

Abstract
CD4 glycoprotein on the surface of T cells helps in the immune response and is the receptor for HIV infection. The structure of a soluble fragment of CD4 determined at 2.3 Å resolution reveals that the molecule has two intimately associated immunoglobulin-like domains. Residues implicated in HIV recognition by analysis of mutants and antibody binding are salient features in domain D1. Domain D2 is distinguished by a variation on the β-strand topologies of antibody domains and by an intra-sheet disulphide bridge.
CD4, a cell-surface glycoprotein found primarily on T lymphocytes, is required to shape the T-cell repertoire during thymic development and to permit appropriate activation of mature T cells1. T cells that recognize antigens associated with class II major histocompatibility complex (MHC) molecules, mainly T helper cells, express CD4. Evidence is accumulating that CD4 and the T-cell receptor coordinately engage class II molecules on antigen-presenting cells to mediate an efficient cellular immune response, and that engaged CD4 may transmit a signal to an associated cytoplasmic tyrosine kinase, p56lck.
CD4 belongs to the immunoglobulin superfamily of molecules which generally serve in recognition processes2,3. The sequence of CD44,5 indicates that it consists of a large (~370 residues) extracellular segment composed of four tandem immunoglobulin-like domains, a single transmembrane span, and a short (38 residues) C-terminal cytoplasmic tail. The first domain (D1) shares several features with immunoglobulin variable domains, but the sequence similarities between immunoglobulins and the other extracellular domains (D2, D3 and D4) are more remote.
In humans, CD4 can be subverted from its normal immuno-supportive role to become the receptor for infection by the human immunodeficiency virus (HIV)1,6,7. Recombinant soluble CD4 proteins bind to the HIV envelope glycoprotein gp120, and can thus inhibit viral infection and virus-mediated cell fusion in vitro (refs8, 9 and references therein). Domain D1 suffices for high-affinity binding to gp120 (ref. 8), and the analysis of substitution mutants further limits the sites of interaction to discrete regions in the domain8,10–18.
Crystals of whole soluble CD4 (sCD4) molecules have been grown19,20 but there has been limited success in achieving adequate diffraction order. The high solvent content and weak diffraction of several characterized polymorphs of human sCD4 are compatible with an extended, flexible molecule19. From the pattern of proteolytic cleavages that generate stable fragments (refs21–23 and unpublished results), the main flexibility seems to be at the D2 to D3 junction. We have now crystallized a truncated derivative of CD4 that diffracts well, and here we report its atomic structure. This recombinant fragment8 as secreted from Chinese hamster ovary (CHO) cells consists of residues 1–183 of human CD4 plus two missense residues, Asp-Thr; and it is unglycosylated. This molecule, which we refer to as D1D2, is as active as sCD4 in binding to gp120 (dissociation constant Kd ≃ 3 nM) and retains all antibody epitopes mapped to these domains of CD4 (ref.8 and unpublished results). Others have crystallized similar fragments from the N-terminal half of sCD424,25 and the structure of one is reported in the accompanying paper25.
Here we describe the D1D2 structure in comparison with that of immunoglobulin domains, provide a geometrical definition for HIV recognition sites, and discuss implications of the structure for normal CD4 function and evolution of the immunoglobulin family. We find that the domains of CD4 are indeed immunoglobulin-like, although there are significant differences from the antibody analogues. The primary sites for HIV interaction are on loops that protrude from the variable-like D1 domain in analogy with immunoglobin complementarity-determining regions (CDRs). The D2 domain, which is intimately associated with D1, resembles constant domains but it is distinguished by a strand topology that is variable-like.
Structure determination
D1D2 protein secreted from CHO cells was purified to homogeneity by following procedures similar to those used for sCD4 (ref.26). Crystals of this protein grown from polyethylene glycol (PEG) and stabilized at pH 8.2 (Table 1) belong to space group C2 and have unit cell dimensions of a = 83.71 Å, b = 30.07 Å, c = 87.54 Å, β = 117.3°. They contain one D1D2 molecule per asymmetric unit and 50% solvent. In searching for derivatives, we soaked crystals into stabilization medium doped with various heavy-atom compounds.
TABLE 1.
Statistics from the crystallographic analysis
| (a) Diffraction data | ||||||
| Derivative | dmin(Å) | X-ray line/wavelength (Å) | No. of measurements | No. of reflections | Data coverage (%) | Rmerge |
| Native | 2.3 | CuKα/1.542 | 24,407 | 7,882 | 92 | 0.061 |
| K2Pt(NO2)4 | 2.5 | CuKα/1.542 | 40,130 | 6,757 (12,667) | 99 (93) | 0.057 (0.053) |
| 2.7 | AuLβ1/1.084 | 33,914 | 5,314 (9,978) | 97 (97) | 0.069 (0.062) | |
| 2.5 | AuLβ2/1.068 | 43,005 | 6,639 (12,505) | 97 (92) | 0.072 (0.065) | |
| 2.7 | AuLα1/1.276 | 30,724 | 5,432 (10,210) | 99(99) | 0.062 (0.054) | |
| K2PtCI6 | 2.8 | CuKα/1.542 | 22,033 | 4,786 (8,751) | 96 (88) | 0.094 (0.086) |
| (b) MIR analysis | ||||||||||
| dmin (Å) | 11.1 | 7.7 | 5.9 | 4.8 | 4.0 | 3.4 | 3.0 | 2.7 | Total | |
| K2Pt(NO2)4 | ||||||||||
| r.m.s. fH/Eiso | 2.18 | 1.69 | 2.01 | 1.48 | 1.04 | 1.12 | 1.24 | 1.29 | 1.32 | |
| r.m.s. ΔFcalc/Eano | 0.85 | 0.88 | 1.08 | 1.08 | 0.79 | 0.74 | 0.49 | 0.37 | 0.63 | |
| K2PtCI6 | ||||||||||
| r.m.s. fH/Eiso | 1.54 | 1.72 | 1.92 | 1.52 | 1.13 | 1.19 | 1.23 | — | 1.35 | |
| r.m.s. ΔFcalc/Eano | 0.45 | 0.54 | 0.58 | 0.39 | 0.24 | 0.21 | 0.14 | — | 0.23 | |
|
|
0.81 | 0.71 | 0.75 | 0.71 | 0.58 | 0.57 | 0.51 | 0.37 | 0.53 |
| (c) MAD analysis | (d) Solvent flattening | ||||||||
| Observed ratio | Shell/cycle | R | Δϕ | ||||||
| λ (Å) | 1.542 | 1.084 | 1.068 | 1.276 | 1/1 | 0.31 | 28.2 | ||
| 1/5 | 0.24 | 35.5 | |||||||
| 1.542 | 0.043 | 0.066 | 0.057 | 0.069 | 3/5 | 0.23 | 37.0 | ||
| 1.084 | 0.036 | 0.029 | 0.030 | 7/5 | 0.22 | 43.8 | |||
| 1.068 | 0.038 | 0.036 | 13/10 | 0.21 | 63.4 | ||||
| 1.276 | 0.037 |
| (e) Refinement | |||||||
| Step | Program | Data range (Å) | B-value refinement | Rebuilding | H2O | R | Δd (Å) |
| 0 | PROLSQ | 10.0–2.7 | No | No | 0 | 0.447 | 0.044 |
| 1 | PROLSQ | 5.0–2.7 | No | No | 0 | 0.271 | 0.025 |
| 2 | PROLSQ | 10.0–2.3 | Yes | Yes | 0 | 0.273 | 0.024 |
| 3 | XPLOR | 10.0–2.3 | No | Yes | 0 | 0.268 | 0.024 |
| 4 | PROLSQ | 10.0–2.3 | Yes | Yes | 0 | 0.250 | 0.023 |
| 5 | PROLSQ | 10.0–2.3 | Yes | Yes | 106 | 0.208 | 0.020 |
| 6 | XPLOR/PROLSQ | 10.0–2.3 | Yes | Yes | 72 | 0.196 | 0.018 |
Crystallization. D1D2 was purified in sequential steps of ion-exchange chromatography26 involving S-Sepharose capture at pH 5.2, Q-Sepharose passage at pH 9.0 and a final elution from S-Sepharose at pH 6.0 in 0.6 M NaCI. Crystallization was carried out at 20 °C by equilibration against reservoirs of 20–25% PEG 3350 starting from hanging droplets composed of protein at ~20 mg ml−1 in PBS at pH 7.0 and equal volumes from the reservoir. Collected crystals were transferred to a stabilization medium (30% PEG 3350, 80 mM Tris buffer, pH 8.2). Typical Crystals have dimensions of 0.5 × 0.3 × 0.2 mm. Diffraction data for MIR phasing and for refinement were measured with CuKα X rays generated by a Rigaku RU200 rotating anode and detected on two multiwire chambers on the Mark III system at UCSD. All data were collected at or near 20 °C. The inverse beam method was used to collect Bijvoet mates; that is, settings were changed from (ω, χ, ϕ) to (ω, −χ, ϕ + π) after rotations at each crystal orientation. Data for MAD phasing were collected on the Mark II area detector system. A graphite monochromator was used to select specific characteristic lines produced from a gold-plated anode installed on an Elliot GX-6 generator. The monochromator was tuned to 50% intensity at one side or the other of the unresolved Lβ1/Lβ2 doublet for selection of a primary monochromatic component. The UCSD area detector processing package was used for the reduction of all data. Values given inside of parentheses for numbers of reflections and Rmerge are without merging of Friedel mates (Rmerge=Σ|/obs−/avg|/Σ/avg) MIR analysis. Heavy-atom positions were determined from Patterson syntheses. The refinement of heavy-atom parameters and the phase calculations were performed with a version of program PHARE modified by Z. Otwinowski (personal communication) for an improved error treatment. Relative occupancies for the K2Pt(NO2)4 and K2PtCI6 derivatives were (1.00,0.94) and (0.53, 0.51, 0.25, 0.22, 0.19), respectively. The first two heavy-atom sites of the K2PtCI6 derivative are in common with the sites of the K2Pt(NO2)4 derivative. Parameters cited in the table are fH calculated heavy-atom structure-factor contributions; ΔFcalc, calculated Bijvoet difference; Eiso, r.m.s. isomorphous closure error; Eano, r.m.s. anomalous closure error; and , mean figure-of-merit. MAD analysis. The MADSYS system of programs54 was used to produce initial MAD phases. Phase combination between MIR and MAD was performed by the combination of ABCD coefficients55. But, in this case it was necessary to modify the original MAD refinement formulation54 so as to produce phases for the native non-anomalously scattering component56 of the platinyl CD4 complex. The previously used method for extracting ABCD coefficients55 proved to be ineffective in this case; instead, these phasing coefficients were extracted directly from MAD phases and figures-of-merit57. MIR and MAD phases were combined at 20.0–3.5 Å; MIR phases were used alone for the 3.5–2.7 Å data. Observed anomalous diffraction difference ratios are given for the data between 20.0–3.5 Å. Bijvoet (diagonal elements of matrix) and dispersive (off-diagonal elements) difference ratios represent r.m.s. (ΔF±h)/r.m.s. (F) and r.m.s. (ΔFΔλ/r.m.s. (F), respectively. Solvent flattening was performed following procedures used in the structure analysis of myohemerythrin58. After interpretation of the first domain, a molecular envelope was delineated which included 58% of the asymmetric unit of the crystal (as compared with 50% for the true solvent content) to ensure that ail protein density was included. After subtraction of the mean solvent density and truncation of the lowest density values within the molecular boundary, the contents of the modified density function were Fourier-inverted. Phasing coefficients from the inversion were reduced by 0.5 before combination with ABCD values from MIR. A truncation level to eliminate the lowest 20% of molecular points (as compared with 0% and 10%) was chosen as giving the best definition for known features in domain D1. The phase refinement was carried out starting from the 20.0–4.5 Å shell, and including higher angle reflections successively until finally reaching 2.7 Å spacings. Five cycles of solvent flattening were carried out for each shell between shell 1 and shell 12, and 10 cycles were run for shell 13. R, Crystallographic R value between observed structure factor magnitudes and calculated values from the solvent-flattened map. Δϕ, Phase differences between phases from MIR plus MAD and phases from the solvent flattened map. Refinement. Selected points along the course of refinement are listed. For all steps, the data having Fobs > 4σ(Fobs) were included. Step 0 refers to the unrefined starting model after adjustment of the scale factor only. From step 2 onward, the data between 10.0 and 5.0 Å were included which helped to achieve better connectivities in 2Fobs − Fcalc maps. The final model at 2.3 Å resolution is based on the 5,689 reflections (64%) which met the 4σ criterion. This partially refined model includes the 1,420 atoms from residues 1–173 plus the 72 water sites. Thermal parameters were restrained as typified by an r.m.s. discrepancy of 1.17 Å in B values between bonded main-chain atoms. R=Σ |Fobs−Fcalc|/Σ Fobs; Δd, r.m.s. bond deviation from ideality (Å).
Diffraction data for the structure determination (Table 1) were measured at an area detector facility27 where we collected CuKα data for the native protein and for candidate derivatives. Observable intensities could be recorded to 1.9 Å spacings from fresh native crystals, and even more strongly from the K2Pt(NO2)4 derivative. We could also measure multiwavelength anomalous diffraction (MAD) data from this platinum derivative using characteristic gold emission lines which bracket the PtLIII absorption edge28. Phases were evaluated by both multiple isomorphous replacement (MIR) and MAD methods (Table 1). Electron density maps from MIR phasing (including anomalous data) at 2.7 Å resolution and from MAD phasing at 3.5 Å resolution both showed similar features. We then probabilistically combined MIR and MAD phase information to produce a combined map at 2.7 Å resolution (Fig. 1a) which showed distinct solvent channels and an apparent two-domain structure. A complete chain-tracing consistent with the amino-acid sequence for the first domain was possible, but the second domain remained difficult to interpret. By using a hand-drawn molecular envelope, we then performed solvent flattening and density truncation to improve the map further (Fig. 1b). This enabled us to trace the chain through the second domain. The C-terminal 12 residues (174–185) could not be seen.
FIG. 1.

Electron-density distributions used in the structural determination. The portion displayed in each panel includes the segment Phe 26-His 27- Trp28-Lys29 with the partially refined model 5 (yellow) superimposed on the density (blue), a, Experimental map at 2.7 Å resolution based on combined MIR plus MAD phase information; = 0.58. b, Experimental map at 2.7 Å resolution after phase refinement by solvent flattening and density truncation; = 0.86. c, Refined 2|Fobs|−|Fcalc| map with model phases after refinement at 2.3 Å resolution; R = 0.208.
A complete model was built into the solvent-flattened map as displayed by FRODO29 program on a graphic workstation. Fragments from well-refined structures were fitted onto Cα guide points measured from a minimap and used as the starting point for building. Refinement made use of PROLSQ30 and XPLOR31 programs and included several manual rebuildings. Resolution of the analysis was gradually extended to 2.3 Å. After analysis of model 5 (Table 1) at R = 0.208, we discovered on comparing manuscripts that this model and one developed by Wang et al.25 differed in the alignments of sequence in two strands (E of D1 and B of D2) and in conformations at residues 1, 103–108, 134–139 and 151–154. We then tested a model with residues 66–73 and 112–120 positioned in the former places of 64–71 and 114–122, respectively, and with the 134–139 region also rebuilt. Further refinement produced model 6 (Table 1) with an R value of 0.196 and stereochemical ideality typified by an r.m.s. deviation of 0.018 Å from ideal bond lengths. Study of this still incompletely refined model confirms the revised alignment in D1 and supports the changes made in D2, although several loops in D2 remain ill-defined. All conclusions drawn from model 5 remain unchanged, but the precise numerical quantities and Figs 2b, 4 and 5a were redone from model 6.
FIG. 2.
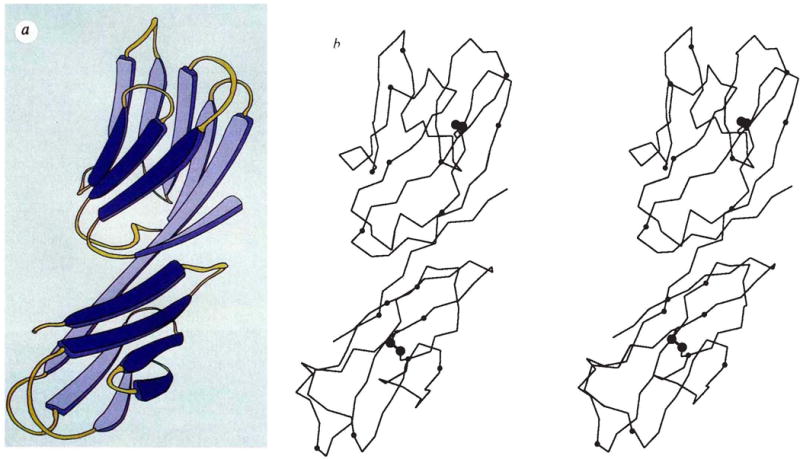
Backbone structure of the D1D2 fragment of CD4. a, Schematic diagram (copyright Yarmolinski and Hendrickson, 1990). b, Stereodiagram of the α-carbon backbone. Positions of residue numbers divisible by ten are indicated by small spheres, sulphur atoms in disulphide bridges are indicated by larger spheres. The point of view is down the Z″ axis after rotations about X(100°), Z′(−30°), Y′(−90°) and X″ (10°) starting with the frame having X, Y, Z along a, b and c*.
FIG. 4.
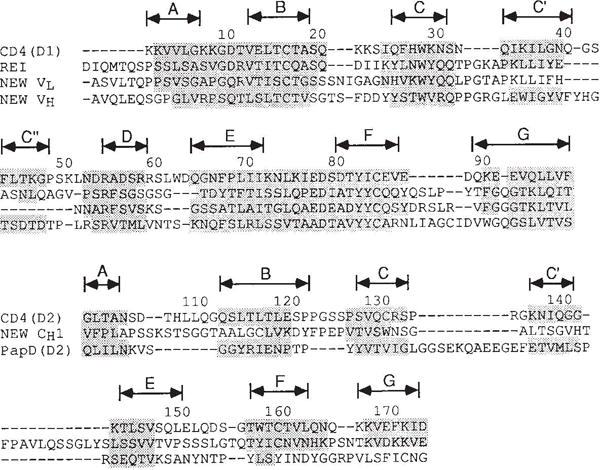
Structural alignment of the amino-acid sequences of CD4 domains with other immunoglobulin-related domains. Shaded residues have Cα positions within 2.5 Å of corresponding CD4 positions after optimal superposition of all shaded residues for a given pair of domains. (Exceptions up to 2.60 Å were allowed for residues in the middle of strands.) Each superposition relates a certain number of positions, N, within the specified 2.5 Å limit, and these match at a certain r.m.s. discrepancy, Δ. For the match of D1 with Rei (VLκ), N = 72 and Δ = 1.22 Å; for D1 versus New (VLλ), N = 66 and Δ = 1.12 Å; and for D1 versus. New (VH), N = 68 and Δ = 1.13 Å. For the match of D2 with New (CH1), N = 33 and Δ = 1.59 Å; for D2 versus PapD(D2), N = 32 and Δ = 1.26 Å; for D2 versus PapD(D1), N = 39 and Δ = 1.55 Å; and for D2 versus Rei (VLκ), N = 30 and Δ = 1.68 Å. The alignments of D2 versus PapD(D1) and D2 versus Rei (VLκ) are not shown.
FIG. 5.
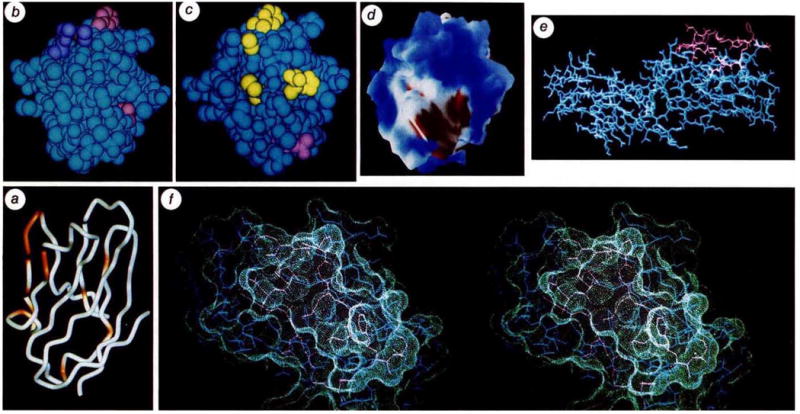
Images of CD4 relating to sites of interaction with HIV. a, Cα backbone of D1 with sites implicated by mutational and structural analysis to affect high-affinity gp120 binding (see Table 2). Non-buried residues that show reduction in binding without global disruption of structure (as judged by antibody binding) are coloured red-orange (29, 41, 42, 43, 44, 45, 47, 49, 52, 58, 59, 77, 81 and 85) on a backbone drawn by WORM (L. Andrews). The orientation is as in Figs 2 and 3. b, Van der Waals’ surface of D1 with side chains of gp120-sensitive residues coloured. Those showing marked reductions are in pink (43, 44, 85, are visible) and those showing moderate effects are in purple (59, 42, are visible). The point of view is from above a, looking down at the tips of CDR-like loops, c, Van der Waals’ surface of D1 with side chains of antibody epitopes coloured. Residues identified with epitopes of the Leu3a family and shown in purple (24, 25, 27, 42, 43, visible) and those identified with the L71 family are shown in yellow (88, 89, visible). The view is roughly as in b. d, Electrostatic potential surface computed with DELPHI33 at neutral pH, and displayed with AAK (A. Nicholls and B. Honig) at the levels of the solvent-accessible surface with a probe radius of 1.4 Å. Blue represents positive potential, red negative, and white neutral. The negative patch is associated with residues 85, 87 and 88. The view is approximately as in b. e, An all-atom representation of D1D2. Residues in the gp120-binding region from residue 41 to 59 are drawn in red. The direction of view is roughly as in Fig. 2, but the molecule has been rotated by ~90° about this view axis. This view illustrates the exposed nature of Phe 43. The actual conformation of this phenyl side chain in the crystal is at least partly determined by lattice interactions, but its highly exposed nature would also be expected to persist in other conformations accessible in solution, f, Stereoview of the molecular surface in the major gp120-binding region of D1. Atoms are drawn as in e, and are enveloped by the surface in contact with a probe sphere of 1.4 Å radius as displayed by QUANTA (Polygen). The view is taken after rotation from e by ~90° about the horizontal axis.
Overall structure
The D1D2 structure is of the all-β type. It consists of two intimately associated domains, each of which is a β sandwich folded with immunoglobulin-strand topology. The polypeptide folding is shown in Fig. 2. The first domain (D1) comprises residues 1–98 disposed in nine β strands, and the second domain (D2) contains residues 99–173 and has seven β strands. These domain boundaries are in striking correspondence with intron boundaries immediately after residues 100 and 177 in the gene structure5. The suggestion that J-like regions follow these introns is not borne out by the structure.
The last strand of D1 continues straight into the first strand of D2, running for a length of 49 Å from residues 88–103. This tandem association of domains leads to a rod-shaped molecule of dimensions roughly 25 × 25 × 60 Å. By comparing the solvent-accessible surface area of D1D2 with that of the separated domains, we found that 310 Å2 from each domain is buried in the interface. This compares with 110 Å2 for the VH-CH interface of Fab New32. Interdomain contacts are largely hydrophobic. Residues involved in interdomain contacts include Val 3, Leu 5, Ile 76, Leu 96, Val 97 and Phe 98 from D1, and Glu 119, Pro 121, Gln 163, Val 168 and Phe 170 from D2. The surface of D1D2 has many positively charged amino acids (the calculated pI of residues 1–183 is 10.0), and the electrostatic potential surface33 computed from the model is mostly positive at neutral pH. There are, however, two prominent patches of negative potential: one is associated with Glu 85, Glu 87 and Asp 88 and the other is from Asp 105, Asp 153 and Asp 173.
The D1D2 crystal lattice contains diad axes that could accommodate a dimeric molecule. However, contacts across screw axes dominate the lattice interactions and the only diad-mediated interface (between D1 domains) seems unlikely to persist in solution. The lattice interactions of D1 are more extensive than those of D2 which is reflected in molecular mobilities of the domains: the average B value is 19.9 Å2 for D1 compared with 37.8 Å2 for D2. This accounts for our greater difficulty in tracing the D2 chain.
CD4, like many members of the immunoglobulin superfamily, is a single-chain cell-surface receptor composed of tandemly repeated domains. Some of these domains have been assigned as V-like and others as C-like or in the ‘C2 set’34. The D1D2 domain of CD4 contains both types and, apart from the similarity of CD5 with the PapD bacterial chaperone protein35, this is the first three-dimensional structure for such members of the family.
Variable-like domain D1
As anticipated by sequence comparisons, D1 is similar to the variable (V) domains of antibodies. A schematic drawing of the folding pattern with standard immunoglobulin designations for the secondary structural elements is shown in Fig. 3. Strands B, D and E make up one β sheet, and strands A, C, C′, C″, F and G are in the other sheet of the β sandwich. In immunoglobulin Vdomains, the first half of strand A is hydrogen-bonded to strand B of the outer sheet but the second half switches to the inner sheet. Strand A of D1 is foreshortened and occupies only the ‘inner-sheet’ position, hydrogen-bonded in parallel with strand G. D1 preserves the inter-sheet disulphide bridge and several other elements of the hydrophobic core characteristic of the immunoglobulin framework. Starting from these alignments we have superimposed D1 onto representative immunoglobulin V domains from Bence-Jones protein Rei (VLκ)36, and Fab New (VLλ and VH)32. The structurally aligned sequences are shown in Fig. 4. D1 superimposes remarkably well onto the β-sheet framework of all V domains, with a best match to Rei bringing 72 Cα atoms to an r.m.s. discrepancy of 1.22 Å from exact superposition.
FIG. 3.
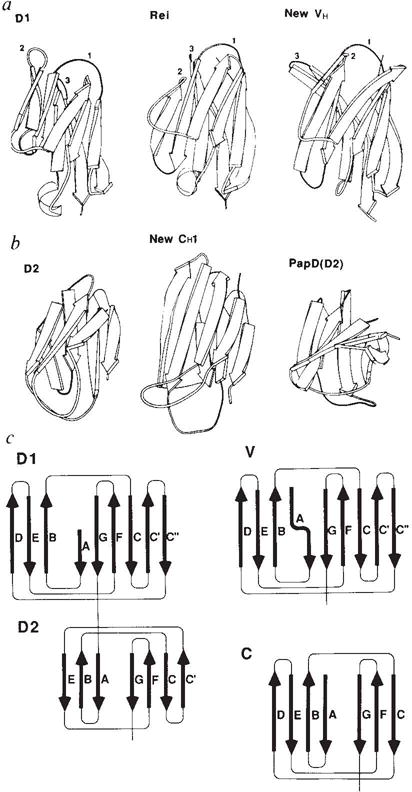
Schematic comparisons between domains of CD4 and other immunoglobulin-folded domains, a, Ribbon diagrams comparing D1 with a variable λ light-chain domain and a variable heavy-chain domain. The point of view is as in Fig. 2; the CDR loops of immunoglobulins and their analogues in D1 are indicated by numerals, b, Ribbon-diagram comparisons of D2 with a constant domain and with domain D2 of the bacterial chaperone protein PapD. c, Topology diagrams and strand nomenclature for the β sheets in CD4 and in variable and constant domains of immunoglobulins.
In striking contrast with the conserved core of β strands, several loops between strands in CD4 are quite different from those in immunoglobulin V domains. In particular, loop CC′ is shortened by four residues and loop FG (immunoglobulin CDR3) is shortened by four to six residues from canonical immunoglobulin lengths. These loops mediate VH-VL dimerization in immunoglobulins, an interaction that does not occur with CD4. One addition, important for gp 120 binding, is the lengthening of loop C′C″ (immunoglobulin CDR2) by three residues in CD4 compared with κ light chains such as Rei. In this regard, CD4 more closely resembles VH domains even though overall it superposes best with Rei. This CDR2-like loop interacts with Trp 62, not found in antibodies, and juts out at the tip. Few of the loop changes in D1 were detected in previous alignments with Rei, and consequently structural predictions based on Rei37 have failed to capture the essence of CD4. The salience of the CDR2 analogue of CD4 and the diminutive nature of the CC′ loop and the CDR3 analogue are evident in Figs 2 and 3.
Distinctive domain D2
Domain D2 of CD4 has a tertiary folding that can readily be recognized as resembling immunoglobulin constant domains. Again following standard immunoglobulin nomenclature (Fig. 3), one β sheet of the sandwich-like structure contains strands A, B and E, and the other sheet has strands C, C′, F and G. Despite the general similarity of D2 to constant domains, details of the folding are considerably divergent. First, the size of D2 is small (75 residues) compared with that of immunoglobulin constant domains (~100 residues). This manifests itself in shorter strand lengths. A second variation is that strand C′, which corresponds sequentially to the D strand of normal constant domains, joins the β sheet consisting of strands C, F and G, rather than being hydrogen-bonded to strand E in the other sheet. In this respect, D2 is V-like. A final striking difference from immunoglobulins is that D2 has its disulphide bond between strands in the same sheet, that is strands C and F (Fig. 2), rather than between sheets as is usual. This feature, which is unusual but not unprecedented38, had been predicted34; however, strand C′ was not anticipated. The hydrophobic core of immunoglobulin domains is preserved in D2 despite the unusual disulphide connection (Fig. 4). Leu 116 on strand B occupies the position of a normal cysteine partner for Cys 159 on strand F. The inward orientation of the intra-sheet disulphide bridge makes it an important member of the hydrophobic core even though it does not join the sheets.
D2 can be structurally aligned with the CH1 domain of Fab New (Fig. 4), but the resulting spatial superposition is not as close as for D1 comparisons with V domains. Only six of the seven strands superimpose at a 2.5 Å stringency for matches, and this gives 33 matches with an r.m.s. discrepancy of 1.59 Å. D2 superimposes somewhat less well with the Rei variable domain (30 matches within 2.5 Å for 1.68 Å r.m.s. discrepancy). In fact, D2 is actually more similar to the domains of PapD35 than to immunoglobulin domains. As in the D2 topology, PapD domains also exhibit sheet switching (partially in D1(PapD), fully in D2(PapD)), and 39 Cα atoms of D2 (CD4) superimpose on D1(PapD) with an r.m.s. discrepancy of 1.55 Å. Drawings of the aligned structures are shown in Fig. 3.
Binding site for gp120
The initial molecular event in HIV infection is the binding of gp120 on the viral envelope to CD4 on the cell surface. The high affinity of this interaction, at least for several laboratory strains of the HIV-1 virus39, has permitted a detailed mapping of the binding site. More than 200 mutant CD4 recombinants have been constructed and tested for gp120 affinity (refs8, 10–18, J. Arthos et al., manuscript submitted, and unpublished results). Some of these mutant proteins have also been used to map the CD4 epitopes of a battery of monoclonal antibodies. In Table 2 we distil the mutation results to those point mutants in D1 which impair gp120 binding without causing extensive disruptions of structure as judged by antibody binding. Analyses have been reported for mutations at 80 of the 98 residues in D1, but only 19 positions among them have impact on gp120 binding without apparent global conformational change. Thirteen of these are in the span from 38 to 59. Completely buried residues can be discounted as direct participants in binding because their exposure would require major conformational change, which is without precedent for immunoglobulin domains. We have calculated the degree of exposure for each residue in D1 (Table 2), and find that three of the gp120-sensitive residues, Gly 38, Ala 55 and Phe 67, are inaccessible. It seems unlikely that either Lys 29 or Gln 64 is critical for binding because there is no effect for mutations at the more exposed neighbours of Lys 29 or for single or multiple mutations at 64, except alanine. Thus only the alanine replacements at 77, 81 and 85 fall outside of the 41–59 span. The distribution of binding mutants is shown in Fig. 5 a, b.
Table 2.
Residues implicated in HIV recognition by point mutation in D1 of CD4*

|
Within the 98 amino-acid residues of the D1 domain, there is no mutational information on residues 3–6, 12, 14, 16, 18, 26, 69–71, 76, 83, 84 and 97. However, the binding of gp120 to Rhesus CD410 and to the human–rat chimaeric protein eliminates 12, 14, 18 and 76 from this list. Mutations which caused global alteration of structure, as evidenced by reduced binding of several anti-CD4 monoclonal antibodies, were not considered15,16. Such mutations were the only data for 13 amino acids and thus led to exclusion of residues 7, 13, 28, 36, 37, 54, 57, 62, 78, 79, 82, 93 and 95. Thus, in total, 25 amino acids are excluded from consideration,
Fractional solvent accessibility was calculated as the ratio of solvent-accessible surface area for atoms of an amino-acid residue X in the protein to that area obtained after reducing the structure to a Gly-X-Gly tripeptide59. Values cited are for side-chain atoms except in the case of glycine residues where main-chain values are given.
Mutants are identified by single-letter code. Mutational effects are symbolized by. blackening in steps proportional to the quantitational sensitivity of the assays as reported. Only single amino-acid substitutions are included. The results from double substitutions largely confirm these data. Certain double mutations are of particular note: A, G41S/F43C severely disrupts binding15, emphasizing the significance of F43; B, L51M/A55S does not affect binding15, indicating that the severe effect by the A55F substitution is conformational in nature; C, E87G/K90G15 and D88N/Q89R8 do not affect binding, further indicating that this region is not involved in initial binding to g120.
Relative effects of the Indicated mutations on virus10 or gp12015 binding to cell surface CD4 or gp120 binding to soluble CD4 proteins8,12,16 The results shown are an interpretation of the primary data and comparisons among the results from the different laboratories are approximate.
Relative ability of cells bearing CD4 mutant proteins to form syncytium with cells expressing viral envelope proteins10,17 or of soluble CD4 proteins to inhibit syncytium8.
The Q40A mutation increased binding affinity to gp120 twofold.
The importance of the 41–59 region in HIV binding is corroborated by a chimaeric CD4 in which the human sequence from 36 to 62 has been inserted into rat CD4 (A. F. Williams, personal communication). This imparts full gp120 binding activity on the normally unreactive rat CD4 molecule. It also shows that 33 additional side chains which differ in rat and human CD4, including Gln 64, are not absolutely essential for the binding4. But, Glu 77, Thr 81 and Glu 85 are among the residues conserved between rodents and primates. Thus, whereas the 41–59 region is positively identified as being involved in gp120 binding, participation of the 77–85 region cannot be excluded.
Antibody binding studies also help to define the gp120 binding site (refs10, 40, and our unpublished results), pointing especially to the CDR2-like region. Two major groups of D1-binding monoclonal antibodies have been described and their epitopes are illustrated in Fig. 5c. The epitope of one cross-blocking group, typified by Leu3a, maps to the CDR2-like loop and usually includes portions of the CDR1-like loop. These antibodies also block gp120 binding. The epitope of the second group, typified by monoclonal L71, includes the CDR3-like loop and sometimes a portion of the strand G. These antibodies inhibit viral infection and cell fusion, but do not efficiently block gp120 binding; some even form ternary complexes with gp120 and CD4 (A. Trunch et al., manuscript submitted).
The CDR2-like region that is strongly implicated in gp120 binding is also very prominent in the CD4 structure (Figs 3 and 5). The C′–C″ hairpin loop at Gln 40–Phe 43 is almost completely exposed. Most strikingly, the hydrophobic side chain of Phe 43 juts out into the solvent as shown in Fig. 5e, f. The involvement of a phenyl group in binding is also suggested by peptide inhibitor studies41,42. If the 77–85 span were also to be directly involved in gp120 binding, this would present a puzzle because this site is on a face nearly opposite from the CDR2-like region (the vector from the molecular centre of D1 through Cα43 makes an angle of 152° with the similar vector through Cα81), and no intervening residues have been implicated in gp120 recognition.
Fusion determinants
After the initial binding event, entry of virus into the cell occurs through fusion of cellular and viral envelope membranes. In addition, HIV-infected cells fuse with uninfected CD4+ cells to form syncytia, a process mediated by interactions between HIV envelope protein expressed on the infected cell surface and CD4 on the uninfected cell. Syncytium formation is thought to mimic viral entry and could be an important mode of viral spreading. Chimpanzee CD4, which binds to HIV but does not support syncytium formation, has Gly replacing Glu at residue 87. This replacement in human CD4 abolishes syncytia formation while preserving gp120 binding17 and, curiously, normal viral infection kinetics. Residues 88 and 89 are also implicated in this process by mutation (Table 2). These residues lie at the β-turn tip of the CDR3-like loop in D1 and are part of a patch of negative potential on the otherwise mostly positive D1D2 surface (Fig. 5d). The CDR3-like loop is spatially separated from the CDR2-like loop implicated in high-affinity binding to gp120 (Cα87 is 17 Å from Cα43). In accord, monoclonal antibodies of the L71 family block syncytium formation although they permit gp120 binding to CD4. So, it seems that the CDR3-like loop is necessary for secondary interactions between the viral envelope and CD4 before fusion. This could relate to CD4-induced release of gp120 from virus and infected cells43–45 and exposure of a fusogenic domain on the membrane envelope protein gp41.
Immune response interactions
The observation that CD4+ lymphocytes react only with class II-bearing target cells suggests that CD4 associates directly with class II MHC, and it is thought that CD4 may also have a role in signal transduction. There is evidence from cellular assays to support the association of CD4 with class II MHC molecules46, the T-cell receptor47, and the p56lck kinase48, as well as other CD4 molecules49. Except for the interaction of CD4 with p56lck, however, these associations are necessarily weak and thus difficult to measure. Mutagenesis in vitro has been used to identify the sites on CD4 that interact with class II MHC molecules. On the basis of assays of either cell adhesion18,50 or interleukin-2 production18, large separated expanses of the CD4 molecule, involving regions on D1, D2 and D3, have been implicated in the CD4-MHC interaction. These findings are not easy to reconcile with the structure of CD4. Some of the disruptive mutations occur in contact or bridging regions between the domains. Perhaps some of the other disruptions reflect complications from the diverse components of the cellular system rather than direct binding. For example, self-associations of CD419 might be involved in signal transduction and possibly in class II binding.
Evolutionary implications
There is little doubt that CD4 and immunoglobulins have evolved from a common ancestor. Intron and domain boundaries coincide and, although the sequences are highly divergent, superimposable strand topologies and a common hydrophobic core are preserved. It seems likely, however, that evolution to the antibody family, with its vast repertoire of dimeric receptor units, was a later event, and that CD4 is more prototypical of immunoglobulin superfamily receptors which are often monomeric and nonpolymorphic. The absence from CD4 of J-like regions, which impart diversity in antibodies, is consistent with this. The progenitor of the diverse immunoglobulin superfamily of the present may have its vestige in the gene structure of D1, which is split by an intron (at position 47) as are genes for other immunoglobulin-like domains51,52. A quasi-diad axis, perpendicular to the sheets and passing midway between strands B and E on one sheet and between strands C and F on the other, can be used to superimpose successive strands in the first exon on those in the second exon. An immunoglobulin progenitor produced by a gene duplication event53 might have been V-like, but a D2-like progenitor that would evolve to V and C domains is also a possibility.
Whatever the course of evolution, it is remarkable that molecules that function in recognition are often members of the immunoglobulin gene family. Clearly the immunoglobulin fold provides a facile framework from which a variety of loops with distinctive characteristics can be elaborated. These loops can have distinct functions, as in binding to gp120 at the CDR2-like loop and affecting fusion from the CDR3-like loop, and such modularity might have evolutionary advantage. Similarly, the quasi-symmetric nature of the immunoglobulin motif, with N and C termini at opposite ends, facilitates the concatenation of tandem, flexibly linked modules that can have distinct roles. In the case of CD4, one domain might be involved in class II binding whereas another domain might effect self-associations of the kind found in crystalline polymorphs19, and these could be essential for signal transduction. Indeed, in an alignment of D3 with D1, dimerization loops of immunoglobulin V domains would not be foreshortened in D3 as they are in D1. Such modules could evolve separately and be recombined by exon shuffling to integrate complex recognition and effector functions.
Acknowledgments
We thank M. Skinner, D. Sullivan, V. Ashford and C. Neilsen for help in data collection; colleagues at SmithKIine Beecham for help in protein preparation and purification; C. Ogata for helpful discussions; Z. Qtwinowski for his phasing program; C. Brändén and M. Amzel for supplying PapD and Fab New coordinates, respectively; A. Williams, Q. Sattentau, P. Bjorkman and D. Capon for communicating results before publication, and R. Yarmolinski for her drawing. This study was supported in part by the National Institutes of Health (W.A.H.) and by the Markey Foundation (N.-h.X.); the area detector facility at UCSD is an NIH Research Resource (N.-h.X.). Atomic coordinates have been deposited in the Protein Data Bank with entry name 1CD4 for release one year after publication of this paper.
References
- 1.Robey E, Axel R. Cell. 1990;60:697–700. doi: 10.1016/0092-8674(90)90082-p. [DOI] [PubMed] [Google Scholar]
- 2.Maddon PJ, et al. Cell. 1985;42:93–104. doi: 10.1016/s0092-8674(85)80105-7. [DOI] [PubMed] [Google Scholar]
- 3.Williams AF, Barclay AN. A Rev Immun. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 4.Clark SJ, Jefferies WA, Barclay AN, Gagnon J, Williams AF. Proc natn Acad Sci USA. 1987;84:1649–1653. doi: 10.1073/pnas.84.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maddon PJ, et al. Proc natn Acad Sci USA. 1987;84:9155–9159. doi: 10.1073/pnas.84.24.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgleish AG, et al. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 7.Klatzmann D, et al. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 8.Arthos J, et al. Cell. 1989;57:469–481. doi: 10.1016/0092-8674(89)90922-7. [DOI] [PubMed] [Google Scholar]
- 9.Capon DJ, et al. Nature. 1988;337:525–531. [Google Scholar]
- 10.Peterson A, Seed B. Cell. 1988;54:65–72. doi: 10.1016/0092-8674(88)90180-8. [DOI] [PubMed] [Google Scholar]
- 11.Landau NR, Warton M, Littman DR. Nature. 1988;334:159–162. doi: 10.1038/334159a0. [DOI] [PubMed] [Google Scholar]
- 12.Clayton LK, et al. Nature. 1988;335:363–366. doi: 10.1038/335363a0. [DOI] [PubMed] [Google Scholar]
- 13.Mizukami T, Fuerst TR, Berger EA, Moss B. Proc natn Acad Sci USA. 1988;85:9273–9277. doi: 10.1073/pnas.85.23.9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao BH, et al. J biol Chem. 1989;264:5182–5186. [Google Scholar]
- 15.Brodsky MH, Warton M, Myers RM, Littman DR. J Immun. 1990;144:3078–3086. [PubMed] [Google Scholar]
- 16.Ashkenazi A, et al. Proc natn Acad Sci USA. 1990;87:7150–7154. doi: 10.1073/pnas.87.18.7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camerini D, Seed B. Cell. 1990;60:747–754. doi: 10.1016/0092-8674(90)90089-w. [DOI] [PubMed] [Google Scholar]
- 18.Lamarre D, et al. Science. 1989;245:743–746. doi: 10.1126/science.2549633. [DOI] [PubMed] [Google Scholar]
- 19.Kwong PD, et al. Proc natn Acad Sci USA. 1990;87:6423–6427. doi: 10.1073/pnas.87.16.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis SJ, et al. J molec Biol. 1990;213:7–10. doi: 10.1016/S0022-2836(05)80116-0. [DOI] [PubMed] [Google Scholar]
- 21.Richardson NE, et al. Proc natn Acad Sci USA. 1988;85:6102–6106. doi: 10.1073/pnas.85.16.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibegbu CC, et al. J Immun. 1989;142:2250–2256. [PubMed] [Google Scholar]
- 23.Fleaiey D, et al. J exp Med. 1990;172:1233–1242. [Google Scholar]
- 24.Chamow SM. Biochemistry. 1990;29:9885–9891. doi: 10.1021/bi00494a019. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, et al. Nature. 1990;348:411–418. doi: 10.1038/348411a0. [DOI] [PubMed] [Google Scholar]
- 26.Carr S, et al. J biol Chem. 1989;264:21286–21295. [PubMed] [Google Scholar]
- 27.Xuong Ngh, Sullivan D, Nielsen C, Hamlin R. Acta crystallogr. 1985;B41:267–269. [Google Scholar]
- 28.Ashford V, Dai X, Nielsen C, Sullivan D, Xuong Ngh. Acta crystallogr. 1990;A46:C-15. [Google Scholar]
- 29.Jones TA. J appl Crystallogr. 1978;11:268–272. [Google Scholar]
- 30.Hendrickson WA, Konnert JH. In: Computing in Crystallography. Diamond R, Ramaseshan S, Venkatesan K, editors. Indian Institute of Science; Bangalore: 1980. pp. 13.01–13.23. [Google Scholar]
- 31.Brünger AT, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 32.Saul FA, et al. J biol Chem. 1978;253:585–597. [PubMed] [Google Scholar]
- 33.Gilson M, Sharp K, Honig B. J comp Chem. 1987;9:327–335. [Google Scholar]
- 34.Williams AF, Davis SJ, He Q, Barclay AN. Cold Spring Harb Symp quant Biol. 1989;54:637–647. doi: 10.1101/sqb.1989.054.01.075. [DOI] [PubMed] [Google Scholar]
- 35.Holmgren A, Brändén C-l. Nature. 1989;342:248–251. doi: 10.1038/342248a0. [DOI] [PubMed] [Google Scholar]
- 36.Epp O, et al. Eur J Biochem. 1974;45:513–524. doi: 10.1111/j.1432-1033.1974.tb03576.x. [DOI] [PubMed] [Google Scholar]
- 37.Bates PA, McGregor MJ, Islam SA, Sattentau Q, Sternberg MJE. Protein Eng. 1989;3:13–21. doi: 10.1093/protein/3.1.13. [DOI] [PubMed] [Google Scholar]
- 38.Wilson IA, Skehel, J J, Wiley DL. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 39.Daar ES, Xi LL, Moudgil T, Ho DD. Proc natn Acad Sci USA. 1990;87:6574–6578. doi: 10.1073/pnas.87.17.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sattentau QJ, et al. J exp Med. 1989;170:1319–1334. doi: 10.1084/jem.170.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lifson JD, et al. Science. 1988;241:712–716. doi: 10.1126/science.2969619. [DOI] [PubMed] [Google Scholar]
- 42.Finberg RW, et al. Science. 1990;249:287–291. doi: 10.1126/science.2115689. [DOI] [PubMed] [Google Scholar]
- 43.Kirsh R, et al. AIDS Res hum Retroviruses. 1990;6:1209–1212. doi: 10.1089/aid.1990.6.1209. [DOI] [PubMed] [Google Scholar]
- 44.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. Science. doi: 10.1126/science.2251501. in the press. [DOI] [PubMed] [Google Scholar]
- 45.Hart TK, et al. Proc natn Acad Sci USA. in the press. [Google Scholar]
- 46.Doyle C, Strominger JL. Nature. 1987;330:256–259. doi: 10.1038/330256a0. [DOI] [PubMed] [Google Scholar]
- 47.Chuck RS, Cantor CR, Tse DB. Proc natn Acad Sci USA. 1990;87:5021–5025. doi: 10.1073/pnas.87.13.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner JM, et al. Cell. 1990;60:755–765. doi: 10.1016/0092-8674(90)90090-2. [DOI] [PubMed] [Google Scholar]
- 49.Veillette A, Bookman MA, Horak EM, Samelson LE, Bolen JB. Nature. 1989;338:257–259. doi: 10.1038/338257a0. [DOI] [PubMed] [Google Scholar]
- 50.Clayton LK, Sieh M, Pious DA, Reinherz EL. Nature. 1989;339:548–551. doi: 10.1038/339548a0. [DOI] [PubMed] [Google Scholar]
- 51.Owens GC, Edelman GM, Cunningham BA. Proc natn Acad Sci USA. 1987;84:294–298. doi: 10.1073/pnas.84.1.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lemke G, Lamar E, Patterson J. Neuron. 1988;1:73–83. doi: 10.1016/0896-6273(88)90211-5. [DOI] [PubMed] [Google Scholar]
- 53.Bourgois A. Immunochemistry. 1975;12:873–876. doi: 10.1016/0019-2791(75)90244-x. [DOI] [PubMed] [Google Scholar]
- 54.Hendrickson WA. Trans Am crystallogr Assoc. 1985;21:11–21. [Google Scholar]
- 55.Pähler A, Smith JL, Hendrickson WA. Acta crystallogr. 1990;A46:537–540. doi: 10.1107/s0108767390002379. [DOI] [PubMed] [Google Scholar]
- 56.Karle J. Int J quant Chem Symp. 1980;7:357–367. [Google Scholar]
- 57.Hendrickson WA. Acta crystallogr. 1971;B27:1472–1473. [Google Scholar]
- 58.Hendrickson WA, Klippenstein GL, Ward KB. Proc natn Acad Sci USA. 1975;72:2160–2164. doi: 10.1073/pnas.72.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheriff S, Hendrickson WA, Stenkamp RE, Sieker LC, Jensen LH. Proc natn Acad Sci USA. 1985;82:1104–1107. doi: 10.1073/pnas.82.4.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]