the silica framework.24b Mesopore interconnectivity also plays a role in controlling esterification activity, with interconnectivity between the hexagonal cylindrical mesopores of PrSO3H/KIT‐6 offering superior mass transport and active site accessibility to non‐interconnected PrSO3H/SBA‐15.20g Mesopore expansion (from ≈5 to 14 nm),14 and macropore incorporation23 offer alternative approaches to enhance the esterification activity of PrSO3H/SBA‐15 for long chain fatty acid esterification.
With respect to bio‐oil upgrading through catalytic esterification, most studies have employed only model compounds owing to the complex nature of real pyrolysis bio‐oils7a and the associated analytical challenge. We previously reported the application of PrSO3H/SBA‐15 for acetic acid esterification of model bio‐oils.26, 28 Here, we report the synthesis and application of HSPG‐derived mesoporous PrSO3H/SBA‐15, and a macroporous counterpart, for the esterification of simple carboxylic acids (C3, C6, and C12), and the upgrading of thermal fast pyrolysis bio‐oil derived from woodchips.
Results and Discussion
Catalyst characterization
The successful synthesis of an ordered mesoporous skeleton for SBA‐15 and a macroporous–mesoporous (MM) skeleton for MM‐SBA‐15 (with a mean macropore diameter of ≈200 nm, close to that of the polystyrene colloidal hard template, Figure S1 in the Supporting Information) supports was confirmed by TEM. An ordered, 2D hexagonal mesopore channel network was observed for the former, and a well‐defined interconnecting macropore‐mesopore network for the latter (Figure S2). Formation of the desired p6mm pore architecture for both SBA‐15 and MM‐SBA‐15 was confirmed by low angle X‐ray diffraction (Figure S3), which revealed reflections characteristic of hexagonally ordered mesostructures. Both supports retained hexagonal close packed pore architectures following functionalization by propylsulfonic acid in a H2O/NaCl mixture (the HSPG method). However, a shift in the diffraction peaks to higher angle was observed post‐functionalization owing to mesopore contraction.23 Mesopore generation (and retention after sulfonation) was further evidenced by N2 porosimetry, which showed type IV isotherms with H1 hysteresis loops for all materials (Figure S4). The textural properties of PrSO3H/SBA‐15 and PrSO3H/MM‐SBA‐15 are summarized in Table 1. The BET surface areas decreased after sulfonic acid grafting over both silicas owing to micropore blockage, which was apparent as a dramatic drop in the micropore area and pore volume. These changes were accompanied by a decrease in pore diameter and an increase in wall thickness, suggesting the uniform grafting of sulfonic acid groups throughout both pore networks without distortion of their unit cells. Previous studies have shown the macropores in such hierarchical frameworks are open and interconnected by bottleneck pore openings.23, 29
Table 1.
Physicochemical properties of mesoporous SBA‐15 and macroporous–mesoporous SBA‐15 and their sulfonic acid analogues.
| Sample | Surface area | d p | V total | V micropore | Wall thickness | Unit cell parameter | S loading | Acid loading |
|---|---|---|---|---|---|---|---|---|
| [m2 g−1][a] | [nm][b] | [cc g−1] | [cc g−1][c] | [nm] | [nm] | [wt %][d] | [mmol g−1][e] | |
| SBA15 | 879 | 5.5 | 1.17 | 0.08 | 5.5 | 11.0 | – | – |
| PrSO3H/SBA15 | 379 | 3.8 | 0.49 | 0.01 | 7.3 | 11.1 | 5.8 | 1.5 |
| MM‐SBA‐15 | 357 | 4.5 | 0.55 | 0.02 | 5.9 | 9.0 | – | – |
| PrSO3H/MM‐SBA‐15 | 186 | 3.4 | 0.24 | 0.00 | 7.2 | 9.2 | 5.5 | 1.6 |
[a] BET, [b] BJH, [c] t‐plot, [d] CHNS, [e] propylamine adsorption/TGA‐MS.
Diffuse reflectance infrared fourier transform spectra (DRIFTS) of the parent silicas showed bands at 700–1400 cm−1 and 3000–3800 cm−1, which were indicative of framework Si‐O‐Si and surface silanols, respectively (Figure S5).15 Additional bands appeared at approximately 2960‐2830 cm−1after sulfonation of both materials, which were attributed to CH2 vibrations of the propyl backbone, and a new CH2−Si band centered at 1360 cm−1. CHNS elemental analysis of the sulfonated silicas revealed that both contained approximately 6 wt % sulfur (Table 1), which represented a five‐fold increase over conventional toluene grafting,14, 23 in good agreement with our preliminary results using the HSPG method.24a S 2p XP spectra of both sulfonic‐acid‐functionalized materials in Figure S6 reveal two distinct S chemical environments; a low binding energy centered at 164.5 eV associated with unoxidized thiol, and a higher energy doublet arising from sulfonic acid groups centered at 168.9 eV.30 Quantitative XPS analysis (Table S1) showed that approximately 85 % of S was incorporated as sulfonic acid groups. Thermogravimetric analysis (Figure S7 b) highlighted two major weight losses; one below 100 °C, which was attributed to physisorbed water; and the second between 250–650 °C owing to propylsulfonic acid decomposition.31 The bulk S content estimated from this second loss feature was approximately 5 wt % in accordance with elemental analysis. Acid properties of both sulfonated silica were subsequently probed through pyridine and propylamine adsorption. DRIFT spectra of pyridine‐titrated materials (Figure S8) evidenced only Brønsted acid sites.26 Temperature‐programmed analysis of reactively formed propene from chemisorbed propylamine confirmed that PrSO3H/SBA‐15 and PrSO3H/MM‐SBA‐15 possessed similar acid strengths and loadings (Figure S9 and Figure S10). Therefore, the incorporation of macropores into the SBA‐15 architecture had minimal impact on silica functionalization; the propylsulfonic acid functions grafted over silica surfaces in PrSO3H/SBA‐15 and PrSO3H/MM‐SBA‐15 catalysts were chemically identical. Therefore, any differences in TOFs between the two catalysts must arise solely from diffusion phenomena. However, despite their similar acid site loadings, the surface coverage of acid sites was higher over the macroporous material (which possessed a lower surface area). Note that the higher S loadings accessible through the HSPG method offer acid loadings of approximately 1.5 mmol g−1, approximately twice those obtained through sulfonic acid grafting in toluene (0.6–0.8 mmol g−1).2a Molecular dynamics simulations and adsorption calorimetry revealed that cooperative effects between silanol and sulfonic acid functions can weaken their acidity in PrSO3H/MCM‐41 owing to hydrogen bonding and associate sulfonate reorientation.32 However, such effects only operated for low sulfonic acid loadings, and were absent on crowded surfaces such as those employed in this work; hence, cooperative effects were not expected to influence the catalytic performance.
Esterification of model carboxylic acids
The catalytic performance of mesoporous and macroporous–mesoporous sulfonic acid silicas was evaluated in the esterification of propanoic (C3), hexanoic (C6), and lauric acids (C12) with methanol to explore the influence of the macropores on the reactivity under previously optimized conditions.2a Because both catalysts possessed similar acid loadings and strength, any differences in activity must arise from their pore architecture. Both sulfonic acid catalysts were active for methylic esterification of the C3, C6, and C12 acids (Figure S11), which were 100 % selective to their corresponding methyl esters. The rate of esterification decreased with increasing alkyl chain length owing to polar and steric effects.33
The associated turnover frequencies (TOFs) for carboxylic acid esterification were similar over both catalysts for the C3 and C6 acids (Figure 1), whereas the TOF for lauric acid over the hierarchical PrSO3H/MM‐SBA‐15 was twice that observed for the purely mesoporous PrSO3H/SBA‐15 (Figure S12). This rate enhancement for the bulky lauric acid esterification could be explained in terms of improved sulfonic acid accessibility through (i) faster in‐pore diffusion of the reactant/ester product; (ii) shorter mesopore channel lengths owing to truncation by macropores; and (iii) an increased number of mesopore openings, which may boost the sulfonic acid density at mesopore entrances.23
Figure 1.
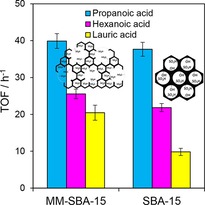
TOF for esterification of various carboxylic acids over PrSO3H/SBA‐15 and PrSO3H/MM‐SBA‐15 catalysts. (Reaction conditions: 25 mg catalyst, 5 mmol acid, acid/MeOH molar ratio=1:30, 60 °C).
