

Abstract
Chalcogen bonding is a little explored noncovalent interaction similar to halogen bonding. This manuscript describes the first application of selenium‐based chalcogen bond donors as Lewis acids in organic synthesis. To this end, the solvolysis of benzhydryl bromide served as a halide abstraction benchmark reaction. Chalcogen bond donors based on a bis(benzimidazolium) core provided rate accelerations relative to the background reactivity by a factor of 20–30. Several comparative experiments provide clear indications that the observed activation is due to chalcogen bonding. The performance of the chalcogen bond donors is superior to that of a related brominated halogen bond donor.
Keywords: chalcogen bonding, chalcogens, Lewis acids, noncovalent interactions, solvolysis
In recent years, the application of previously hardly explored interactions, such as anion–π1 and halogen bonding,2 in solution has received increased interest. Closely related to halogen bonding is chalcogen bonding (ChB), that is, the attractive interaction between an electrophilic chalcogen substituent Ch (S, Se, or Te) and a Lewis base LB (Figure 1).3 Such Lewis acids R/R′−Ch are typically, albeit somewhat confusingly, called “chalcogen bond donors” (despite their function as electron acceptors).
Figure 1.

Definition of chalcogen bonding. LB=Lewis base.
Several components likely contribute to the overall interaction energy. Similarly to halogen bonding, the electronic distribution of heavier chalcogen atoms is anisotropic, with reduced electron density in the elongation of the R−Ch axes. In suitably polarized compounds, a region of positive electrostatic potential (“σ‐hole”)4 is formed, which interacts favorably with a negatively polarized Lewis base. Furthermore, electron‐withdrawing groups R lower the energy of the σ*‐orbital of the R−Ch bond and increase its coefficient on the Ch substituent. Thus chalcogen bonding may also be described as an n→σ* charge transfer interaction5 between the chalcogen bond donor and the nonbonding lone pair of the Lewis base. Finally, dispersion contributions will also be relevant for heavier chalcogens.
Owing to its electronic origin, chalcogen bonding is highly directional,5, 6 as reasonably strong chalcogen bonding requires R−Ch⋅⋅⋅LB angles of approximately 180°. Even though it is typically weaker than halogen bonding,7 chalcogen bonding has two distinct advantages. First, the second substituent R′ on the chalcogen atom, which is orientated at 90° relative to the Ch⋅⋅⋅LB interaction, interacts more directly with the substrate than the backbone R substituent of halogen bond donors R−X. Second, if both substituents R and R′ are sufficiently electronegative, there are two perpendicular electrophilic axes on the chalcogen substituent.
In the solid state, chalcogen bonding has been applied in a few cases to construct supramolecular assemblies such as nanotubes,8a,8b nanosheets,8c wires,8d and macrocycles.8e In solution, fundamental studies and applications are arguably even more rare and focus mostly on anion recognition. Investigated systems include a mixed telluronium/boron Lewis acid,9a benzotelluradiazoles as monodentate receptors,9b and tellurophene derivatives as bidentate ones.9c Very recently, Beer et al. also reported the use of seleno‐ and tellurotriazol(ium) motifs in anion‐binding rotaxanes.10 Even though sulfur‐based chalcogen bond donors are expected to form weaker interactions than selenium‐ or tellurium‐based ones, an appropriately designed bidentate dithienothiophene (DTT) derivative has been used by Matile and co‐workers for anion transport.11
As Lewis acids based on “unconventional” weak interactions such as anion–π12 and halogen bonding13 have by now been introduced in organic synthesis and organocatalysis, a similar approach should be feasible for chalcogen bonding. The few currently known examples related to this concept focus almost exclusively on intramolecular binding as a tool to rigidify structural motifs.14
In contrast, the first application of chalcogen‐bonding‐based organocatalysis by intermolecular coordination and activation of a substrate has recently been published by Matile and co‐workers.15 In this case, DTT derivatives catalyzed the reduction of quinolone derivatives.
Herein, we present the first application of selenium‐based chalcogen bond donors as noncovalent activators, utilizing a C−X activation (“anion binding”) benchmark reaction.16 Such proof‐of‐principle studies pose two main challenges: 1) As chalcogen bonds are rather weak, other interactions will likely also contribute, and it is difficult to ascribe the action of an activator to chalcogen bonding as the main cause. 2) It is often difficult to rule out the action of impurities, most importantly hidden traces of acid.
As a consequence, a relatively simple test reaction, the solvolysis of benzhydryl bromide (1) in wet acetonitrile, was chosen (Scheme 1). This transformation, which we have already applied in fundamental studies on halogen bonding, has been shown to be immune to hidden acid catalysis.16 In addition, it has virtually no background reaction at room temperature and is easy to follow by 1H NMR spectroscopy.
Scheme 1.
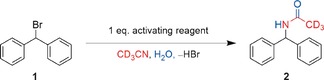
Anion‐binding benchmark reaction.
For the design of strong chalcogen‐bonding‐based activators, we decided to rely on cationic backbones R in order to achieve a strong polarization of (at least one) R−Ch bond.17 More precisely, the bis(benzimidazolium)‐based backbone structure of 4 (Scheme 2) was selected as the corresponding halogen bond donor had shown a relatively strong Lewis acidity.18 The trifluoromethyl group in these compounds prevents rotation of the benzimidazolium groups and enables characterization by 19F NMR spectroscopy. As the chalcogen, selenium was selected as it should provide a stronger Lewis acidity than sulfur while being less prone to decomposition than tellurium. Ideally, the substrates should coordinate to chalcogen bond donors 4 in a bidentate fashion, as predicted by gas‐phase calculations (see the Supporting Information). As selenium is smaller than iodine, it remained uncertain whether this binding motif would indeed be realized in solution. Finally, simple alkyl groups were introduced as second substituents on the chalcogen, namely octyl or isopropyl moieties for good solubility, and methyl groups for crystallization studies.
Scheme 2.
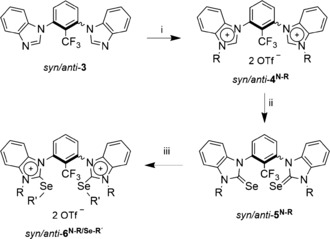
Synthesis of chalcogen bond donors. Reagents and conditions: i) ROTf, CH2Cl2 (R=Me, Oct); ii) Se, Cs2CO3, MeOH; iii) ROTf, CH2Cl2 (R=Me, Oct, iPr). Selected yields: syn/anti‐4N‐Oct: 84 %; syn‐5N‐Me: 80 %; anti‐5N‐Me: 87 %; syn‐5N‐Oct: 33 %; anti‐5N‐Oct: 63 %; syn‐6N‐Oct/Se‐iPr: 95 %; anti‐6N‐Oct/Se‐iPr: 90 %. See also the Supporting Information.
The synthesis of the chalcogen bond donors is depicted in Scheme 2. Starting from an (inseparable) syn/anti mixture of 4N‐Me or 4N‐Oct, selenation was achieved with cesium carbonate and elemental selenium.19 In both cases, the resulting selenated isomers could be separated by column chromatography (4N‐Me: 38 % syn, 62 % anti; 4N‐Oct: 26 % syn, 74 % anti). Subsequent alkylation with methyl, octyl, or isopropyl triflate proceeded with good to excellent yields and provided the desired cationic chalcogen bond donors (see the Supporting Information). All chalcogen bond donors are stable under air and moisture and show no signs of decomposition when kept in acetonitrile solution even after three months (according to 1H and 19F NMR analysis).
X‐ray structural analysis of compound anti‐6N‐Me/Se‐Me revealed two dications and four triflates in the unit cell (Figure 2). All four selenium centers form chalcogen bonds (in elongation of the Cbenzimidazolium−Se bonds) to oxygen atoms of triflate. The corresponding Se⋅⋅⋅O distances range from 2.94 to 3.24 Å, which are all markedly below the sum of the van der Waals radii of both elements (3.42 Å).20 The C−Se⋅⋅⋅O angles (163–173°) are in general agreement with the expected linearity, with one slight exception (151°), which is likely due to additional packing effects. Overall, the crystal structure clearly confirms the expected σ*‐acidity of the carbon–selenium bonds.
Figure 2.
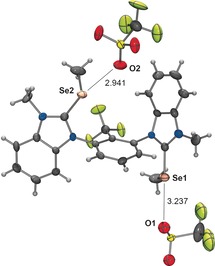
X‐ray structural analysis of anti‐6N‐Me/Se‐Me. One ion pair of two in the unit cell is shown. Ellipsoids set at 50 % probability. Selected bond lengths [Å] and angles [°]: C–Se2 1.891, C–Se1 1.900; C‐Se2‐O2 163, C‐Se1‐O1 173.
With these promising findings in hand, several possible activators were tested in the benchmark reaction mentioned above (Scheme 1). All reactions were reproduced at least twice with only minor variations. In all cases, a clean transformation into amide 2 was observed. Even after 140 h, the background reactivity amounted to only 10 % yield of 2 (Table 1, entry 1).
Table 1.
Effect of various chalcogen bond donors and reference compounds on the anion‐binding benchmark reaction of Scheme 1.
| Entry | Activating reagent | Equivalents[a] | Yield [%][b] |
|---|---|---|---|
| 1 | – | – | 10 |
| 2 | syn/anti‐4N‐Oct | 1.0 | 11 |
| 3 | syn‐6N‐Oct/Se‐iPr | 1.0 | 64 |
| 4 | anti‐6N‐Oct/Se‐iPr | 1.0 | 45 |
| 5 | syn‐5N‐Oct | 1.0 | <5[c] |
| 6 | iPrBr | 1.0 | <5 |
| 7 | 7H | 2.0 | 16 |
| 8 | 8 | 2.0 | <5[c] |
| 9 | 9Se‐Oct | 2.0 | 34 |
| 10 | 9Se‐iPr | 2.0 | 45 |
| 11 | 7I | 2.0 | 48[d] |
| 12 | syn‐10I | 1.0 | >95[e] |
| 13 | syn‐10Br | 1.0 | 35 |
[a] Equivalents of activating reagent (relative to 1). [b] Yield of 2 after 140 h at room temperature determined by 1H NMR analysis (see the Supporting Information). [c] Low solubility in acetonitrile. [d] Yield after 96 h. [e] Quantitative yield of 2 after 24 h.
Next, several potentially bidentate activating reagents were employed. The (thus far inseparable) syn/anti mixture of the non‐selenated reference compound 4N‐Oct resulted in only 11 % product formation (entry 2) and thus provided no noticeable activation of 1.
In contrast, all selenated (cationic) derivatives induced a marked increase in the yield of 2. Compound syn‐6N‐Oct/Se‐Me, for instance, led to a yield of approximately 60 % after 96 h. However, NMR spectra of the reaction showed clear signs of activator decomposition by dealkylation of the selenium center, as MeBr formation was observed. Titration experiments with bromide confirmed that this chalcogen bond donor is not stable under the reaction conditions. The same is true, albeit to a somewhat lesser extent, for the octylated variant syn‐6N‐Me/Se‐Oct, and thus both were not considered further. As dealkylation will likely occur through an SN2 mechanism, we reasoned that a secondary alkyl substituent on selenium should provide more stability. Indeed, activator candidate syn‐6N‐Oct/Se‐iPr showed only minor signs of decomposition (4 % after 140 h according to 19F NMR analysis) and was thus considered to be suitable for further activation experiments. Amide 2 was formed in 64 % yield (entry 3; for a stack plot, see the Supporting Information). The NMR spectra indicate that the slight decomposition of syn‐6N‐Oct/Se‐iPr over time is again due to dealkylation with formation of isopropyl bromide. To rule out any activity of syn‐5N‐Oct and iPrBr, which would have to be catalytic, both were also tested and provided less than 5 % of product 2 (Table 1, entries 5 and 6).
All findings presented thus far provide strong indications that the activity of syn‐6N‐Oct/Se‐iPr is based on chalcogen bonding. Acid catalysis can be ruled out in this reaction, and the otherwise identical non‐selenated compound (which should form anion–π interactions that are at least as strong) is completely inactive. Thus the selanylalkyl group must constitute the active site, and X‐ray structural analysis of anti‐6N‐Me/Se‐Me as well as DFT calculations (see the Supporting Information) clearly show that chalcogen bonding is its binding mode.21
The corresponding anti isomer of 6N‐Oct/Se‐iPr was somewhat less active (45 % yield of 2; entry 4), but the difference to the syn isomer was not very significant. This seems to indicate that syn‐6N‐Oct/Se‐iPr does not bind to bromide in a clean bidentate fashion as a more pronounced effect might be expected in this case.
Subsequently, several simple monodentate benzimidazolium derivatives (Figure 3) were used as potential activating reagents to further elucidate any effect of the backbone structure of anti‐6N‐Oct/Se‐iPr on the activity of the individual selenobenzimidazolium moieties. Two equivalents of these species were used in the test reactions to provide the same number of active centers as with the bifunctional chalcogen bond donors described before.
Figure 3.
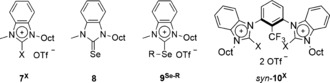
Further reference compounds (X=H, Br, I; R=Oct, iPr).
Similarly to the previous findings, the selenated derivatives 9Se‐Oct (34 % yield; entry 9) and 9Se‐iPr (45 % yield; entry 10) were markedly more active than the non‐selenated reference compound 7H (16 %; entry 7) and the non‐alkylated precursor 8 (<5 %; entry 8). Thus, as the activity of two equivalents of 9Se‐iPr is identical to that of one equivalent of anti‐6N‐Oct/Se‐iPr, there seems to be no additional effect of the backbone of the latter on its chalcogen bonding subunits.
Finally, a direct comparison of the activation by chalcogen bonding with the already established one by halogen bonding was aspired. To this end, closely related halogenated analogues were also used, and in the case of 7I (48 % yield after 96 h; entry 11), the performance was somewhat superior to the one of 9Se‐iPr. The difference was more pronounced for the bidentate variants as the use of syn‐10I led to quantitative product formation after 24 h.22 This difference might at least partially be due to the less strained bidentate binding of bromide by the iodinated Lewis acid. However, an arguably fairer comparison is the one to the halogen of the same period, and syn‐10Br is indeed even slightly less active (35 %, entry 13) than syn‐6N‐Oct/Se‐iPr.
Yield/time profiles for selected reactions are presented in Figure 4. Based on the initial slopes of product formation, the reaction rate can be estimated to be about one order of magnitude higher in the presence of syn‐10Br compared to the background reaction (k rel=9). The rate acceleration by the chalcogen bond donors, in turn, is about twice (anti‐6N‐Oct/Se‐iPr, k rel=23) or three times (syn‐6N‐Oct/Se‐iPr, k rel=34) that of syn‐10Br.
Figure 4.
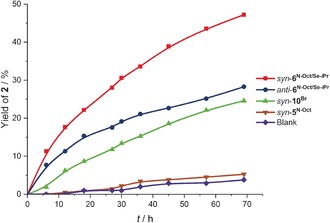
Yield/time profiles of selected reactions.
In conclusion, the first intermolecular use of selenium‐based chalcogen bond donors as Lewis acids in organic synthesis has been presented. Using a suitable benchmark reaction for halide binding reactivity and several comparative experiments, strong indications for chalcogen bonding as the actual mode of action were obtained, most notably the fact that the corresponding non‐selenated reference compound was inactive. Even though the observed effect is less strong than the activity of bidentate iodine‐based halogen bond donors, further detailed investigations into the use of chalcogen bonding in solution will likely provide the basis for more sophisticated mixed catalyst systems in which chalcogen bonding could play an important role.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (638337).
P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz, S. M. Huber, Angew. Chem. Int. Ed. 2017, 56, 12009.
References
- 1.See, for example:
- 1a. Zhao Y., Cotelle Y., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 4270–4277; [DOI] [PubMed] [Google Scholar]
- 1b. Giese M., Albrecht M., Rissanen K., Chem. Commun. 2016, 52, 1778–1795. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478–2601; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Erdélyi M., Chem. Soc. Rev. 2012, 41, 3547–3557. [DOI] [PubMed] [Google Scholar]
- 3.See, for example:
- 3a. Bleiholder C., Werz D. B., Köppel H., Gleiter R., J. Am. Chem. Soc. 2006, 128, 2666–2674; [DOI] [PubMed] [Google Scholar]
- 3b. Nakanishi W. in Handbook of Chalcogen Chemistry: New Perspective in Sulfur, Selenium and Tellurium, The Royal Society of Chemistry, 2007, pp. 644–668; [Google Scholar]
- 3c. Cozzolino A. F., Elder P. J. W., Vargas-Baca I., Coord. Chem. Rev. 2011, 255, 1426–1438. [Google Scholar]
- 4. Murray J. S., Lane P., Clark T., Politzer P., J. Mol. Model. 2007, 13, 1033–1038. [DOI] [PubMed] [Google Scholar]
- 5. Rosenfield R. E., Parthasarathy R., Dunitz J., J. Am. Chem. Soc. 1977, 99, 4860–4862. [Google Scholar]
- 6. Guru Row T. N., Parthasarathy R., J. Am. Chem. Soc. 1981, 103, 477–479. [Google Scholar]
- 7.
- 7a. Iwaoka M., Tomoda S., J. Am. Chem. Soc. 1996, 118, 8077–8084; [Google Scholar]
- 7b. Iwaoka M., Komatsu H., Katsuda T., Tomoda S., J. Am. Chem. Soc. 2002, 124, 1902–1909. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Werz D. B., Gleiter R., Rominger F., J. Am. Chem. Soc. 2002, 124, 10638–10639; [DOI] [PubMed] [Google Scholar]
- 8b. Gleiter R., Werz D. B., Rausch B. J., Chem. Eur. J. 2003, 9, 2676–2683; [DOI] [PubMed] [Google Scholar]
- 8c. Yi Y., Fa S., Cao W., Zeng L., Wang M., Xu H., Zhang X., Chem. Commun. 2012, 48, 7495–7497; [DOI] [PubMed] [Google Scholar]
- 8d. Kremer A., Fermi A., Biot N., Wouters J., Bonifazi D., Chem. Eur. J. 2016, 22, 5665–5675; [DOI] [PubMed] [Google Scholar]
- 8e. Ho P. C., Szydlowski P., Sinclair J., Elder P. J. W., Kübel J., Gendy C., Lee L. M., Jenkins H., Britten J. F., Morim D. R., Vargas-Baca I., Nat. Commun. 2016, 7, 11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Zhao H., Gabbaï F. P., Nat. Chem. 2010, 2, 984–990; [DOI] [PubMed] [Google Scholar]
- 9b. Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S., Taylor M. S., J. Am. Chem. Soc. 2015, 137, 4126–4133; [DOI] [PubMed] [Google Scholar]
- 9c. Garrett G. E., Carrera E. I., Seferos D. S., Taylor M. S., Chem. Commun. 2016, 52, 9881–9884. [DOI] [PubMed] [Google Scholar]
- 10. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Felix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122–3133. [DOI] [PubMed] [Google Scholar]
- 11. Benz S., Macchione M., Verolet Q., Mareda J., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 9093–9096. [DOI] [PubMed] [Google Scholar]
- 12. Liu L., Cotelle Y., Klehr J., Sakai N., Ward T. R., Matile S., Chem. Sci. 2017, 8, 3770–3774, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bulfield D., Huber S. M., Chem. Eur. J. 2016, 22, 14434–14450. [DOI] [PubMed] [Google Scholar]
- 14. Mukherjee A. J., Zade S. S., Singh H. B., Sunoj R. B., Chem. Rev. 2010, 110, 4357–4416. [DOI] [PubMed] [Google Scholar]
- 15. Benz S., López-Andarias J., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 812–815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 830–833. [Google Scholar]
- 16. Walter S. M., Kniep F., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2011, 50, 7187–7191; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7325–7329. [Google Scholar]
- 17.For earlier examples of cationic chalcogen bond donors, see:
- 17a. Weiss R., Schlierf C., Schloter K., J. Am. Chem. Soc. 1976, 98, 4668; [Google Scholar]
- 17b. Weiss R., Marolt P., Synthesis 1980, 227; [Google Scholar]
- 17c. Laitinen R., Steudel R., Weiss R., J. Chem. Soc. Dalton Trans. 1986, 1095. [Google Scholar]
- 18. Jungbauer S. H., Huber S. M., J. Am. Chem. Soc. 2015, 137, 12110–12120. [DOI] [PubMed] [Google Scholar]
- 19. Bhabak K. B., Satheeshkumar K., Jayavelu S., Mugesh G., Org. Biomol. Chem. 2011, 9, 7343. [DOI] [PubMed] [Google Scholar]
- 20. Bondi A., J. Phys. COrg. Biomol. Chem. 2011, 9, 7343hem. 1964, 68, 441. [Google Scholar]
- 21.In further experiments to rule out active impurities, different batches of elemental selenium were used and different materials were employed for column chromatography, with virtually identical results in all cases.
- 22.Titration experiments with NOct4Br in acetonitrile also showed that syn-6N-Oct/Se-iPr binds more weakly to bromide (K=340 m −1) than syn-10I (K=3.5×106 m −1; Ref. [18]); see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary