

Abstract
Beta-amyloid (Aβ) deposition in brain accumulates as a function of age in people with Down syndrome (DS) with subsequent development into Alzheimer disease neuropathology, typically by 40 years of age. In vivo imaging using the Pittsburgh Compound B (PiB) ligand has facilitated studies linking Aβ, cognition, and dementia in DS. However, there are no studies of PiB binding across the lifespan in DS. The current study describes in vitro 3H-PiB binding in the frontal cortex of autopsy cases with DS compared to non-DS controls. Tissue from 64 cases included controls (N=25) and DS (N=39). In DS, 3H-PiB binding was significantly associated with age. After age 40 years in DS, 3H-PiB binding rose dramatically along with increasing individual variability. 3H-PiB binding correlated with the amount of Aβ42. Using fixed frontal tissue and fluorescent 6-CN-PiB, neuritic and cored plaques along with extensive cerebral amyloid angiopathy (CAA) showed 6-CN-PiB binding. These results suggest that cortical PiB binding as shown by positron emission tomography imaging reflects plaques and CAA in DS brain.
Keywords: Aging, Alzheimer disease, Beta-amyloid, Plaques, Neurofibrillary tangles, Trisomy 21, Thioflavine S, 3H-X-34
1. Introduction
Alzheimer disease (AD) is the most common cause of dementia in the elderly and affects 1 in 9 people over the age of 65 years (http://www.alz.org/facts/). There are currently no biomarkers for AD that can clearly distinguish people who will develop the disease from those who will not. The presence of beta-amyloid (Aβ) plaques and tau neurofibrillary tangles determined at autopsy are defining characteristics for a pathological diagnosis of AD. However, the development of in vivo ligands that selectively bind to Aβ, allows these lesions to be visualized and quantified in people using positron emission tomography (PET), significantly accelerating biomarker development (Cohen and Klunk, 2014;Johnson, et al., 2012;Mintun, et al., 2006;Sperling, et al., 2014). The first of these ligands, Pittsburgh Compound B (PiB) (Klunk, et al., 2004) has now been used in a large number of clinical studies in patients with AD and can detect Aβ plaques in early disease (Cohen and Klunk, 2014).
3H-PiB and 6-CN-PiB binding in vitro has been described in autopsy cases of AD in the general population (Bacskai, et al., 2007;Beckett, et al., 2012;Ikonomovic, et al., 2012;Ikonomovic, et al., 2008;Klunk, et al., 2007). Aβ40 and Aβ42 positive plaques as well as vascular Aβ bind PiB in vitro. PiB binding was more robust in compact or cored plaques and less so with diffuse plaques. Typically neurofibrillary tangles did not bind PiB except for possible weak binding to extracellular “ghost” tangles, which may be due to associated Aβ (Ikonomovic, et al., 2008). Finally, PiB binding correlates early in disease with postmortem insoluble Aβ measures and with plaque loads. In one case that was PET imaged in life with PiB and then came to autopsy, there was a significant overlap in the regional distribution of the in vivo plaque binding and in vitro PiB binding (Bacskai, et al., 2007).
Down syndrome (DS) or trisomy 21, is the most common genetic cause of intellectual disability and is associated with a neurologic phenotype that includes the development of AD neuropathology by the age of 40 years (Head, et al., 2016;Lott, 2012;Lott and Dierssen, 2010). Further, autopsy studies indicate that in DS there is an age-associated progression of AD neuropathology with initial deposits of Aβ and subsequent formation of neurofibrillary tangles (Head, et al., 2016;Hof, et al., 1995;Leverenz, 1998;Wisniewski, et al., 1985). Even so, the age of onset of dementia may be delayed by almost a decade (over 50 years of age) after AD neuropathology is present and a subset of people with DS appear to not develop dementia even at very old ages (Lai, 1989;Schupf, 2002).
PiB PET imaging studies in people with DS show that binding is age-dependent (Annus, et al., 2016;Handen, et al., 2012;Hartley, et al., 2014;Landt, et al., 2011;Lao, et al., 2016). Two of these studies included measures of impaired cognition and dementia status and found a positive significant correlation with PiB load (Annus, et al., 2016;Hartley, et al., 2014). Striatal PiB is the earliest site of binding with age in DS, typically observed after 35 years of age (Annus, et al., 2016;Handen, et al., 2012;Lao, et al., 2016) and is similar to reports of patients with presenilin-1 mutations (Klunk, et al., 2007;Koivunen, et al., 2008;Villemagne, et al., 2009). Subsequently, with increasing age, more brain regions become affected including the neocortex (Annus, et al., 2016).
The age-dependency of PiB binding in DS in vivo may be a biomarker for AD neuropathology that can be used as an outcome measure in clinical trials. We hypothesized that 3H-PiB binding would increase as a function of age in DS in frontal cortex. Thus, we measured 3H-PiB binding biochemically in frontal cortex homogenates from autopsy cases with and without DS. We used the highly fluorescent PiB derivative, 6-CN-PiB, which has similar binding properties as PiB (Ikonomovic, et al., 2008;Mathis, et al., 2003) in fixed tissue to visualize plaques and, if present, cerebral amyloid angiopathy (CAA) to determine what types of pathology PiB binding represents in vivo.
2. Materials and methods
2.1 Autopsy cases
Frozen frontal cortex (Brodmann area 46) was obtained from 64 cases in total from the University of Kentucky Alzheimer Disease Center, the Alzheimer Disease Research Center at the University of California at Irvine, and the NIH NeuroBioBank. Human tissue collection and handling conformed to each University’s Institutional Review Board guidelines.
Cases ranged from 1 to 66 years of age (Table 1). Control cases were selected to match for age and postmortem interval (PMI) to match the DS cases. Both males and females were included in the study, but given the challenges of matching cases, we did not match for gender. The level of premorbid intellectual disability or cognitive status was not available in most cases and thus it was not possible to use these variables in the analysis.
Table 1.
Case demographics
| Characteristic | DS | Control |
|---|---|---|
| N | 39 | 25 |
| Mean Age (range) years | 44.1 (1–66) | 36.4 (1–66) |
| PMI (hours) | 9.6 | 15.2 |
| Female (%) | 20 (59) | 10 (40) |
The PMI was different across groups, with the control cases showing an overall longer PMI (t(62)=3.0 p=.004) relative to DS cases. Correlation co-efficients were thus adjusted for PMI when necessary.
2.2 3H-PiB binding
3H-PiB binding was used to measure fibrillar Aβ and was assessed in homogenates of frontal cortex (Beckett, et al., 2012;Matveev, et al., 2014). Instead of the 10 μM concentrations typically used for histological assay, which will also bind to low affinity sites, we used 1.2 nM PiB, a concentration closer to the Kd of the PiB analog CN-PiB for Aβ pathology and to the in vivo concentration of 11C-PiB used to visualize high affinity PiB binding in human AD brain. For binding studies, 20 μL of a PBS homogenate containing 0.166 mg wet weight tissue were added to each of three wells of a 96-well polypropylene plate (Costar 3365). 200 μL of 1.2 nM 3H-PiB (cat. VT 278 specific radioactivity = 70.2 Ci/mmol, Vitrax [Placentia, CA]) in PBS + 5% v/v EtOH was added to two of the wells (total binding) and to the third well 200 μL of 1.2 nM 3H-PiB + 1 μM unlabeled competitor BTA-1 (non-specific binding) was added. The plate was sealed with plastic film. Samples were incubated for 2 hours at room temperature without shaking, transferred to a 96-well Millipore Multiscreen HTS Hi Flow FB (GF/B) filter plate, and filtered on a multi-well plate vacuum manifold (Millipore Corporation, Bedford, MA). The filters were washed three times with 200 μL of PBS + 5% v/v EtOH, dried, removed from the plate, and placed in scintillation vials with 2 mL of BudgetSolve scintillation fluid and counted for 3H. Specific binding for each sample was calculated as total binding (mean CPM of the two filters from wells containing radioactive PiB) minus non-specific binding (CPM value from the well containing radioactive PiB + 1 μM nonradioactive BTA-1 competitor).
2.3 3H-X-34 binding
3H-X-34 binding (Matveev, et al., 2014) was used to measure a combination of fibrillar Aβ and neurofibrillary tangles and was performed with 10 μl of a PBS homogenate containing 0.166 mg wet weight tissue similar to 3H-PiB binding, with 5 nM 3H-X-34, 23 Ci/mmol, custom titrated by Vitrax) (Matveev, et al., 2014) with 10 μM Congo Red as the nonradioactive X-34 competitor.
2.4 Aβ ELISA
To measure total soluble and insoluble Aβ40 and Aβ42 in frontal cortex, we used an ELISA. The methods for tissue extraction and Aβ measurements have been published previously (Beckett, et al., 2010). Briefly, frozen cortical samples were serially extracted to obtain fractions of different assembly states of Aβ. The tissue was homogenized in a Dounce homogenizer in ice cold phosphate buffered saline (PBS, pH 7.4) containing 1× complete protease inhibitor cocktail (PIC) (Amresco, Solon, OH), and centrifuged at 20,800 × g for 30 minutes at 4°C. Following centrifugation, the supernatant was collected for subsequent measures of PBS soluble Aβ and the pellets were sonicated (10 × 0.5 sec pulses at 100W, Fisher Sonic Dismembrator) in room temperature 2% SDS in PBS with PIC followed by centrifugation (as above, but at 14°C). The supernatant was again collected to measure SDS soluble Aβ, and the remaining pellets were sonicated in 70% formic acid (FA) followed by centrifugation at 20,800 × g for 1 hour at 4°C. The supernatant collected was used to measure insoluble Aβ. Aβ was measured in these extracts using a standard, well-characterized two-site sandwich ELISA as described previously [47]. Briefly, an Immulon 4HBX plate (Dynex-Thermo Fisher) was coated with 0.5 μg/well of antibody, incubated overnight at 4° C, then blocked with a solution of Synblock (AbD Serotec, Raleigh, NC), as per the manufacturer’s instructions. Antigen capture was performed using monoclonal antibody Ab9 (against Human Aβ(1–16)). Antigen detection was performed using biotinylated antibodies 13.1.1. (end specific for Aβ(x-40)), and 12F4 (end specific for Aβ(x-42); Covance, Princeton, NJ).
To measure insoluble Aβ, formic acid-soluble supernatant was initially neutralized by a 1:20 dilution in TP buffer (1 M Tris base, 0.5 M Na2HPO4), followed by a further dilution as needed (1:100 to 1:400) in Antigen Capture buffer (AC) (0.02 M sodium phosphate buffer, 0.4 M NaCl, 2mM EDTA, 0.4% Block Ace (AbD Serotec), 0.05% NaN3, 0.2% BSA, 0.05% CHAPS, pH 7). SDS-soluble fractions were diluted (1:20) in AC buffer alone and PBS fractions were diluted 1:4 in AC buffer alone. A peptide standard curve of Aβ was run on the same plate for comparison, and standards and samples were run at least in duplicate; Aβ values were determined by interpolation relative to the standard curve. Plates were washed 2–4 times between assay steps with standard PBS containing 0.05% Tween-20 (2–4×) followed by PBS (2–4×). Plates were developed with TMB reagent (Kirkegaard & Perry Laboratories, Gaithersburg, MD), stopped with 6% o-phosphoric acid, and read at 450 nm using a BioTek multiwell plate reader.
2.5 CN-PiB binding in vitro and comparison to AD neuropathology
To visually characterize plaque and vascular binding of PiB in vitro, we used fixed tissue sections from the frontal cortex of two control cases (19 years and 51.3 years), DS (19.8 years), and DS with AD (DSAD) case (51.4 years) and the fluorescent cyano-PiB (CN-PiB). Sections were first mounted on slides and allowed to dry prior to incubation in 100 nM CN-PiB for 1 hour at RT using a similar protocol as published previously (Ikonomovic, et al., 2008) but at a lower concentration closer to the CN-PiB Kd to focus on high affinity imaging ligand-relevant binding. Slides were washed in PBS for 3× for 2 minutes then incubated briefly (30s) in TrueBlack™ lipofuscin autofluorescence quencher (Biotium, Hayward, CA). After three more 2-minute washes in PBS, slides were coverslipped using Everbrite™ mounting media (Biotium). Images were captured using an Olympus BX51 microscope with a Q Color 5 digital camera. A second set of sections was first incubated in CN-PiB as described above but prior to Trueblack quenching for autofluorescence, slides were incubated in 0.5% thioflavine-S (Sigma-Aldrich, St. Louis, MO) in 50% ethanol, differentiated in 50% ethanol, washed, and then exposed to TrueBlack. Sections were coverslipped using Everbrite™.
2.6 Statistical analysis
SPSS for Windows and independent t-tests were used to test for PMI differences across the 2 groups (Control, DS). A stepwise linear regression was used to determine which outcome measure best predicted age in DS cases. Spearman rank correlations were used to measure the strength of association between specific 3H-PiB binding, 3H-X-34 binding, and age in DS.
3. Results
3.13H-PiB binding
We tested the hypothesis that specific 3H-PiB binding levels would vary as a function of age in DS. Fig. 1A shows a significant association between age at death in DS autopsy cases and the amount of 3H-PiB binding that was not linear but rather increased dramatically in cases over 40 years of age. A significant Spearman rank correlation between specific 3H-PiB binding and age in DS was observed (r=0.54 p<0.0005 n=39), which remained after controlling for PMI (r=0.35 p<0.03) but not in controls (selected to be pathology free). Additional individual variability may be due to the presence or severity of dementia, however these data were not available for this study.
Fig. 1.
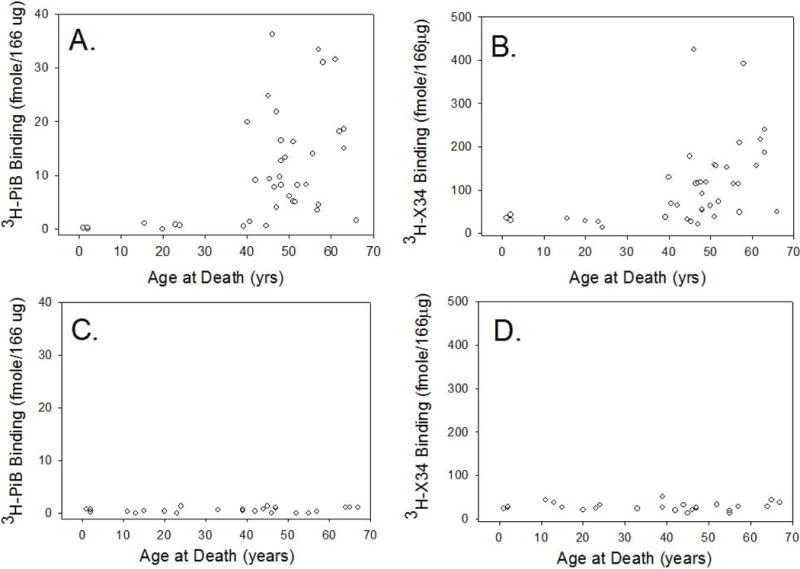
Age-Specific 3H-PiB and 3H-X-34 frontal cortex binding in DS. In DS, the extent of 3H-PiB binding in the frontal cortex increased with age, with an exponential increase over the age of 40 years (A). 3H-X-34 binding was also significantly increased with age (B). This is in contrast to 3H-PiB (C) and 3H-X-34 (D) binding in similarly aged control cases showing no change as a function of age.
3.2 3H-X-34 binding
We next tested the hypothesis that the highly-fluorescent Congo Red derivative, 3H-X-34, which binds to a site on amyloid fibrils in plaques and neurofibrillary tangles and that is distinct from the 3H-PiB binding site, would also vary with age in cases with DS. As with 3H-PiB binding, we observed a significant increase in specific 3H-X-34 binding and age of death (r=0.58 p<.0005 n=39) in DS (but not controls, selected to be pathology free) (Fig.1B). However, this correlation was reduced with the inclusion of PMI when calculating a partial correlation co-efficient (r=0.29 p=0.08).
Predicting 3H-PiB and 3H-X-34 binding
Two independent stepwise linear regressions were used to determine the best predictor of specific 3H-PiB or 3H-X-34 binding with PBS soluble oligomers, PBS, SDS, and FA Aβ40 and Aβ42 levels included in the analysis. The SDS Aβ42 fraction, the amount of oligomers, and the FA fraction of Aβ42 predicted specific 3H-PiB binding (Table 2). In contrast, 3H-X-34 binding was best predicted by PBS oligomers and the two FA fractions (Aβx-40 and Aβx-42) (Table 2). 3H-X-34 also binds to neurofibrillary tangles, which were not systematically measured in the current study. Interestingly, of all the measures that predict age in the DS autopsy series, specific 3H-PiB binding was the best, not oligomers or Aβ measures (r=0.50, F(1,38)=12.47 p=0.001).
Table 2.
Stepwise regression analysis of best predictors for PiB and 3H-X-34binding
| Dependent | Model | R | R Square | Adjusted R Square |
|---|---|---|---|---|
| Specific PiB | SDS Aβ42 | 0.711 | 0.506 | 0.499 |
| SDS Aβ42 + Oligomers | 0.788 | 0.621 | 0.610 | |
| SDS Aβ42 + Oligomers + FA Aβ42 | 0.811 | 0.658 | 0.643 | |
| Specific 3H-X-34 | Oligomers | 0.650 | 0.423 | 0.414 |
| Oligomers + FA Aβ 40 | 0.734 | 0.539 | 0.525 | |
| Oligomers + FA Aβ 40+FA Aβ 42 | 0.760 | 0.577 | 0.558 |
3.5 6-CN-PiB labeling in frontal cortex of DS and control cases
6-CN-PiB binding was not present at detectable levels in control cases (Fig. 2A and B) and in a young case with DS (Fig.2C). The DSAD case showed significant amounts of 6-CN-PiB labeling of plaques (Fig. 2D). In addition, substantial 6-CN-PiB binding to the vasculature was observed in DSAD, consistent with CAA. Double label studies with thioflavine S clearly show that 6-CN-PiB does not bind to neurofibrillary tangles in DSAD brain (Fig. 3A). Thioflavine S binds to the same site as Congo Red and 3H-X-34 and does not disturb 6-CN-PiB binding. At higher magnification, 6-CN-PiB appears to bind to neuritic plaques (Fig.3B). Further, 6-CN-PiB binds to the cores of plaques as thick fibrils with a halo of thioflavine S-positive amyloid fibers on the periphery (Fig. 3C).
Fig. 2.
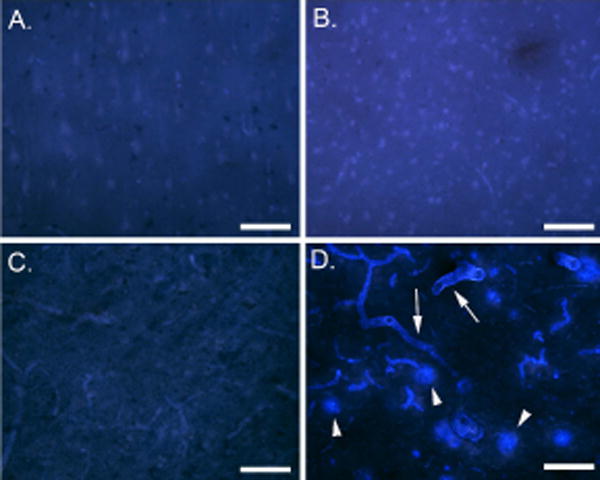
6-CN-PiB binding in the frontal cortex of DS and control cases. 6-CN-PiB binding was not observed in A. a 19-year old control case, B. a 19.8-year old DS case and, C. a 51-year old middle aged control case. In contrast, D. shows abundant 6-CN-PiB binding in plaques (arrowheads) and in CAA (arrows) in a 51.4 year old person with DS. Bar = 300 μm.
Fig. 3.
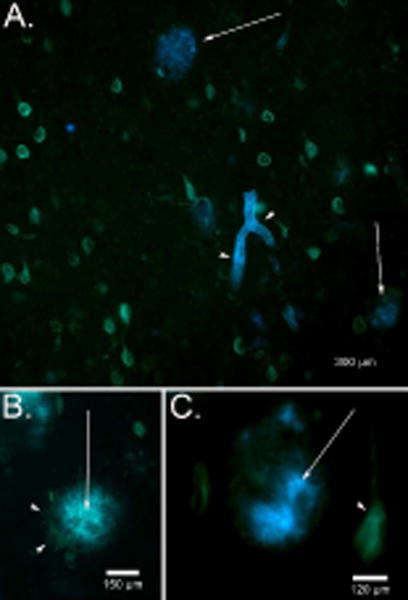
Double fluorescence labeling for 6-CN-PiB and thioflavine S in DS. (A) In a 51.4 year old DS autopsy case, 6-CN-PiB (blue) bound to plaques (arrows) and CAA (arrowheads) and could be distinguished from thioflavine S labeling of neurofibrillary tangles (green). (B) A higher magnification of a single 6-CN-PiB positive plaque (arrow) shows the presence of thioflavine S positive dystrophic neurites (arrowheads) in the periphery suggesting a neuritic plaque. (C) In another 6-CN-PiB positive plaque (arrow), fibrils can be seen forming a positive core of the plaque, whereas thioflavine S-positive fibrils were associated with the periphery (arrow) and were also present in neurofibrillary tangles (arrowheads).
4. Discussion
The current study describes age-associated high affinity specific 3H-PiB and 3H-X-34 binding in autopsy cases with DS, which expands existing PiB PET studies in vivo in people with DS to now include a broad range of ages from 1 to 66 years. Instead of the 10 μM concentrations of ligand used for histology, which will also bind to low affinity sites, we use 100 nM CN-PiB, a concentration closer to the Kd of CN-PiB for Aβ pathology and to the in vivo concentration of 11C-PiB used to visualize high affinity CN-PiB binding in human AD brain. In DS, 3H-PiB binding increases rapidly after 40 years of age with variability between individuals also increasing with age. 3H-PiB binding in DS appears to be best correlated with Aβ42 levels (SDS and FA soluble) rather than Aβ40 peptide levels based on regression analyses. 6-CN-PiB binding in autopsy cases shows a preference for fibrillar, cored, and neuritic plaques along with CAA with no binding at these low concentrations to intracellular neurofibrillary tangles (Ikonomovic, et al., 2008).
In the first report of autopsy correlates of 11C-PiB binding in vivo in sporadic AD, Bacskai and colleagues showed overlap in immunostaining for Aβ and 11C-PiB binding in vitro, including CAA (Bacskai, et al., 2007). Several studies now report 11C-PiB binding in autopsy cases showing that in vitro binding to tissue sections, PiB binds to neuritic plaques and weakly to diffuse plaques. Strong binding is observed in CAA and in some cases labeling of extracellular tangles (Ikonomovic, et al., 2012;Ikonomovic, et al., 2008;Klunk, et al., 2007). In a study of a PET 11C-PiB negative patient with mild cognitive impairment at the time of the PET scan, but elevated CSF tau and reduced CSF Aβ, numerous diffuse Aβ plaques and CAA were observed at autopsy suggesting either that there was insufficient fibrillar Aβ to bind PiB in vivo (Cairns, et al., 2009) or that not all fibrillar forms of Aβ bind to 11C-PiB. In a series of 6 older autopsy cases with PET 11C-PiB imaging, there was a positive correlation between Aβ plaque counts and extent of PiB binding (Driscoll, et al., 2012). Results from the current study also suggest that 3H-PiB binding in vitro in DS frontal cortex is associated with neuritic plaques and CAA with weak or no labeling of diffuse plaques in DS in the small set of cases we could examine. Thus, consistent with studies in sporadic AD and in non-DS autopsy cases, it may also be the case that 6-CN-PiB binds only a subset of Aβ deposits but not those within diffuse deposits in DS. Autopsy studies suggest that diffuse plaques in DS appear prior to neuritic plaques (prior to 40 years of age) (Head, et al., 2016) but PET PiB studies show a later age of onset of binding than that observed at autopsy. In combination, these results may indicate that weak PiB binding precludes diffuse plaque visualization by PET imaging. However, higher concentrations of CN-PiB in vitro may allow it to bind to diffuse plaques in autopsy tissue and assessing more cases between 20–40 years of age will be helpful in future to establish this possible outcome.
In the current study, we show a broad range in 3H-PiB binding in the frontal DS cortex that is strongly age-associated. In addition, by the inclusion of multiple measures of soluble and insoluble Aβ40 and Aβ42 as well as oligomers, regression analyses show that SDS soluble Aβ42 was the strongest predictor of PiB binding in DS but including the amount of oligomers and insoluble Aβ42 improved the prediction of PiB binding. Our study confirms and extends previous biochemical measures of PiB binding in autopsy tissue that suggest binding in vivo by PET imaging is associated with the presence of insoluble Aβ (Kadir, et al., 2011). Studies in autopsy tissue from the frontal cortex and hippocampus show significantly higher amounts of PiB binding in clinically characterized AD cases compared with age-matched clinically normal controls, although there can be some overlap (Ni, et al., 2013). Further, total Aβ40 and Aβ42 measured by ELISA also correlates with PiB binding in vitro in a case series of 5 AD and 5 control brains (Ni, et al., 2013). In a large case series that included controls, preclinical AD, and AD samples, PiB binding correlated with oligomeric Aβ, SDS soluble Aβ, and FA soluble Aβ (Beckett, et al., 2012).
The age-dependency of our biochemical measures of PiB binding in frontal cortex in DS cases suggests a dramatic increase in PiB binding after the age of 40 years, which parallels age-associated increases in insoluble Aβ42 (Cenini, et al., 2012). Previous studies of PiB imaging in people with DS have included volunteers from 25 years and older and we now provide data from autopsy samples as young as one year of age. Our in vitro binding assay data are consistent with PET 11C-PiB imaging studies in people with DS typically showing positive binding in cortex in participants over the age of 35 years (Annus, et al., 2016;Handen, et al., 2012;Hartley, et al., 2014;Landt, et al., 2011;Lao, et al., 2016). However, a caveat with our study is that we have a gap in DS autopsy cases when Aβ typically begins to accumulate (25–40 years). This can be a challenge with DS autopsy studies that are limited by the number of cases available. Further, we are not able to determine if clinical status may be a better predictor of 3H-PiB binding in vitro given that these data were not available on the current cases. Further, there as of yet is no consensus on how best to characterize severity of dementia in people with DS as standardized tests such as the MMSE, do not apply well in this cohort.
Combined with the results of the current study, PiB binding in DS frontal cortex appears to reflect primarily fibrillar Aβ42 as well as possibly extensive CAA, which may be exacerbated in DS (Wilcock, et al., 2016). However, it should be stressed that in vitro, 3H-PiB binding shows equivalent binding to both Aβ40 and Aβ42 (Klunk, et al., 2005). It may also be the case that 3H-PiB binding reflects that there is more Aβ42 than Aβ40 in brain. The significant individual variability we observe in DS autopsy studies particularly over 40 years of age may be due to the presence of dementia or to AD disease severity as suggested in a study of sporadic AD (Beckett, et al., 2012) or in disease duration.
We have focused on only the frontal cortex in the current study, which is a limitation given that the earliest signs of PET 11C-PiB binding in DS are in the striatum (Annus, et al., 2016;Handen, et al., 2012;Lao, et al., 2016). In these PET 11C-PiB binding studies, striatal binding occurs between 36–40 years of age. A similar study using striatal samples would be very interesting in this cohort of at-risk individuals and may reveal an earlier age of onset of PiB binding in vitro; it would require more cases between 20–40 years. Unfortunately, we do not as yet have frozen samples to make a similar comparison in this brain region. Future studies will evaluate both the binding of 3H-PiB in frozen samples and CN-PiB in fixed striatal samples with similar hypotheses. We may predict that striatal 3H-PiB would have an earlier age of onset of increase in concentration than frontal cortex. It would also be critical to determine if PiB is binding only to plaques and CAA in the striatum or if other pathological features may be leading to enhanced binding. Ideally, cognitively characterized autopsy cases would be included in future studies and may help account for individual variability but these cases are a challenge to acquire currently.
Acknowledgments
The authors are grateful to Dr. William Klunk at the University of Pittsburgh for providing unlabeled X-34. 6-CN-PiB was synthesized by Dr. Fanxing Zeng at the Emory University PET Center. Funding for the current study was from the National Institutes of Health (NIH)/National Institutes of Child Health and Development, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD064993 (EH, FAS), NIH R21NS080576-01A1 (HL, HPS), BrightFocus A20140445 (HL) and NIH/National Institutes on Aging (NIA) P50AG16573, NIH/NIA R01AG21912 and NIH R01HD065160 (ITL, ED). A subset of the brain tissues used here were obtained from the NIH NeuroBioBank (https://neurobiobank.nih.gov/). We thank Ms. Paula Thomason for her careful editing of this manuscript.
References
- Annus T, Wilson LR, Hong YT, Acosta-Cabronero J, Fryer TD, Cardenas-Blanco A, Smith R, Boros I, Coles JP, Aigbirhio FI, Menon DK, Zaman SH, Nestor PJ, Holland AJ. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016;12(5):538–45. doi: 10.1016/j.jalz.2015.07.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, Irizarry MC, Klunk WE, Mathis CA, Dekosky ST, Greenberg SM, Hyman BT, Growdon JH. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64(3):431–4. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Beckett TL, Niedowicz DM, Studzinski CM, Weidner AM, Webb RL, Holler CJ, Ahmed RR, LeVine H, 3rd, Murphy MP. Effects of nonsteroidal anti-inflammatory drugs on amyloid-beta pathology in mouse skeletal muscle. Neurobiology of disease. 2010;39(3):449–56. doi: 10.1016/j.nbd.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett TL, Webb RL, Niedowicz DM, Holler CJ, Matveev S, Baig I, LeVine H, 3rd, Keller JN, Murphy MP. Postmortem Pittsburgh Compound B (PiB) binding increases with Alzheimer’s disease progression. J Alzheimers Dis. 2012;32(1):127–38. doi: 10.3233/JAD-2012-120655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, Reinwald LT, Carter D, Felton A, Holtzman DM, Mintun MA, Klunk WE, Morris JC. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009;66(12):1557–62. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, LeVine H, 3rd, Lott IT, Schmitt FA, Butterfield DA, Head E. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822(2):130–8. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Klunk WE. Early detection of Alzheimer’s disease using PiB and FDG PET. Neurobiol Dis. 2014;72(Pt A):117–22. doi: 10.1016/j.nbd.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, Kraut MA, Ferrucci L, Mathis CA, Klunk WE, O’Brien RJ, Davatzikos C, Wong DF, Resnick SM. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol. 2012;124(6):823–31. doi: 10.1007/s00401-012-1025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8(6):496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SL, Handen BL, Devenny DA, Hardison R, Mihaila I, Price JC, Cohen AD, Klunk WE, Mailick MR, Johnson SC, Christian BT. Cognitive functioning in relation to brain amyloid-beta in healthy adults with Down syndrome. Brain. 2014;137(Pt 9):2556–63. doi: 10.1093/brain/awu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E, Lott IT, Wilcock DM, Lemere CA. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr Alzheimer Res. 2016;13(1):18–29. doi: 10.2174/1567205012666151020114607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Perl DP, Sparks DL, Mehta N, Morrison JH. Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down’s syndrome. Arch Neurol. 1995;52:379–91. doi: 10.1001/archneur.1995.00540280065020. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Price JC, Hamilton RL, Mathis CA, Paljug WR, Debnath ML, Cohen AD, Mizukami K, DeKosky ST, Lopez OL, Klunk WE. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123(3):433–47. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(4):a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadir A, Marutle A, Gonzalez D, Scholl M, Almkvist O, Mousavi M, Mustafiz T, Darreh-Shori T, Nennesmo I, Nordberg A. Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain. 2011;134(Pt 1):301–17. doi: 10.1093/brain/awq349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25(46):10598–606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27(23):6174–84. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen J, Verkkoniemi A, Aalto S, Paetau A, Ahonen JP, Viitanen M, Nagren K, Rokka J, Haaparanta M, Kalimo H, Rinne JO. PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain. 2008;131(Pt 7):1845–53. doi: 10.1093/brain/awn107. [DOI] [PubMed] [Google Scholar]
- Lai F, Williams MD. A prospective study of Alzheimer Disease in Down Syndrome. Arch Neurol-Chicago. 1989;46:849–53. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- Landt J, D’Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, Hong YT, Menon DK, Baron JC, Zaman SH. Using positron emission tomography and Carbon 11-labeled Pittsburgh Compound B to image Brain Fibrillar beta-amyloid in adults with down syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68(7):890–6. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- Lao PJ, Betthauser TJ, Hillmer AT, Price JC, Klunk WE, Mihaila I, Higgins AT, Bulova PD, Hartley SL, Hardison R, Tumuluru RV, Murali D, Mathis CA, Cohen AD, Barnhart TE, Devenny DA, Mailick MR, Johnson SC, Handen BL, Christian BT. The effects of normal aging on amyloid-beta deposition in nondemented adults with Down syndrome as imaged by carbon 11-labeled Pittsburgh compound B. Alzheimers Dement. 2016;12(4):380–90. doi: 10.1016/j.jalz.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz JB, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: A regional quantitative analysis. Experimental Neurology. 1998;150:296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–21. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9(6):623–33. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46(13):2740–54. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Matveev SV, Kwiatkowski S, Sviripa VM, Fazio RC, Watt DS, LeVine H., 3rd Tritium-labeled (E,E)-2,5-bis(4′-hydroxy-3′-carboxystyryl)benzene as a probe for beta-amyloid fibrils. Bioorganic & medicinal chemistry letters. 2014;24(23):5534–6. doi: 10.1016/j.bmcl.2014.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev SV, Spielmann HP, Metts BM, Chen J, Onono F, Zhu H, Scheff SW, Walker LC, LeVine H., 3rd A distinct subfraction of Abeta is responsible for the high-affinity Pittsburgh compound B-binding site in Alzheimer’s disease brain. J Neurochem. 2014;131(3):356–68. doi: 10.1111/jnc.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Ni R, Gillberg PG, Bergfors A, Marutle A, Nordberg A. Amyloid tracers detect multiple binding sites in Alzheimer’s disease brain tissue. Brain. 2013;136(Pt 7):2217–27. doi: 10.1093/brain/awt142. [DOI] [PubMed] [Google Scholar]
- Schupf N, Sergievsky GH. Genetic and host factors for dementia in Down’s syndrome. British journal of psychiatry. 2002;180:405–10. doi: 10.1192/bjp.180.5.405. [DOI] [PubMed] [Google Scholar]
- Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron. 2014;84(3):608–22. doi: 10.1016/j.neuron.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Ataka S, Mizuno T, Brooks WS, Wada Y, Kondo M, Jones G, Watanabe Y, Mulligan R, Nakagawa M, Miki T, Shimada H, O’Keefe GJ, Masters CL, Mori H, Rowe CC. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66(12):1537–44. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Schmitt FA, Head E. Cerebrovascular contributions to aging and Alzheimer’s disease in Down syndrome. Biochim Biophys Acta. 2016;1862(5):909–14. doi: 10.1016/j.bbadis.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski K, Wisniewski H, Wen G. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17:278–82. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]