Esterification of thermal pyrolysis bio‐oil
The performance of both sulfonic acid silicas was also assessed for the upgrading of a bio‐oil produced by thermal fast pyrolysis of oak woodchips at a bench‐scale, continuous fluidized bed reactor at 500 °C. Some physicochemical properties of the parent biomass feedstock are presented in Table S2, and of the crude bio‐oil in Table S3. Although the bio‐oil possessed a similar calorific value to the woodchips, the volumetric energy density of the former was significantly higher than that of the original biomass, whose density was only 600–900 kg m−3. The bio‐oil contained 23 wt % water, typical of fast pyrolysis bio‐oil,6b, 34 although the total acid number (TAN) of 61.6 mg KOH g−1 measured by the Modified D664A acid number titration method35 was relatively low.34
Figure 2 compares TOFs for total acid removal (as determined by KOH titration) through catalytic esterification with methanol, and the corresponding reaction profiles for total acid conversion (Figure 2 inset). The PrSO3H/MM‐SBA‐15 catalyst was almost three times more active in terms of TOF, and converted twice the amount of acid than the PrSO3H/SBA‐15 after 6 h. Because the pyrolysis oil contains numerous bulky compounds as described in Table 2, Table 3, and Table S4, we attributed the superior performance of the hierarchical catalyst to improved active site accessibility akin to that for lauric acid esterification. The carboxylic acid constituents of fast pyrolysis bio‐oils may drive low level (<5 %) autocatalytic esterification.36 This was consistent with a control experiment in the absence of any sulfonic acid catalyst, which revealed <8 % total acid conversion of the pyrolysis bio‐oil. Hence, autocatalysis exerted minimal impact on our results.
Figure 2.
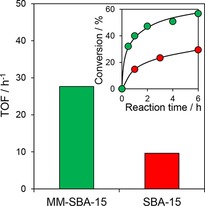
Effect of support architecture on the TOFs of sulfonic acid catalyzed bio‐oil esterification. Inset: acid conversion profiles for bio‐oil esterification using sulfonic acid catalysts. (Reaction conditions: 9.2 g bio‐oil ≈10 mmol acid, 12.1 mL MeOH (acid/MeOH molar ratio=1:30), 100 mg catalyst, 85 °C).
Table 2.
Compositions of crude and upgraded bio‐oils following treatment with PrSO3H/MM‐SBA‐15 catalyst.
| Group | Crude bio‐oil/ Area [%] | Upgraded bio‐oil/ Area [%] |
|---|---|---|
| aromatic hydrocarbons | 1.8 | 1.9 |
| aliphatic hydrocarbons | 0.4 | 2.1 |
| phenolic compounds | 25.8 | 7.8 |
| furanic compounds | 0.6 | 1.4 |
| organic acids | 19.7 | 0.9 |
| esters | 1.9 | 11.8 |
| alcohols | 1.1 | 26.1 |
| ethers | 1.0 | 6.5 |
| aldehydes | 5.2 | 0.4 |
| ketones | 10.8 | 2.9 |
| sugars and anhydro sugars | 26.6 | 13.5 |
| unidentified | 5.3 | 24.7 |
Table 3.
Esters present in crude and upgraded thermal fast pyrolysis bio‐oils following treatment with PrSO3H/MM‐SBA‐15 catalyst.
| Crude bio‐oil | Esterified bio‐oil |
|---|---|
| acetic acid, methyl ester | acetic acid, methyl ester |
| formic acid, 2‐propenyl ester | butanedioic acid, dimethyl ester |
| ethanedioic acid, diethyl ester | hexanoic acid, methyl ester |
| propanoic acid, ethenyl ester | 9‐octadecenoic acid (Z)‐, methyl ester |
| ethyl homovanillate | butanedioic acid, methyl‐, dimethyl ester |
| methyl propionate | |
| octanoic acid, methyl ester | |
| levulinic acid, methyl ester | |
| nonanoic acid, methyl ester |
The chemical composition of the crude and upgraded bio‐oil following catalytic treatment by PrSO3H/MM‐SBA‐15 were analyzed in detail by GC×GC–time‐of‐flight mass spectrometry (GC×GC–ToFMS), and the resulting 2D chromatograms are shown in Figure 3. For both the crude and upgraded bio‐oils, the chromatographic space was divided into six discreet molecular groups: acids and esters; aldehydes and ketones (including furanoics and cyclic carbonyls); hydrocarbons (saturated and unsaturated non‐aromatic); aromatic hydrocarbons; phenolic compounds; and sugars. Compounds that could not be identified by the library and/or did not meet the required identification criteria (as detailed in the Supporting Information) were classified as “unidentified”. A more detailed classification of each molecular group and their relative chromatographic area is presented in Table 2. Almost complete loss of organic acids (from 19.7 to 0.9 %) and a significant decrease in phenolics, ketones, aldehydes, and sugars was observed following catalytic upgrading, accompanied by a significant increase in ester and alcohol components, consistent with esterification. Additional details on the removal/formation of specific phenolics, ethers, and carbonyls is presented in Table S4. Acetic acid was the major organic acid in both the crude and upgraded bio‐oils. Esters with relative areas >0.1 in the crude and upgraded bio‐oils are presented in Table 3.
