

Abstract
Aims
Amyloid beta (Aβ) accumulation in the walls of leptomeningeal arteries as cerebral amyloid angiopathy (CAA) is a major feature of Alzheimer's disease. In this study, we used global quantitative proteomic analysis to examine the hypothesis that the leptomeningeal arteries derived from patients with CAA have a distinct endophenotypic profile compared to those from young and elderly controls.
Methods
Freshly dissected leptomeningeal arteries from the Newcastle Brain Tissue Resource and Edinburgh Sudden Death Brain Bank from seven elderly (82.9 ± 7.5 years) females with severe capillary and arterial CAA, as well as seven elderly (88.3 ± 8.6 years) and five young (45.4 ± 3.9 years) females without CAA were used in this study. Arteries from four patients with CAA, two young and two elderly controls were individually analysed using quantitative proteomics. Key proteomic findings were then validated using immunohistochemistry.
Results
Bioinformatics interpretation of the results showed a significant enrichment of the immune response/classical complement and extracellular matrix remodelling pathways (P < 0.05) in arteries affected by CAA vs. those from young and elderly controls. Clusterin (apolipoprotein J) and tissue inhibitor of metalloproteinases‐3 (TIMP3), validated using immunohistochemistry, were shown to co‐localize with Aβ and to be up‐regulated in leptomeningeal arteries from CAA patients compared to young and elderly controls.
Conclusions
Global proteomic profiling of brain leptomeningeal arteries revealed that clusterin and TIMP3 increase in leptomeningeal arteries affected by CAA. We propose that clusterin and TIMP3 could facilitate perivascular clearance and may serve as novel candidate therapeutic targets for CAA.
Keywords: clusterin, complement pathway, extracellular matrix remodelling, leptomeningeal arteries, proteomics, TIMP3
Introduction
The deposition of amyloid‐β (Aβ) peptides in the walls of cerebral arteries as cerebral amyloid angiopathy (CAA) is a major feature of Alzheimer's disease and may contribute to cognitive decline 1, 2. CAA predominantly affects the leptomeningeal and cortical arteries especially in the occipital lobe, while capillaries are less frequently and veins rarely involved 3, 4, 5. In the majority of cases there is no overproduction of Aβ in the vessel wall, suggesting that the deposition of Aβ in the walls of cerebral arteries is a result of a failure of elimination of neuronally derived Aβ 6. Increasing age and possession of at least one apolipoprotein ε4 (APOE4) allele are risk factors for CAA and both have been suggested to impair cerebral Aβ clearance systems, thereby reducing Aβ elimination from the brain 7, 8, 9, 10. We have demonstrated that Aβ and other solutes are eliminated along the basement membranes of capillaries and arteries, effectively the lymphatic drainage of the brain 11. Experimental work involving intraparenchymal injections of tracers demonstrated that the biochemical structure and morphology of the basement membranes of capillaries and arteries change with age and with possession of APOE4 genotype, resulting in failure of efficient clearance of Aβ 12, 13, 14. The exact targets for the facilitation of perivascular clearance of Aβ are not clear.
Proteomics allows the in‐depth and global assessment of gene products at the protein level as they occur in a variety of biological specimens, including cell lines, tissue, blood and proximal fluids. The advanced use of liquid chromatography combined with mass spectrometry permits the identification of thousands of proteins with ultra‐high precision and sensitivity, not available by any other analytical approach. Using stable isotope isobaric reagents allow such proteomes to be profiled in parallel across multiple biological or clinical states under identical analytical conditions, a feature referred to as the multiplex advantage 15, 16, 17, 18, 19, 20, 21, 22, 23. For example, such a strategy allows the comparison of a given in vitro or in vivo model under a given homeostatic state (that is physiological condition) relative to a perturbation state (that is pathological condition or exposure to a stimulus) under exactly the same experimental conditions.
This study employed isobaric quantitative proteomic analysis of fresh frozen human leptomeningeal arteries from young and elderly subjects and patients with CAA to test the hypothesis that leptomeningeal arteries derived from patients with CAA have a unique endophenotypic profile compared to those from young and elderly controls.
Materials and methods
Isolation of human leptomeningeal arteries
Human fresh frozen post mortem leptomeningeal arteries from the Newcastle Brain Tissue Resource and MRC Sudden Death Brain & Tissue Bank (Edinburgh) were used for this study. CAA cases were diagnosed post mortem by JA, according to published criteria including the neuritic Braak stages 24, Thal amyloid phases 25, CERAD scores 26, NIA‐AA scores 27 and McKeith criteria 28 and showed varying degrees of Alzheimer's disease pathology. For CAA we used a recently developed staging system, which assesses meningeal and parenchymal CAA separately and also scores capillary CAA 1, 2. All CAA cases had severe CAA as they showed widespread circumferential Aβ in meningeal and cortical arterial vessels as well as Aβ depositions in capillary walls. None of the cases was diagnosed with CAA during their lifetime. The cases from the MRC Sudden Death Brain & Tissue Bank (Edinburgh) had no neurological disease during life and no significant neuropathological changes post mortem. We excluded cases with arteriolosclerosis/lipohyalinosis from this cohort. Samples were collected and prepared in accordance with the National Research Ethics Service‐approved protocols. Leptomeningeal arteries in the occipital regions were removed from the frozen coronal slices from brains of young females (45.4 ± 3.9 years; n = 5), elderly females without CAA (88.3 ± 8.6 years; n = 7) and females with severe CAA (82.6 ± 7.5 years; n = 7) (Table 1). Only female subjects were included in the present study as it has been shown that sex‐dependent differences exist in CAA 29, 30, 31. The frozen coronal slices were placed at −20°C overnight to acclimatize from the −70°C storage prior to dissection in a cold cabinet at −12°C. Arteries were identified based on their morphology of a vessel and they were distinguished from veins by the thicker wall and leptomeningeal sheet as they penetrate the cortex. The abundant presence of vascular smooth muscle actin confirmed they were arteries. Selected vessels were eased with a micro‐scalpel from the meningeal surface of the gyri and sulci, removed and placed in pre‐cooled tubes to avoid thawing. These specimens were then snap frozen at −80°C.
Table 1.
Details of post mortem samples
| Sample # | Study group | Age (years) | Used in proteomic analysis | Braak stage | Thal amyloid phase | Post mortem delay (h) | Cause of death | Duration of dementia (years) | CAA inflammation/vasculitis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Young control | 51 | Yes | 0 | Not applicable | 81 | Metastatic carcinoma | 0 | Not applicable |
| 2 | Young control | 46 | Yes | 0 | Not applicable | 49 | Myocardial infarction; coronary artery thrombosis; coronary artery atherosclerosis | 0 | Not applicable |
| 3 | Young control | 45 | No | 0 | Not applicable | 93 | Coronary artery atherosclerosis | 0 | Not applicable |
| 4 | Young control | 40 | No | 0 | Not applicable | 77 | Bronchial asthma | 0 | Not applicable |
| 5 | Young control | 45 | No | 0 | Not applicable | 40 | Suspension by ligature | 0 | Not applicable |
| 6 | Elderly control | 79 | Yes | IV | 3 | 9 | Old age, dementia with Parkinson's disease | 9 | Mild, some vessels with perivascular infiltrate |
| 7 | Elderly control | 88 | Yes | III | 0 | 22 | Aspiration pneumonia; total anterior circulation stroke | Not available | Not remarkable |
| 8 | Elderly control | 74 | No | III | 1 | 53 | Heart failure and lung cancer | Not available | Not remarkable |
| 9 | Elderly control | 94 | No | II | 1 | 15 | Left ventricle failure; ischaemic heart disease | Not available | Not remarkable |
| 10 | Elderly control | 95 | No | III | 0 | 66 | Ischaemic bowel disease (inoperable) | Not available | Not remarkable |
| 11 | Elderly control | 96 | No | II | 3 | 114 | Stroke and left ventricular failure | 2 (mild) | Not remarkable |
| 12 | Elderly control | 92 | No | VI | 5 | 74 | Pneumonia | >2 | Not remarkable |
| 13 | CAA case | 93 | Yes | VI | 5 | 53 | Stroke, general deterioration | 13 | Mild, some vessels with perivascular infiltrate |
| 14 | CAA case | 73 | Yes | IV | 5 | 47 | Frontal lobe dementia | 1.3 | Not remarkable |
| 15 | CAA case | 76 | Yes | VI | 3 | 37 | not applicable | 8 | Not remarkable |
| 16 | CAA case | 87 | Yes | VI | 5 | 54 | Aspiration pneumonia secondary to stroke | 8 | Not remarkable |
| 17 | CAA case | 86 | No | VI | 5 | 47 | not applicable | 6 | Not remarkable |
| 18 | CAA case | 77 | No | VI | 2 | 63 | Aspiration pneumonia | 14 | Not remarkable |
| 19 | CAA case | 88 | No | VI | 5 | 84 | Bronchopneumonia | 15 | Not remarkable |
CAA, cerebral amyloid angiopathy.
Quantitative proteomic analysis on human leptomeningeal arteries
For the proteomic analysis, samples from two young and two elderly subjects and four patients with CAA were randomly selected from the cohort (Table 1). The justification for this number of CAA cases was to compensate for their innate tissue heterogeneity and to ensure a statistical power of over 0.7, factoring in a representative 30% measurement error and a fold change >1.5 between replicate observations, as reported in a recent simulation study 32. Samples were dissolved in dissolution buffer (0.5 M triethylammonium bicarbonate/0.05% sodium dodecyl sulphate), homogenized using the FastPrep system (Savant Bio, Cedex, Fr) and then subjected to pulsed probe sonication (Misonix, Farmingdale, NY, USA). Lysates were centrifuged (16 000 g, 10 min, 4°C) and supernatants were measured for protein content using the Direct Detect™ Spectroscopy system (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions. From each lysate volume (adjusted to the highest volume of 40 μl) containing 100 μg final protein content was subjected to reduction, alkylation, trypsin proteolysis and eight‐plex isobaric tag for relative and absolute quantitation (iTRAQ AbSciex, San Hose, CA, USA) labelling per supplier's specifications (ABSciex, San Hose, CA, USA). Labelled peptides were pooled and fractionated with high‐pH reversed‐phase (RP) chromatography using the Waters, XBridge C8 column (150 × 3 mm, 3.5 μm particle) with the Shimadzu LC‐20AD HPLC (Shimadzu, Kyoto, Japan). Each resulting fraction was LC‐MS analysed with low‐pH RP capillary chromatography (PepMap C18, 50 μm ID × 50 cm L, 100 Å pore, 3.5 μm particle) and nanospray ionization FT‐MS (Ultimate 3000 UHPLC – LTQ‐Velos Pro Orbitrap Elite, Thermo Scientific, Bremen, DE) as reported previously 19, 20, 23 (Figure 1 a).
Figure 1.
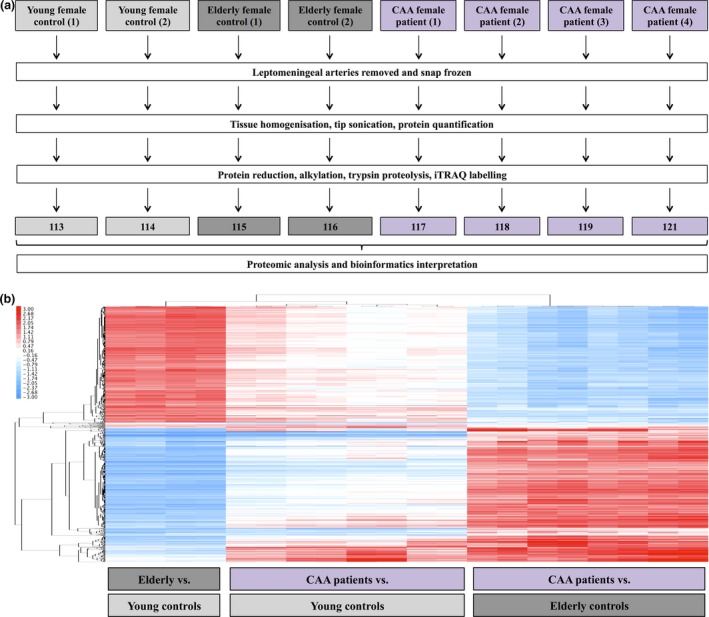
(a) Experimental pipeline of proteomics experiment. (b) Heatmap of differentially expressed proteins in leptomeningeal arteries of elderly controls compared to young controls, cerebral amyloid angiopathy (CAA) patients compared to young controls and CAA patients compared to elderly controls.
Unprocessed raw files were submitted to Proteome Discoverer 1.4 for target decoy searching with SequestHT for tryptic peptides as reported by the authors 19, 20, 23. Quantification ratios were normalized on the median value and log2 transformed. A protein was considered modulated in leptomeningeal arteries from elderly subjects vs. young controls or those affected by CAA type 1 relative to these from young and elderly controls when its log2 ratio was above or below ±1 SD across all analysed samples per category as reported previously 23.
Hierarchical clustering analysis visualized in heatmap format was generated using Gene Cluster (version 3.0) and Java Treeview (version 1.1.6r4). MetaCore (GeneGo, St. Joseph, MI, USA) and DAVID (http://david.abcc.ncifcrf.gov) were applied to identify prebuilt processed networks and gene ontology terms over‐represented in the modulated proteome. False discovery rate (FDR) and Fisher's exact corrected P‐values ≤0.05 were considered significant.
Immunohistochemistry
The immunochemistry validation of key proteomic findings was performed in all 19 subjects (young female controls: n = 5, elderly female controls: n = 7, females with CAA type 1: n = 7). Three sections of occipital cortex from each of the cases were immunostained. After dewaxing in xylene and rehydration through graded alcohols, antigen retrieval was performed by immersing slides in citrate buffer, microwaving on medium power for 25 min and subsequently cooling. This was followed by incubation in pepsin for 5 min (1 mg/ml 0.2 M HCl). The tissue was blocked in 3% H2O2 and 15% goat serum. Occipital cortex from each of the cases was incubated in clusterin (Abcam: Cambridge, UK, ab42673, rabbit polyclonal, dilution 1:500), or tissue inhibitor of metalloproteinases 3 (TIMP3) (Abcam, Ab93637, rabbit polyclonal, dilution 1:100) overnight at 4°C followed by biotinylated goat anti‐rabbit antibodies (Vector BA1000 dilution 1:200) and ABC peroxidase enzyme complex (Vector PK4000, dilution 1:500). Reaction was detected using diaminobenzidine with glucose oxidase enhancement. Images were captured an Olympus: Southend‐on‐Sea, Essex, UK, BX51 microscope fitted with Olympus CC‐12 colour microscope camera.
Double immunofluorescence was performed for Aβ and TIMP3. Prior to the antigen retrieval previously described, pre‐treatment was required which consisted of 5 min in formic acid at 37°C. Tissue was blocked in 15% goat serum followed by incubation in primary antibodies overnight at 4°C. Aβ was detected using mouse monoclonal anti‐Aβ IgG2b Clone 4G8, antibody (BioLegend: London, UK, 800701; dilution 1:100). The secondary antibody for Aβ was goat anti‐mouse IgG2b, AlexaFluor 647 (A‐21242), and for TIMP 3 and clusterin was goat anti‐rabbit IgG AlexaFluor 594 (A‐27096). These were obtained from Thermo Fisher Scientific and dilution 1:200. Images were captured and examined with a Leica SP8 confocal microscope. The specificity of the immunohistochemistry staining was confirmed by omitting the primary antibody.
Results
Quantitative proteomic analysis
The proteomic analysis resulted in the profiling of 5957 proteins (peptide FDR confidence ≥ 99%) (Table S1). A total of 1364 proteins were differentially expressed in arteries from elderly relative to young subjects (Table S2), 280 in arteries from CAA cases relative to young controls (Table S3) and another 983 in arteries from CAA cases relative to elderly controls (Table S4). The hierarchical clustering analysis of differentially expressed proteins between groups revealed that leptomeningeal arteries derived from CAA patients compared to those from young and elderly controls had a distinct proteomic profile from arteries derived from elderly compared to young subjects (Figure 1 b).
In silico bioinformatics analysis showed that the immune response/classical complement pathway (P = 5.0E‐11; 5.007E‐2; 1.168E‐10 in elderly vs. young controls; CAA vs. young controls; CAA vs. elderly controls respectively) (Figure 2) and extracellular matrix remodelling (P = 3.3E‐8; 6.349E‐6; 2.317E‐8 in elderly vs. young controls; CAA vs. young controls; CAA vs. elderly controls respectively) (Figure 3) were significantly over‐represented processes. For both pathways, the expression levels of most proteins were found to decrease in arteries from elderly vs. young controls, whereas they increased in arteries from CAA patients compared to young and elderly controls.
Figure 2.
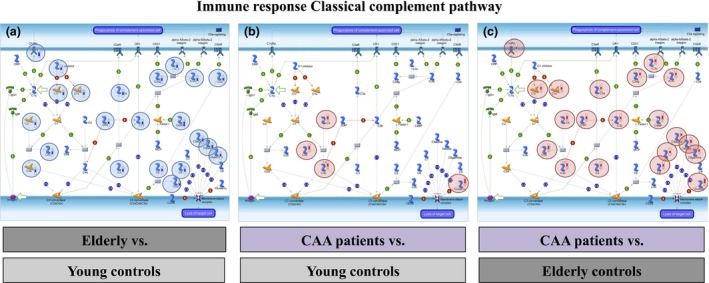
The immune response/classical complement pathway was significantly enriched in the differentially expressed proteome of leptomeningeal arteries from elderly vs. young controls (P = 5.0E‐11) (a), CAA patients compared to young controls (P = 5.007E‐2) (b) and cerebral amyloid angiopathy patients compared to elderly controls (P = 1.168E‐10) (c).
Figure 3.
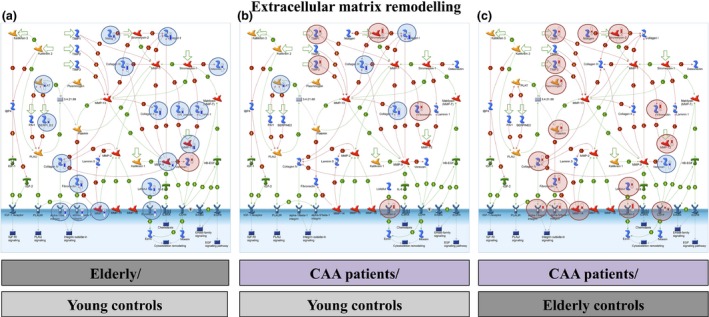
The extracellular matrix remodelling pathway was significantly enriched in the differentially expressed proteome of leptomeningeal arteries from elderly compared to young controls (P = 3.3E‐8) (a), cerebral amyloid angiopathy (CAA) patients compared to young controls (P = 6.349E‐6) (b) and CAA patients compared to elderly controls (P = 2.317E‐8) (c).
The expression of clusterin (apolipoprotein J) and TIMP3 from the immune response/classical complement and the extracellular matrix remodelling pathways, respectively, were up‐regulated in arteries from patients with CAA compared to both young and elderly controls [clusterin: iTRAQ mean log2 ratio (SD) = 2.30 (0.45) and 2.87 (0.44) in CAA vs. young and CAA vs. elderly controls respectively] [TIMP3: iTRAQ mean log2 ratio (SD) = 1.63 (0.89) and 2.48 (0.90) in CAA vs. young and CAA vs. elderly controls respectively].
Immunohistochemistry
Clusterin was found to co‐localize with Aβ in the occipital cortex of CAA cases, but not in the young or elderly controls (Figure 4). The pattern of expression for the immunocytochemistry of TIMP3 was weak in arteries from young controls, increased in elderly controls and was strong in CAA patients (Figure 5). TIMP3 and clusterin were found to co‐localize with Aβ in the leptomeningeal vessels of the occipital cortex from CAA cases (Figure 6).
Figure 4.
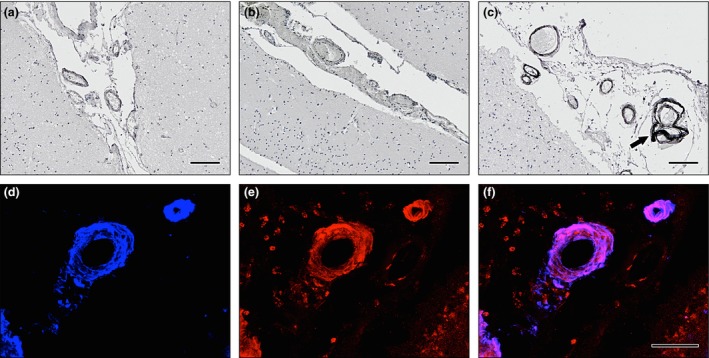
Immunohistochemistry of clusterin. DAB with haematoxylin counterstain in (a) young and (b) elderly controls and (c) cerebral amyloid angiopathy (CAA). The intensity of immunostaining of clusterin is increased in the leptomeningeal vessels present in the sulci in elderly control cases compared to young cases and in CAA compared to elderly control cases. Immunofluorescence for Aβ and clusterin in leptomeningeal arteries in CAA (d–e). Aβ immunofluorescence (blue) in (d) is present in the whole thickness of the arterial wall in a concentric manner; clusterin immunofluorescence (red) in (e) is also present throughout the thickness of the arterial wall; co‐localization (pink) of Aβ and clusterin occupies most of the thickness of the arterial walls in (f). Scale bars: (a–c) = 100 μm/(d–f) = 50 μm.
Figure 5.
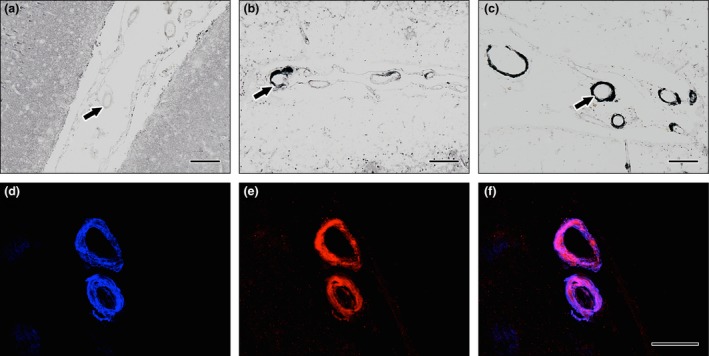
Immunohistochemistry of tissue inhibitor of metalloproteinases 3 (TIMP3) in leptomeningeal arteries. DAB with haematoxylin counterstain in (a) young and (b) elderly controls and (c) cerebral amyloid angiopathy (CAA). The intensity of immunostaining of TIMP3 is increased in the leptomeningeal vessels present in the sulci of elderly control cases compared to young and in CAA cases compared to elderly. Immunofluorescence for Aβ and TIMP3 in leptomeningeal arteries in CAA (d–e). Aβ immunofluorescence (blue) in (d) is present in the whole thickness of the arterial wall in a concentric manner; TIMP3 immunofluorescence (red) in (e) is also present throughout the thickness of the arterial wall; co‐localization (pink) of Aβ and TIMP3 occupies most of the thickness of the arterial walls, especially concentrated in the tunica media, with less in the endothelium and outer layers of the wall (f). Scale bars: (a–c) = 100 μm/(d–f) = 50 μm.
Figure 6.
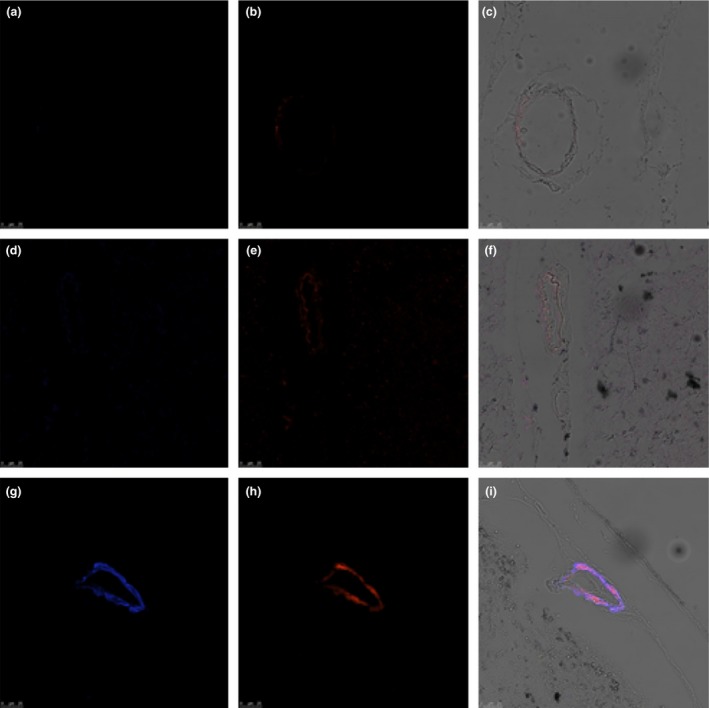
Confocal microscopy images showing distribution of tissue inhibitor of metalloproteinases 3 (TIMP3) (blue) and Aβ (red) in leptomeningeal arteries from young (a–c) and elderly females (d–f) and patients with cerebral amyloid angiopathy (CAA) (g–i). Co‐localization of Aβ and TIMP3 is observed in CAA, on transmission merged images (c–i). Images obtained with ×20 objective. False colour applied to channels.
Discussion
Our study showed that the global endophenotypic profile of leptomeningeal arteries from elderly female patients with severe CAA was different from that of age‐matched and young controls. The immune response/classical complement and extracellular matrix remodelling pathways were significantly enriched in the differentially expressed proteome of arteries between patients with CAA compared to young and elderly controls. Most proteins participating in these pathways were up‐regulated in leptomeningeal arteries from patients with CAA compared to these from controls, possibly reflecting a pro‐inflammatory response in arteries affected by CAA, which could have in turn triggered tissue remodelling processes. The inflammatory profile of CAA is well characterized 33, 34 and previous studies have described an increased activation of the complement system in cerebral amyloid plaques as well as deposition of complement components in CAA affected cerebral arteries 35, 36, 37. Extracellular matrix components can influence the deposition of Aβ thus contributing to Alzheimer's disease progression 38, 39. Conversely, Aβ accumulation damages the integrity of existing extracellular matrix, which affects brain microvascular functions during the early stages of Alzheimer's disease 40, 41, 42.
The study results show that clusterin co‐localizes with Aβ within the walls of leptomeningeal arteries and its expression levels increase in leptomeningeal arteries from patients with CAA compared to those from young and elderly controls. Clusterin (apolipoprotein J or ApoJ) is a disulphide‐linked heterodimeric glycoprotein that activates microglia, initiating an inflammatory cascade 43. Genome‐wide association studies of sporadic Alzheimer's disease, in which Aβ accumulates both in cortical plaques and CAA, have highlighted the importance of common genetic variations in the gene encoding clusterin 44. Experimental work suggests that clusterin regulates Aβ fibril formation 45 and plays a major role in the clearance of Aβ42–ApoJ complexes, via LRP2 46, 47, 48. Although the predominant species of Aβ in CAA is Aβ40, with progressive failure of perivascular clearance of interstitial fluid, there is also accumulation of Aβ42 49. Clusterin appears to be sequestered with Aβ species in the vascular amyloid deposits in sporadic CAA, as well as in the white matter abnormalities in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) 50, 51. A recent study found a significant positive correlation between clusterin concentration and regional levels of insoluble Aβ42 52. It is therefore possible that the up‐regulation of clusterin observed in the CAA arteries, is due to either entrapment of the Aβ–ApoJ complex in the perivascular drainage pathways, or a compensatory up‐regulation of ApoJ to clear the excess Aβ42 that cannot be eliminated normally.
In this study, we demonstrated that the expression of TIMP3 in the brain is restricted to the walls of leptomeningeal arteries and increases in CAA. Homeostasis of the extracellular matrix in the brain is maintained by the balanced action of matrix metalloproteinases that degrade extracellular matrix and by tissue inhibitors of metalloproteinases (TIMP) proteins. Human TIMP3 is a 25‐kDa protein that contains disulphide bonds and is expressed in normal central nervous system 53. In a study by Hoe et al. 54, TIMP3 expression was found to increase in human brains affected by Alzheimer's disease (AD). Furthermore, this study showed that TIMP3 prevents α‐cleavage of amyloid precursor protein (APP), whereas it promotes β‐cleavage of APP thus contributing to elevated Aβ levels in AD. TIMP3 preserves the integrity of extracellular matrix in arteries as the absence of TIMP3 in knock‐out mice results in pathological arterial vasodilation 55. Our results showed that expression of TIMP3 in the brain is restricted to the walls of leptomeningeal, thus antagonistically targeting TIMP‐3 could also facilitate perivascular drainage of Aβ. Examining this hypothesis was beyond the scope of the present study and constitutes a future objective.
In conclusion, this proteomic study demonstrates the activation of inflammatory and extracellular matrix remodelling pathways in human leptomeningeal arteries from CAA patients compared to these from cognitively normal young and elderly controls. Furthermore, we observed increased levels of clusterin and TIMP3 in leptomeningeal arteries from CAA patients compared to young and elderly controls and co‐localization of these two proteins with Aβ in the occipital cortex of the CAA cases. Future work will test the hypothesis that clusterin and TIMP3 could facilitate perivascular clearance and represent novel therapeutic targets for CAA.
Author contributions
AM performed the proteomic experiments, interpreted the results and wrote manuscript. MG performed the immunohistochemistry experiments and interpreted the results. CHW performed the bioinformatics analysis. CS and JARN interpreted the results and edited the manuscript. MJ, RK and JA provided the samples and edited the manuscript. SDG designed the proteomic experiments, supervised their execution, interpreted the results and wrote manuscript. ROC conceived the study, funded the study, designed the immunohistochemistry experiments, interpreted the results and wrote manuscript.
Conflicts of interest
None declared.
Supporting information
Table S1. Total proteome (peptide FDR confidence > 99%) (log2 ratio).
Table S2. Differentially expressed proteins in leptomeningeal arteries from elderly vs. young controls (log2 ratio).
Table S3. Differentially expressed proteins in leptomeningeal arteries from CAA patients vs. young controls (log2 ratio).
Table S4. Differentially expressed proteins in leptomeningeal arteries from CAA patients vs. age‐matched controls (log2 ratio).
Acknowledgements
We are indebted to Mr. Roger Allsopp, Mr. Derek Coates and Hope for Guernsey for their fund raising and vision in establishing a clinical mass spectrometry laboratory at the University of Southampton. This study was funded by the BBSRC, Rosetrees Trust and the Wessex Cancer Trust and Medical Research, UK. The authors are grateful to the support of King Saud University, Deanship of Scientific Research Chair, Prince Mutaib Bin Abdullah Chair for Biomarkers of Osteoporosis, College of Science, as well as the Visiting Professor Program of King Saud University, Riyadh, Saudi Arabia. Tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council (grant number G0400074) and by Brains for Dementia Research, a joint venture between Alzheimer's Society and Alzheimer's Research UK.
Manousopoulou A., Gatherer M., Smith C., Nicoll J. A. R., Woelk C. H., Johnson M., Kalaria R., Attems J., Garbis S. D. and Carare R. O. (2016) Neuropathology and Applied Neurobiology 43, 492–504 Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy
References
- 1. Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011; 37: 75–93 [DOI] [PubMed] [Google Scholar]
- 2. Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, Yamada M, McCarron M, Minett T, Matthews F, Greenberg S, Mann D, Kehoe PG. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post‐mortem brain tissue. Am J Neurodegen Dis 2014; 3: 19–32 [PMC free article] [PubMed] [Google Scholar]
- 3. Alafuzoff I, Thal DR, Arzberger T, Bogdanovic N, Al‐Sarraj S, Bodi I, Boluda S, Bugiani O, Duyckaerts C, Gelpi E, Gentleman S, Giaccone G, Graeber M, Hortobagyi T, Höftberger R, Ince P, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovács GG, Meyronet D, Monoranu C, Nilsson T, Parchi P, Patsouris E, Pikkarainen M, Revesz T, Rozemuller A, Seilhean D, Schulz‐Schaeffer W, Streichenberger N, Wharton SB, Kretzschmar H. Assessment of beta‐amyloid deposits in human brain: a study of the BrainNet Europe Consortium. Acta Neuropathol 2009; 117: 309–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol 2003; 29: 106–17 [DOI] [PubMed] [Google Scholar]
- 5. Tian J, Shi J, Smallman R, Iwatsubo T, Mann DM. Relationships in Alzheimer's disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagen. Neuropathology Appl Neurobiol 2006; 32: 332–40 [DOI] [PubMed] [Google Scholar]
- 6. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008; 18: 253–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica‐Worms D, Holtzman DM. P‐glycoprotein deficiency at the blood‐brain barrier increases amyloid‐beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005; 115: 3285–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta‐peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004; 43: 333–44 [DOI] [PubMed] [Google Scholar]
- 9. Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta‐degrading enzymes in Alzheimer's disease. Brain Pathol 2008; 18: 240–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tarasoff‐Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol 2015; 11: 457–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carare RO, Bernardes‐Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 2008; 34: 131–44 [DOI] [PubMed] [Google Scholar]
- 12. Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, Weller RO, Carare RO. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid‐beta from the mouse brain. Aging Cell 2013; 12: 224–36 [DOI] [PubMed] [Google Scholar]
- 13. Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 2011; 121: 431–43 [DOI] [PubMed] [Google Scholar]
- 14. Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid‐beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS ONE 2012; 7: e41636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galanos P, Vougas K, Walter D, Polyzos A, Maya‐Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, Canovas B, Igea A, Ahuja AK, Zellweger R, Havaki S, Kanavakis E, Kletsas D, Roninson IB, Garbis SD, Lopes M, Nebreda A, Thanos D, Blow JJ, Townsend P, Sørensen CS, Bartek J, Gorgoulis VG. Chronic p53‐independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016; 18: 777–89. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giannogonas P, Apostolou A, Manousopoulou A, Theocharis S, Macari SA, Psarras S, Garbis SD, Pothoulakis C, Karalis KP. Identification of a novel interaction between corticotropin releasing hormone (Crh) and macroautophagy. Sci Rep 2016; 6: 23342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White CH, Johnston HE, Moesker B, Manousopoulou A, Margolis DM, Richman DD, Spina CA, Garbis SD, Woelk CH, Beliakova‐Bethell N. Mixed effects of suberoylanilide hydroxamic acid (SAHA) on the host transcriptome and proteome and their implications for HIV reactivation from latency. Antiviral Res 2015; 123: 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delehouzé C, Godl K, Loaëc N, Bruyère C, Desban N, Oumata N, Galons H, Roumeliotis TI, Giannopoulou EG, Grenet J, Twitchell D, Lahti J, Mouchet N, Galibert MD, Garbis SD, Meijer L. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 2014; 33: 5675–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manousopoulou A, Saito S, Yamamoto Y, Al‐Daghri NM, Ihara M, Carare RO, Garbis SD. Hemisphere asymmetry of response to pharmacologic treatment in an Alzheimer's disease mouse model. J Alz Dis 2016; 51: 333–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manousopoulou A, Koutmani Y, Karaliota S, Woelk CH, Manolakos ES, Karalis K, Garbis SD. Hypothalamus proteomics from mouse models with obesity and anorexia reveals therapeutic targets of appetite regulation. Nutr Diabetes 2016; 6: e204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al‐Daghri NM, Al‐Attas OS, Johnston HE, Singhania A, Alokail MS, Alkharfy KM, Abd‐Alrahman SH, Sabico SL, Roumeliotis TI, Manousopoulou‐Garbis A, Townsend PA, Woelk CH, Chrousos GP, Garbis SD. Whole serum 3D LC‐nESI‐FTMS quantitative proteomics reveals sexual dimorphism in the milieu intérieur of overweight and obese adults. J Proteome Res 2014; 13: 5094–105 [DOI] [PubMed] [Google Scholar]
- 22. Hanley CJ, Noble F, Ward M, Bullock M, Drifka C, Mellone M, Manousopoulou A, Johnston HE, Hayden A, Thirdborough S, Liu Y, Smith DM, Mellows T, Kao WJ, Garbis SD, Mirnezami A, Underwood TJ, Eliceiri KW, Thomas GJ. A subset of myofibroblastic cancer‐associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016; 7: 6159–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manousopoulou A, Woo J, Woelk CH, Johnston HE, Singhania A, Hawkes C, Garbis SD, Carare RO. Are you also what your mother eats? Distinct proteomic portrait as a result of maternal high‐fat diet in the cerebral cortex of the adult mouse. Int J Obes (Lond) 2015; 39: 1325–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800 [DOI] [PubMed] [Google Scholar]
- 26. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479–86 [DOI] [PubMed] [Google Scholar]
- 27. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on Aging ; Alzheimer's Association . National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012; 123: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del ST, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez‐Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova‐Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65: 1863–72 [DOI] [PubMed] [Google Scholar]
- 29. Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 2005; 62: 685–91 [DOI] [PubMed] [Google Scholar]
- 30. Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci 2004; 1019: 24–8 [DOI] [PubMed] [Google Scholar]
- 31. Shinohara M, Murray ME, Frank RD, Shinohara M, DeTure M, Yamazaki Y, Tachibana M, Atagi Y, Davis MD, Liu CC, Zhao N, Painter MM, Petersen RC, Fryer JD, Crook JE, Dickson DW, Bu G, Kanekiyo T. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol 2016; 132: 225–34. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levin Y. The role of statistical power analysis in quantitative proteomics. Proteomics 2011; 11: 2565–7 [DOI] [PubMed] [Google Scholar]
- 33. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss‐Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015; 14: 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, Neal JW, Love S, Nicoll JA, Boche D. Inflammatory components in human Alzheimer's disease and after active amyloid‐beta42 immunization. Brain 2013; 136: 2677–96 [DOI] [PubMed] [Google Scholar]
- 35. Gasque P, Neal JW, Singhrao SK, McGreal EP, Dean YD, Van BJ, Morgan BP. Roles of the complement system in human neurodegenerative disorders: pro‐inflammatory and tissue remodeling activities. Mol Neurobiol 2002; 25: 1–17 [DOI] [PubMed] [Google Scholar]
- 36. van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci 2003; 992: 56–71 [DOI] [PubMed] [Google Scholar]
- 37. Tanskanen M, Lindsberg PJ, Tienari PJ, Polvikoski T, Sulkava R, Verkkoniemi A, Rastas S, Paetau A, Kiuru‐Enari S. Cerebral amyloid angiopathy in a 95 + cohort: complement activation and apolipoprotein E (ApoE) genotype. Neuropathol Appl Neurobiol 2005; 31: 589–99 [DOI] [PubMed] [Google Scholar]
- 38. Ariga T, Miyatake T, Yu RK. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer's disease and related disorders: amyloidogenesis and therapeutic strategies–a review. J Neurosci Res 2010; 88: 2303–15 [DOI] [PubMed] [Google Scholar]
- 39. Genedani S, Agnati LF, Leo G, Buzzega D, Maccari F, Carone C, Andreoli N, Filaferro M, Volpi N. beta‐Amyloid fibrillation and/or hyperhomocysteinemia modify striatal patterns of hyaluronic acid and dermatan sulfate: possible role in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 2010; 7: 150–7 [DOI] [PubMed] [Google Scholar]
- 40. Ajmo JM, Bailey LA, Howell MD, Cortez LK, Pennypacker KR, Mehta HN, Morgan D, Gordon MN, Gottschall PE. Abnormal post‐translational and extracellular processing of brevican in plaque‐bearing mice over‐expressing APPsw. J Neurochem 2010; 113: 784–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sykova E, Vorisek I, Antonova T, Mazel T, Meyer‐Luehmann M, Jucker M, Hajek M, Ort M, Bures J. Changes in extracellular space size and geometry in APP23 transgenic mice: a model of Alzheimer's disease. Proc Natl Acad Sci U S A 2005; 102: 479–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol 2015; doi: 10.1111/nan.12295. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 43. Xie Z, Harris‐White ME, Wals PA, Frautschy SA, Finch CE, Morgan TE. Apolipoprotein J (clusterin) activates rodent microglia in vivo and in vitro 1. J Neurochem 2005; 93: 1038–46 [DOI] [PubMed] [Google Scholar]
- 44. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al‐Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O'Donovan M, Owen MJ, Williams J. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009; 41: 1088–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu JT, Tan L. The role of clusterin in Alzheimer's disease: pathways, pathogenesis, and therapy. Mol Neurobiol 2012; 45: 314–26 [DOI] [PubMed] [Google Scholar]
- 46. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 2007; 27: 909–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech 2000; 50: 305–15 [DOI] [PubMed] [Google Scholar]
- 48. Ladu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, Getz GS. Lipoproteins in the central nervous system. An N Y Acad Sci 2000; 903: 167–75 [DOI] [PubMed] [Google Scholar]
- 49. Soontornniyomkij V, Choi C, Pomakian J, Vinters HV. High‐definition characterization of cerebral beta‐amyloid angiopathy in Alzheimer's disease. Hum Pathol 2010; 41: 1601–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Craggs L, Taylor J, Slade JY, Chen A, Hagel C, Kuhlenbaeumer G, Borjesson‐Hanson A, Viitanen M, Kalimo H, Deramecourt V, Oakley AE, Kalaria RN. Clusterin/Apolipoprotein J immunoreactivity is associated with white matter damage in cerebral small vessel diseases. Neuropathol Appl Neurobiol 2016; 42: 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Howlett DR, Hortobagyi T, Francis PT. Clusterin associates specifically with Abeta40 in Alzheimer's disease brain tissue. Brain Pathol 2013; 23: 623–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miners JS, Clarke P, Love S. Clusterin levels are increased in Alzheimer's disease and influence the regional distribution of Aβ. Brain Pathol 2016; doi: 10.1111/bpa.12392. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kishnani NS, Staskus PW, Yang TT, Masiarz FR, Hawkes SP. Identification and characterization of human tissue inhibitor of metalloproteinase‐3 and detection of three additional metalloproteinase inhibitor activities in extracellular matrix. Matrix Biol 1995; 14: 479–88 [DOI] [PubMed] [Google Scholar]
- 54. Hoe HS, Cooper MJ, Burns MP, Lewis PA, van der Brug M, Chakraborty G, Cartagena CM, Pak DT, Cookson MR, Rebeck GW. The metalloprotease inhibitor TIMP‐3 regulates amyloid precursor protein and apolipoprotein E receptor proteolysis. J Neurosci 2007; 27: 10895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Basu R, Lee J, Morton JS, Takawale A, Fan D, Kandalam V, Wang X, Davidge ST, Kassiri Z. TIMP3 is the primary TIMP to regulate agonist‐induced vascular remodelling and hypertension. Cardiovasc Res 2013; 98: 360–71 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Total proteome (peptide FDR confidence > 99%) (log2 ratio).
Table S2. Differentially expressed proteins in leptomeningeal arteries from elderly vs. young controls (log2 ratio).
Table S3. Differentially expressed proteins in leptomeningeal arteries from CAA patients vs. young controls (log2 ratio).
Table S4. Differentially expressed proteins in leptomeningeal arteries from CAA patients vs. age‐matched controls (log2 ratio).