

Abstract
Thousands of genome-wide association studies (GWAS) have been conducted to identify the genetic variants associated with complex disorders. However, only a small proportion of phenotypic variances can be explained by the reported variants. Moreover, many GWAS failed to identify genetic variants associated with disorders displaying hereditary features. The “missing heritability” problem can be partly explained by rare variants. We simulated a causality scenario that gestational ages, a quantitative trait that can distinguish preterm (<37 weeks) and term births, were significantly correlated with the rare variant aggregations at 1000 single-nucleotide polymorphism loci. These 1000 simulated causal rare variants were embedded into randomly selected subsets of 9642 promoter regions from the 1000 Genomes Project genotypic data according to different proportions of causal rare variants within the embedded promoters. Through analysis of the correlations between rare variant aggregations and gestational ages, we found that the embedded promoters as a whole showed weaker genetic association when the proportion of causal rare variants decreased, and no individual embedded promoters showed genetic association when the proportion of causal rare variants was smaller than 0.4. Our analyses indicate that association signals can be greatly diluted when causal rare variants are dispersedly and sparsely distributed in the genome, accounting for an important source of missing heritability.
Keywords: Missing heritability, rare variant, causal variant, simulation, preterm birth
Introduction
Genome-wide association study (GWAS) is a common approach for pinpointing the genetic variants associated with complex disorders.1 According to the GWAS Catalog, thousands of GWAS have been conducted.2 Each GWAS may report several to several tens of genetic variants associated with its investigated disorder. However, the identified genetic variants frequently show only modest effects on the disease risk or quantitative trait variation, which is referred to as the “missing heritability” problem.3 Moreover, GWAS for spontaneous preterm birth, a complex disorder displaying hereditary features,4 have not reported any convincing associated variants.5,6
Many theories have been proposed to explain the missing heritability problem in GWAS. Conventionally, GWAS limit analyses to common variants according to minor allele frequency (MAF) ≥5%. It is possible that low-frequency (0.5% ≤ MAF < 5%) and/or rare (MAF <0.5%) variants account for part of the missing heritability.3,7 In rare mendelian disorders, causal rare variants tend to show high penetrance, whereas in complex disorders, the penetrance levels of rare variants are now believed to be mostly moderate to small.8 Recent studies have reported potentially pathogenic roles of rare variants in schizophrenia.9,10
Due to the rareness problem, analysis of individual rare variants is difficult. Thus, association testing for rare variants often relies on collapsing methods, ie, examining the combined effects of rare variants in a gene or a functional unit so as to amplify association signals.11 Specific forms of rare variant collapsing methods include the BURDEN test12 and the sequence kernel association test (SKAT).13
The effectiveness of most rare variant collapsing methods relies on a large proportion of variants in some scanned genomic regions being causal.11 However, it is not reasonable to simply assume that causal rare variants tend to be clustered within several long chromosomal regions. Short functional elements such as transcript factor binding sites, promoters, enhancers, open chromatins, nucleosome positioning, and histone modifications are dispersedly distributed in the genome, and rare variants across a large number of (say >100) such functional elements may collectively modulate phenotypes. Recent studies have demonstrated that disease risk–associated variants may be enriched in particular epigenetic marks across the entire genome.14–16
Effective rare variant analysis approaches must properly model how rare variants are associated with complex disorders. From a network view, the normal functionality of a life system is contingent on the spatiotemporal harmony of the entire gene networks, whereas on the opposite, multiple small genetic disturbances can collective render rewiring of gene networks, further leading to genetic disorders.17–20 In this sense, hundreds to thousands of rare variants that modulate disease-related pathways can be the causes of some complex disorders. Of note, a large number of causal variants do not mean that any disease individual carries most of the causal variants. The genetics of complex disorders are often heterogeneous,21 indicating that combinations of causal variants in specific disease individuals could be distinct. Effective rare variant analysis approaches should have the capabilities of capturing large numbers of small additive effects and accommodating genetic heterogeneity.
Spontaneous preterm birth (gestational age <37 weeks) is apparently a complex genetic disorder, as a woman’s preterm birth risk is higher if she was born preterm or she has preterm birth history.4 In this study, we designed a scenario that preterm birth was caused by the additive effects of 1000 rare variants. One advantage of selecting preterm birth as the disease model is that preterm birth can be approximated by gestational ages, rendering enhanced statistical power. Through simulations we demonstrated that strong genetic associations can simply become undetectable because of the ineffectiveness of rare variant collapsing methods, shedding lights on an important source of the missing heritability in GWAS. Our study may help to explain why genetic associations have not been detected for preterm birth.5,6 Moreover, our simulation procedure can serve as a framework for examining the effectiveness of rare variant association testing approaches.
Methods
Statistics
The correlation coefficient (r) in this study was always the Pearson correlation coefficient. Both the statistics and P value were computed using the R software. Multiple testing was corrected by the Benjamini-Hochberg method,22 also available in the R software.
Promoter regions
Gene positions (GRCh37) were downloaded from the Ensembl Biomart (http://grch37.ensembl.org/biomart). Promoter regions are defined as −800 to 199 bp (base pairs) of the transcription start sites. For the promoters with overlapped positions, only the left promoter was used. Then, 10 000 promoters were randomly selected for subsequent analyses.
Whole-genome genotypic data
The whole-genome genotypic data of 2504 samples were downloaded from the 1000 Genome Project Web site (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/).23 The rare variants (biallelic single-nucleotide polymorphism loci with MAF <0.5%) within the 10 000 selected promoters were retrieved using VCFtools.24 Then, the promoter regions containing less than 10 rare variants were dropped. The total number of promoters included in the analysis was 9642, consisting of 235 842 rare variants.
Aggregation of rare variants
Aggregation of rare variants is not the count of rare variant loci. For any region or set of regions, the rare variant at position j in individual i is coded by the number of the minor allele:
The rare variant aggregation of the analyzed region(s) in individual i is the summation of all rare variant variables:
Simulation of causal rare variants
We designed a simulation study that 1000 rare variants were strongly associated with preterm birth. We simulated 2504 samples, of which half were preterm birth and the other half were term birth. The phenotype was gestational age, ranging from 21 to 41 weeks. Gestational ages of both preterm (21-36 weeks) and term (37-41 weeks) births were generated according to uniform distributions. Then, a total of 1000 causal rare variants were simulated. For each rare variant locus, we generated a guiding MAF ranging from 0.02% to 0.25%, which was obtained according to an exponential distribution with rate = 2. Then, the specific genotype of this locus in each individual was generated by the following procedure:
The genotype was coded in 0 (homozygous for the major allele), 1 (heterozygous), or 2 (homozygous for the minor allele).
-
The probability of generating the minor allele was determined by guiding MAF × risk factor, where the risk factor ranged from 0.75 to 2 depending on the gestational age:
The genotype always had 2 alleles. For each allele, a random number between 0 and 1 was generated using a uniform distribution. If the random number was smaller than the probability of generating the minor allele as described above, the minor allele (+1) was generated.
Results
Quality of the simulated causal rare variants
The actual MAF of the 1000 simulated causal rare variants ranged from 0.02% to 0.48%, falling within the typical MAF range of rare variants. The actual MAFs were also highly correlated with the guiding MAF (R2 = 0.666, P < 2.2 × 10−16). For each sample, rare variant aggregation was computed. Across all samples, rare variant aggregations were significantly associated with gestational ages (R2 = 0.246, P = 2.671 × 10−155). A plot between rare variant aggregations and gestational ages (Figure 1) confirms this association. Moreover, the plot shows that any individual carries no more than 12 causal rare variants, indicating that genetic heterogeneity is also achieved. Thus, simulation of 1000 causal rare variants for preterm birth was achieved.
Figure 1.
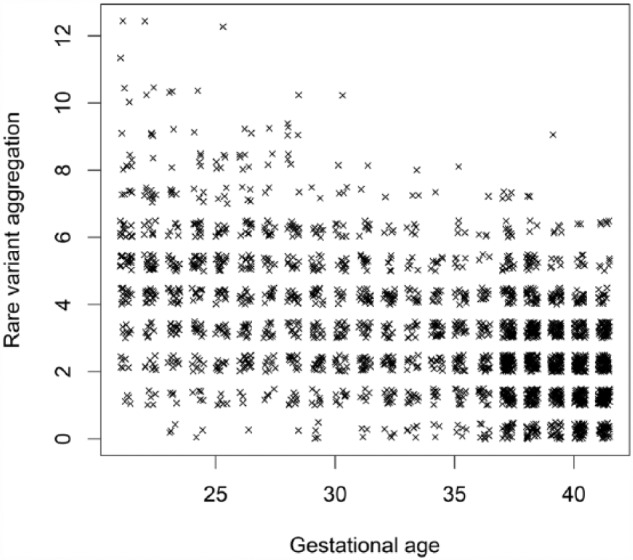
Rare variant aggregations versus gestational ages for the 1000 simulated causal rare variants.
Identifying the simulated causal rare variants from the genome reveals an important source of missing heritability
Identifying causal variants from the entire genome is a core task for association studies. Whole-genome sequencing technologies may generate millions of variants for a study cohort. Thus, this task is very challenging. We further assumed that preterm birth was caused by the 1000 simulated causal rare variants located in the promoter regions of mothers’ whole-blood transcriptome at delivery which consisted of a total of 9642 transcripts. We retrieved the genotypes of 2504 whole-genome sequencing samples from the 1000 Genomes Project and embedded the 1000 simulated causal rare variants into subsets of the 9642 promoter regions from the whole-genome genotypic data. Note that at the embedded locations, the original rare variants were replaced by the simulated causal rare variants. To ameliorate the effect of population stratification, the simulated samples were randomly assigned to 1000 Genomes Project samples.
We assessed the association signals of the 1000 simulated causal rare variants from the whole-genome genotypic data. We generated a series of data sets by varying the proportion of causal rare variants within the embedded promoters. Analysis of individual rare variants is suggested to be impractical due to the rareness problem. Actually, we used the quantitative trait association testing available from the PLINK package25 to assess individual rare variants but did not find any significant rare variant after adjusting for multiple testing. Thus, we assessed genetic associations by correlating promoters’ rare variant aggregations with gestational ages. Under each proportion of causal rare variants, we first computed the number of embedded (affected) promoters and the association signal of all affected promoters as a whole. It is noted that the real association signal (only the 1000 simulated causal rare variants were aggregated) is R2 = 0.246, P = 2.671 × 10−155. As shown in Table 1, when the proportion of causal rare variants decreases, the association signal of all affected promoters becomes weaker. This analysis suggests that the missing heritability is connected to inclusion of noncausal rare variants into the aggregation procedure. However, even with a proportion of 0.1, the association signal is still significant, suggesting that pinpointing the functional elements containing causal rare variants is critical for rare variant collapsing methods.
Table 1.
Association signals under different proportions of causal rare variants in embedded (affected) promoters.
| Proportion of causal rare variants | Association signal of all affected promoters |
Individual promoters identified for association |
||
|---|---|---|---|---|
| No. of affected promoters | Association signal (R2, P value) | No. of positives | No. (%) of true positives | |
| 1 | 44 | 0.240, 7.52 × 10−152 | 31 | 28 (63.6) |
| 0.9 | 45 | 0.234, 7.37 × 10−148 | 24 | 23 (55.1) |
| 0.8 | 53 | 0.225, 6.57 × 10−141 | 21 | 21 (39.6) |
| 0.7 | 61 | 0.209, 1.35 × 10−129 | 25 | 22 (36.1) |
| 0.6 | 69 | 0.183, 2.46 × 10−112 | 10 | 9 (13.0) |
| 0.5 | 84 | 0.168, 4.38 × 10−102 | 9 | 9 (10.7) |
| 0.4 | 111 | 0.140, 3.55 × 10−84 | 2 | 2 (1.8) |
| 0.3 | 143 | 0.098, 6.04 × 10−58 | 0 | 0 (0) |
| 0.2 | 225 | 0.054, 3.69 × 10−32 | 0 | 0 (0) |
| 0.1 | 497 | 0.015, 6.23 × 10−10 | 1 | 0 (0) |
We then scanned individual promoters to examine whether individual promoters could be identified for genetic association, using an adjusted (for all scanned promoters) P value of .05 as the cutoff. The number of true positives (ie, number of affected promoters being identified) was highly dependent on the proportion of causal rare variants (Table 1). When the proportion was very high (≥0.9), more than half of the affected promoters were identified. When the proportion was smaller than 0.8, most of the affected promoters could not be identified, and the approach became totally ineffective when the proportion was smaller than 0.3. This analysis suggests that rare variant collapsing methods are ineffective when causal rare variants are dispersedly and sparsely distributed across the genome.
In summary, our simulation and analyses demonstrate that rare variants could be causes of complex disorders, and the missing heritability problem may result from the ineffectiveness of rare variant collapsing methods.
Discussion
Many theories have been proposed to explain the missing heritability problem in association studies, of which rare variants play important roles.3,7 Causal rare variants were previously suggested to have strong effects.26 However, from the view of gene networks, it is possible that a large number of rare variants with moderate effects can collectively render rewiring of gene networks. Thus, it is reasonable to analyze the additive effects from a large number of rare variants.
In this study, using a simulation approach, we demonstrated that the missing heritability problem can result from the ineffectiveness of rare variant collapsing methods when very few chromosomal regions contain a large proportion of causal rare variants. We used actual promoters instead of simulated promoters to accommodate simulated causal rare variants, which was a real data-based simulation procedure. Real data-based simulations incorporate genomic and population genetic contexts and thus are more realistic than purely simulated data. This strategy was also adopted in one of our previous studies.27
Optimally, any combination of rare variants should be examined for genetic association so that the real association can be eventually identified. However, an exhaustive search is computationally intractable, as a study cohort can have millions of genetic variants. Thus, it is desired to develop novel big data and artificial intelligence approaches to cleverly enhance the scope of examined rare variant combinations. For rare variant association testing, a big challenge is which variants to aggregate.7 Functional annotation of variants such as nonsynonymous, stop-gain/loss, and frameshift may help selection of rare variants for aggregation,7 but this approach may exclude the causal variants within noncoding regions. We suggest an optimization procedure which uses a set of suspected rare variants as the start point and iteratively adds the variants that maximize the association signal of the rare variant aggregation until reaches convergence.
The simulation framework of this study also has implications on the genetic mechanisms of preterm birth. Childbirth is a complicated biological procedure involving multiple pathways such as increased uterine contractility, cervical ripening, and decidua and fetal membrane activation.28 Occurrences of multiple deleterious regulatory rare variants increase the chances of network rewiring in these pathways, which may further lead to enhanced risks of preterm birth.
Footnotes
Peer review:Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 716 words, excluding any confidential comments to the academic editor.
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1. Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 2. MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bezold KY, Karjalainen MK, Hallman M, Teramo K, Muglia LJ. The genomics of preterm birth: from animal models to human studies. Genome Med. 2013;5:34. doi: 10.1186/gm438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Falah N, McElroy J, Snegovskikh V, et al. Investigation of genetic risk factors for chronic adult diseases for association with preterm birth. Hum Genet. 2013;132:57–67. doi: 10.1007/s00439-012-1223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang H, Baldwin DA, Bukowski RK, et al. A genome-wide association study of early spontaneous preterm delivery. Genet Epidemiol. 2015;39:217–226. doi: 10.1002/gepi.21887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014;111:E455–E464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Auer PL, Lettre G. Rare variant association studies: considerations, challenges and opportunities. Genome Med. 2015;7:16. doi: 10.1186/s13073-015-0138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Teng S, Thomson PA, McCarthy S, et al. Rare disruptive variants in the DISC1 Interactome and Regulome: association with cognitive ability and schizophrenia [published online ahead of print June 20, 2017]. Mol Psychiatry. doi:10.1038/mp. 2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cowper-Sal lari R, Zhang X, Wright JB, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 2012;44:1191–1198.doi:10.1038/ng. 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li S, Ovcharenko I. Human enhancers are fragile and prone to deactivating mutations. Mol Biol Evol. 2015;32:2161–2180. doi: 10.1093/molbev/msv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang D, Ovcharenko I. Identifying causal regulatory SNPs in ChIP-seq enhancers. Nucleic Acids Res. 2015;43:225–236. doi: 10.1093/nar/gku1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goh KI, Choi IG. Exploring the human diseasome: the human disease network. Brief Funct Genomics. 2012;11:533–542. doi: 10.1093/bfgp/els032. [DOI] [PubMed] [Google Scholar]
- 18. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu JX, Thomas CE, Brunak S. Network biology concepts in complex disease comorbidities. Nature Rev Genet. 2016;17:615–629. doi: 10.1038/nrg.2016.87. [DOI] [PubMed] [Google Scholar]
- 21. McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 22. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- 23. The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 27. Wang Y, Liu X, Robbins K, Rekaya R. AntEpiSeeker: detecting epistatic interactions for case-control studies using a two-stage ant colony optimization algorithm. BMC Res Notes. 2010;3:117. doi: 10.1186/1756-0500-3-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Institute of Medicine (US). Committee on understanding premature birth and assuring healthy outcomes. In: Behrman RE, Butler AS, eds. Preterm Birth: Causes, Consequences and Prevention. Washington, DC: The National Academies Collection: Reports funded by National Institutes of Health; 2007;169–206. [PubMed] [Google Scholar]