Figure 3.
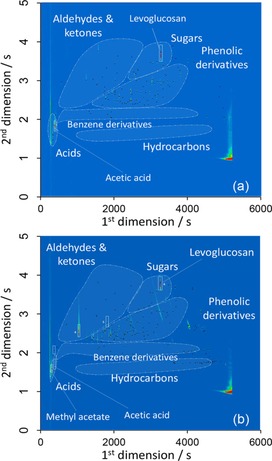
GC×GC–ToFMS chromatogram of a) crude thermal fast pyrolysis bio‐oil and b) bio‐oil after esterification over PrSO3H/MM‐SBA‐15.
Methyl acetate accounted for 10.8 % of the total chromatographic area of the esterified bio‐oil, as compared to only 1.4 % of the crude bio‐oil, alongside a range of methyl and dimethyl esters from C3–C11 compounds. Identifiable ethers were mainly C3–C6 methoxy‐compounds, with 1,1,2,2‐tetramethoxyethane predominant. Considering phenolics, upgrading principally removed methoxy‐phenols, whereas cresol and catechol derivatives were recalcitrant. The increase in alcohols appeared to arise from glycolaldehyde dimethyl acetal (GDA) formation from levoglucosan.37 Previous studies have revealed that levoglucosan can be transformed in alcohol media by acid catalysts to methyl levulinate, through intermediate glycolaldehyde (GA) formation38 (which may itself form glycolaldehyde dimethyl acetal). GA and GDA were detected in the upgraded bio‐oil, supporting this proposed reaction pathway. Future work will address the recyclability of PrSO3H/MM‐SBA‐15 for the esterification of real bio‐oils, wherein we expect strong adsorption of organics that will require the development of low‐temperature regeneration protocols that avoid decomposition of the grafted sulfonate.
In summary, GC×GC–ToFMS analysis confirmed that PrSO3H/MM‐SBA‐15 was an effective catalyst for the esterification of a real thermal pyrolysis bio‐oil, significantly reducing the bio‐oil acidity through esterification of organic acids under mild reaction conditions.
Conclusions
Mesoporous and hierarchical macroporous–mesoporous (MM) propyl sulfonic acid (PrSO3H) silicas were synthesized by hydrothermal saline‐promoted grafting of the pre‐formed architectures. The textural properties of the parent silicas were unperturbed by sulfonation, which resulted in similar sulfonic acid loadings and strengths for both pore networks. The turnover frequencies for catalytic esterification of model C3–C12 carboxylic acids with methanol decreased with alkyl chain length over both materials, however the introduction of 200 nm macropores into the SBA‐15 framework doubled the activity per acid site for the bulkiest lauric acid, which was attributed to enhanced mass transport and active site access, and a higher −PrSO3H surface density. Macropore incorporation also enhanced the esterification activity for the upgrading of a real bio‐oil derived from thermal fast pyrolysis of oak woodchips; the TOF for total organic acid removal increased three‐fold relative to the mesoporous sulfonic acid silica, which was also attributed to superior in‐pore mass transport and active site accessibility. The total acid number was reduced by 57 % over a 6 h reaction at 85 °C using the hierarchical PrSO3H/MM‐SBA‐15 catalyst. GC×GC–time‐of‐flight mass spectrometry (GC×GC–ToFMS) confirmed that catalytic upgrading removed almost all organic acids, and significantly lowered the concentration of reactive, phenolic, aldehyde, and ketone components, accompanied by the formation of carboxylic acids methyl esters and ethers.
Experimental Section
Full details of the catalyst synthesis, bulk and surface characterization (TEM, XRD, N2 porosimetry, DRIFTS, XPS, TGA, pyridine adsorption/DRIFTS, propylamine adsorption/TGA‐MS), and catalytic esterification and bio‐oil analysis protocols are provided in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the EPSRC for financial support (EP/K000 616/2, EP/G007594/4, EP/K036548/2 and EP/N009924/1) and the award of a Leadership Fellowship to AFL. KW thanks the Royal Society for an Industry Fellowship. The British Council is also acknowledged for funding through the Newton Institutional Links Scheme. Support from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 604307 is also greatly acknowledged. Underpinning data can be found at http://doi.org/10.17036/researchdata.aston.ac.uk.00000253 .
J. C. Manayil, A. Osatiashtiani, A. Mendoza, C. M. Parlett, M. A. Isaacs, L. J. Durndell, C. Michailof, E. Heracleous, A. Lappas, A. F. Lee, K. Wilson, ChemSusChem 2017, 10, 3506.
References
- 1.
- 1a. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Eds.: T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, P. M. Midgley), Cambridge University Press, Cambridge, 2013; [Google Scholar]
- 1b. Mullen C. A., Boateng A. A., Energy Fuels 2008, 22, 2104–2109; [Google Scholar]
- 1c. Chheda J. N., Huber G. W., Dumesic J. A., Angew. Chem. Int. Ed. 2007, 46, 7164–7183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7298–7318; [Google Scholar]
- 1d. Lee A. F., Bennett J. A., Manayil J. C., Wilson K., Chem. Soc. Rev. 2014, 43, 7887–7916. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Wilson K., Lee A. F., Philos. Trans. R. Soc. London A 2016, 374, 20150081; [Google Scholar]
- 2b. Lee A. F., Wilson K., Catal. Today 2015, 242, 3–18; [Google Scholar]
- 2c. Huber G. W., Corma A., Angew. Chem. Int. Ed. 2007, 46, 7184–7201; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7320–7338; [Google Scholar]
- 2d. Huber G. W., Iborra S., Corma A., Chem. Rev. 2006, 106, 4044–4098. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Stöcker M., Angew. Chem. Int. Ed. 2008, 47, 9200–9211; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9340–9351; [Google Scholar]
- 3b. Carpenter D., Westover T. L., Czernik S., Jablonski W., Green Chem. 2014, 16, 384–406. [Google Scholar]
- 4. Czernik S., Bridgwater A. V., Energy Fuels 2004, 18, 590–598. [Google Scholar]
- 5. Alcala A., Bridgwater A. V., Fuel 2014, 109, 417–426. [Google Scholar]
- 6.
- 6a. Zhang Q., Chang J., Wang T., Xu Y., Energy Convers. Manage. 2007, 48, 87–92; [Google Scholar]
- 6b. Mohan D., Pittman C. U., Steele P. H., Energy Fuels 2006, 20, 848–889. [Google Scholar]
- 7.
- 7a. Mortensen P. M., Grunwaldt J. D., Jensen P. A., Knudsen K. G., Jensen A. D., Appl. Catal. A 2011, 407, 1–19; [Google Scholar]
- 7b. Graça I., Lopes J. M., Cerqueira H. S., Ribeiro M. F., Ind. Eng. Chem. Res. 2013, 52, 275–287. [Google Scholar]
- 8.
- 8a. Zhang S., Yan Y., Li T., Ren Z., Bioresour. Technol. 2005, 96, 545–550; [DOI] [PubMed] [Google Scholar]
- 8b. Zacher A. H., Olarte M. V., Santosa D. M., Elliott D. C., Jones S. B., Green Chem. 2014, 16, 491–515. [Google Scholar]
- 9. Ciddor L., Bennett J. A., Hunns J. A., Wilson K., Lee A. F., J. Chem. Technol. Biotechnol. 2015, 90, 780–795. [Google Scholar]
- 10.
- 10a. Parlett C. M. A., Wilson K., Lee A. F., Chem. Soc. Rev. 2013, 42, 3876–3893; [DOI] [PubMed] [Google Scholar]
- 10b. Gärtner C. A., Serrano-Ruiz J. C., Braden D. J., Dumesic J. A., ChemSusChem 2009, 2, 1121–1124. [DOI] [PubMed] [Google Scholar]
- 11. Wilson K., Lee A. F., Catal. Sci. Technol. 2012, 2, 884–897. [Google Scholar]
- 12.
- 12a. Keller T. C., Rodrigues E. G., Pérez-Ramírez J., ChemSusChem 2014, 7, 1729–1738; [DOI] [PubMed] [Google Scholar]
- 12b. Rodrigues E. G., Keller T. C., Mitchell S., Perez-Ramirez J., Green Chem. 2014, 16, 4870–4874. [Google Scholar]
- 13. Mullen C. A., Boateng A. A., Goldberg N. M., Lima I. M., Laird D. A., Hicks K. B., Biomass Bioenergy 2010, 34, 67–74. [Google Scholar]
- 14. Dacquin J. P., Lee A. F., Pirez C., Wilson K., Chem. Commun. 2012, 48, 212–214. [DOI] [PubMed] [Google Scholar]
- 15. Pirez C., Wilson K., Lee A. F., Green Chem. 2014, 16, 197–202. [Google Scholar]
- 16.
- 16a. Corma A., Garcia H., Iborra S., Primo J., J. Catal. 1989, 120, 78–87; [Google Scholar]
- 16b. Bedard J., Chiang H., Bhan A., J. Catal. 2012, 290, 210–219; [Google Scholar]
- 16c. Milina M., Mitchell S., Pérez-Ramírez J., Catal. Today 2014, 235, 176–183; [Google Scholar]
- 16d. Peng J., Chen P., Lou H., Zheng X., Bioresour. Technol. 2009, 100, 3415–3418; [DOI] [PubMed] [Google Scholar]
- 16e. Chandra Shekara B. M., Ravindra Reddy C., Madhuranthakam C. R., Jai Prakash B. S., Bhat Y. S., Ind. Eng. Chem. Res. 2011, 50, 3829–3835; [Google Scholar]
- 16f. Nandiwale K. Y., Sonar S. K., Niphadkar P. S., Joshi P. N., Deshpande S. S., Patil V. S., Bokade V. V., Appl. Catal. A 2013, 460, 90–98. [Google Scholar]
- 17.
- 17a. Srilatha K., Issariyakul T., Lingaiah N., Sai Prasad P. S., Kozinski J., Dalai A. K., Energy Fuels 2010, 24, 4748–4755; [Google Scholar]
- 17b. Duan X., Liu Y., Zhao Q., Wang X., Li S., RSC Adv. 2013, 3, 13748–13755; [Google Scholar]
- 17c. Singh S., Patel A., J. Cleaner Prod. 2014, 72, 46–56. [Google Scholar]
- 18.
- 18a. Arata K., Green Chem. 2009, 11, 1719–1728; [Google Scholar]
- 18b. Reddy B. M., Patil M. K., Chem. Rev. 2009, 109, 2185–2208; [DOI] [PubMed] [Google Scholar]
- 18c. Chen X.-R., Ju Y.-H., Mou C.-Y., J. Phys. Chem. C 2007, 111, 18731–18737; [Google Scholar]
- 18d. Rattanaphra D., Harvey A. P., Thanapimmetha A., Srinophakun P., Renewable Energy 2011, 36, 2679–2686. [Google Scholar]
- 19.
- 19a. Liu F., Sun J., Zhu L., Meng X., Qi C., Xiao F.-S., J. Mater. Chem. 2012, 22, 5495–5502; [Google Scholar]
- 19b. Rocha R. P., Pereira M. F. R., Figueiredo J. L., Catal. Today 2013, 218–219, 51–56; [Google Scholar]
- 19c. Yu H., Jin Y., Li Z., Peng F., Wang H., J. Solid State Chem. 2008, 181, 432–438; [Google Scholar]
- 19d. Hara M., Yoshida T., Takagaki A., Takata T., Kondo J. N., Hayashi S., Domen K., Angew. Chem. Int. Ed. 2004, 43, 2955–2958; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3015–3018. [Google Scholar]
- 20.
- 20a. Mbaraka I. K., Shanks B. H., J. Catal. 2005, 229, 365–373; [Google Scholar]
- 20b. Melero J. A., Bautista L. F., Morales G., Iglesias J., Briones D., Energy Fuels 2009, 23, 539–547; [Google Scholar]
- 20c. Mbaraka I. K., Radu D. R., Lin V. S. Y., Shanks B. H., J. Catal. 2003, 219, 329–336; [Google Scholar]
- 20d. Woodford J. J., Dacquin J.-P., Wilson K., Lee A. F., Energy Environ. Sci. 2012, 5, 6145–6150; [Google Scholar]
- 20e. Martín A., Morales G., Martinez F., van Grieken R., Cao L., Kruk M., J. Mater. Chem. 2010, 20, 8026–8035; [Google Scholar]
- 20f. Vinu A., Gokulakrishnan N., Balasubramanian V. V., Alam S., Kapoor M. P., Ariga K., Mori T., Chem. Eur. J. 2008, 14, 11529–11538; [DOI] [PubMed] [Google Scholar]
- 20g. Pirez C., Caderon J.-M., Dacquin J.-P., Lee A. F., Wilson K., Acs Catal. 2012, 2, 1607–1614. [Google Scholar]
- 21. Alcala A., Bridgwater A. V., Fuel 2013, 109, 417–426. [Google Scholar]
- 22. Xiu S., Shahbazi A., Renewable Sustainable Energy Rev. 2012, 16, 4406–4414. [Google Scholar]
- 23. Dhainaut J., Dacquin J.-P., Lee A. F., Wilson K., Green Chem. 2010, 12, 296–303. [Google Scholar]
- 24.
- 24a. Pirez C., Lee A. F., Manayil J. C., Parlett C. M. A., Wilson K., Green Chem. 2014, 16, 4506–4509; [Google Scholar]
- 24b. Pirez C., Reche M. T., Lee A. F., Manayil J. C., dos-Santos V. C., Wilson K., Catal. Lett. 2015, 145, 1483–1490. [Google Scholar]
- 25.
- 25a. Miao S., Shanks B. H., J. Catal. 2011, 279, 136–143; [Google Scholar]
- 25b. Opanasenko M. V., Roth W. J., Cejka J., Catal. Sci. Technol. 2016, 6, 2467–2484. [Google Scholar]
- 26. Manayil J. C., Inocencio C. V. M., Lee A. F., Wilson K., Green Chem. 2016, 18, 1387–1394. [Google Scholar]
- 27. Lee A. F., Wilson K. in Handbook of Green Chemistry (Ed.: P. T. Anastas), Wiley-VCH, Weinheim, 2010. [Google Scholar]
- 28. Manayil J. C., dos Santos V. C., Jentoft F. C., Granollers Mesa M., Lee A. F., Wilson K., ChemCatChem 2017, 9, 2231–2238. [Google Scholar]
- 29.
- 29a. Yuan Z.-Y., Su B.-L., J. Mater. Chem. 2006, 16, 663–677; [Google Scholar]
- 29b. Parlett C. M. A., Isaacs M. A., Beaumont S. K., Bingham L. M., Hondow N. S., Wilson K., Lee A. F., Nat. Mater. 2016, 15, 178–182. [DOI] [PubMed] [Google Scholar]
- 30. Siril P. F., Shiju N. R., Brown D. R., Wilson K., Appl. Catal. A 2009, 364, 95–100. [Google Scholar]
- 31. Wang X., Cheng S., Chan J. C. C., J. Phys. Chem. C 2007, 111, 2156–2164. [Google Scholar]
- 32. Dacquin J.-P., Cross H. E., Brown D. R., Duren T., Williams J. J., Lee A. F., Wilson K., Green Chem. 2010, 12, 1383–1391. [Google Scholar]
- 33. Osatiashtiani A., Durndell L. J., Manayil J. C., Lee A. F., Wilson K., Green Chem. 2016, 18, 5529–5535. [Google Scholar]
- 34. Oasmaa A., van de Beld B., Saari P., Elliott D. C., Solantausta Y., Energy Fuels 2015, 29, 2471–2484. [Google Scholar]
- 35. Roby S. H., Dutta M., Zhu Y., Pathiparampil A., Energy Fuels 2015, 29, 858–862. [Google Scholar]
- 36. Li X., Gunawan R., Lievens C., Wang Y., Mourant D., Wang S., Wu H., Garcia-Perez M., Li C.-Z., Fuel 2011, 90, 2530–2537. [Google Scholar]
- 37. Hilten R., Weber J., Kastner J. R., Energy Fuels 2016, 30, 8357–8368. [Google Scholar]
- 38.
- 38a. Holm M. S., Pagan-Torres Y. J., Saravanamurugan S., Riisager A., Dumesic J. A., Taarning E., Green Chem. 2012, 14, 702–706; [Google Scholar]
- 38b. Hu X., Wu L., Wang Y., Mourant D., Lievens C., Gunawan R., Li C.-Z., Green Chem. 2012, 14, 3087–3098. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary