

Abstract
Isaria cateniannulata is a very important and virulent entomopathogenic fungus that infects many insect pest species. Although I. cateniannulata is commonly exposed to extreme environmental temperature conditions, little is known about its molecular response mechanism to temperature stress. Here, we sequenced and de novo assembled the transcriptome of I. cateniannulata in response to high and low temperature stresses using Illumina RNA-Seq technology. Our assembly encompassed 17,514 unigenes (mean length = 1,197 bp), in which 11,445 unigenes (65.34%) showed significant similarities to known sequences in NCBI non-redundant protein sequences (Nr) database. Using digital gene expression analysis, 4,483 differentially expressed genes (DEGs) were identified after heat treatment, including 2,905 up-regulated genes and 1,578 down-regulated genes. Under cold stress, 1,927 DEGs were identified, including 1,245 up-regulated genes and 682 down-regulated genes. The expression patterns of 18 randomly selected candidate DEGs resulting from quantitative real-time PCR (qRT-PCR) were consistent with their transcriptome analysis results. Although DEGs were involved in many pathways, we focused on the genes that were involved in endocytosis: In heat stress, the pathway of clathrin-dependent endocytosis (CDE) was active; however at low temperature stresses, the pathway of clathrin-independent endocytosis (CIE) was active. Besides, four categories of DEGs acting as temperature sensors were observed, including cell-wall-major-components-metabolism-related (CWMCMR) genes, heat shock protein (Hsp) genes, intracellular-compatible-solutes-metabolism-related (ICSMR) genes and glutathione S-transferase (GST). These results enhance our understanding of the molecular mechanisms of I. cateniannulata in response to temperature stresses and provide a valuable resource for the future investigations.
Introduction
Entomopathogenic hyphomycete fungi, such as Beauveria bassiana and Metarhizium spp., are very common and abundant in ecosystems and many are well known to control insects, nematodes, and plant pathogens [1, 2]. However, they are very susceptible to extreme temperatures (heat and cold) [3–5]. High temperatures affect the efficiency of entomopathogenic fungi as pest control agent by hindering fungal germination [4], growth [6, 7], and virulence [8]. The shelf life of hyphomycetes-based formulations can also be reduced by those factors [9]. Low temperature does not kill conidia of entomopathogenic fungi, but it stops or delays conidial germination [5], and facilitates storage of fungal formulations [3].
Entomopathogenic hyphomycetes have evolved complex molecular mechanisms to protect themselves from extreme environmental temperatures. Many stress-related genes and metabolic pathways have been identified in response to environmental stimuli. Since the fungal cell wall is the first line of defense against harsh environments, cell-wall-major-components-metabolism-related (CWMCMR) genes have been shown to play a critical role [10, 11]. The gene encoding β-1,3-glucan synthase (MaFKS) in M. acridum plays a role in the maintenance of cell wall integrity, hyperosmotic pressure tolerance, and conidiation [12]. Two GPI (glycosylphosphatidylinositol)-anchored protein Ecm33 orthologous genes, Bbecm33 in B. bassiana and Mrecm33 in M. robertsii, have been reported to be essential for cell wall integrity and multi-stress tolerance [13]. Both heat shock proteins (Hsps) and intracellular compatible solutes (e.g., trehalose, mannitol, glycerol) are important anti-stress agents [14, 15]. Under heat stress, in M. anisopliae, 10 Hsp genes and three orthologous trehalose-6-phosphate synthase genes are up-regulated [16]. The expression of Hsp70 gene in B. bassiana is up-regulated by high (38°C) and low (4°C) temperatures, or UV stress [17]. Overexpression of the endogenous Hsp25 gene in M. robertsii improves its tolerance to several stresses, including heat and cold [18]. Both Mas5 and Mdj1, the members of Hsp40 in B. bassiana, play a very important role in environmental adaptation and host infection [19, 20]. The deletion of mannitol-1-phosphate dehydrogenase (MPD) reduces the content of mannitol in B. bassiana, resulting in a decline of conidial tolerance to diverse stresses, including heat [21]. The ribosome pathway, endocytosis pathway and proteasome pathway are active in M. anisopliae under conditions of heat shock stress [16].
The exploration of stress-related genes provides opportunities for genetic improvement of fungal stress tolerance in order to develop high-efficiency and field-persistent mycoinsecticides and/or mycoacaricides. Isaria cateniannulata (Z. Q. Liang) Samson & Hywel-Jones [22] (formerly Paecilomyces cateniannulatus [23]) is a highly virulent entomopathogenic fungus belonging to hyphomycetes [23, 24]. It can infect insect pests such as Lepidoptera [23–25], Coleoptera [23], and Hemiptera [23, 26], as well as mites [27] and nematodes [28]. This fungus is one of the dominant entomopathogenic fungi in several ecosystems including pine plantation [29] and tea plantation [30]. However, I. cateniannulata genomic information and its molecular responses to temperature variation remain unknown.
Fortunately, de novo transcriptome assembly and digital gene expression (DGE) sequencing using the Illumina offer the most cost-effective choice for characterizing non-model organisms without a reference genome [31]. In this study, we present the first comprehensive transcriptome characterization of I. cateniannulata and explore the influence of heat and cold temperatures on gene expressions. Using the Illumina HiSeq2000, we generated over 29 million clean reads and these reads were assembled into 17,514 unigenes without a reference genome. DGE analysis was then employed to further validate the genes and metabolic pathways in response to high or low temperatures in I. cateniannulata. This first transcriptomic report of I. cateniannulata functional genes provides an essential database for developing strategies to enhance resilience of entomopathogenic hyphomycetes to temperature variation.
Materials and methods
Culture of I. cateniannulata
The wild-type strain ICBS918 of I. cateniannulata was used for this study. This strain was isolated from cadaver of Homona coffearia with a high virulence to H. coffearia and Adoxophyes honmai larvae [24]. This strain was cultured on potato dextrose agar (Difco) for 7 d at 26°C under the light/dark cycle of 14h/10h. After this period, three replicates of the culture were exposed separately to three temperatures: 4°C (cold or LT), 26°C (control or NT), and 39°C (heat or HT) for 4 h (for a total of nine replicates). Mycelia were then harvested for total RNA extraction.
RNA isolation, library preparation, and sequencing
Total RNA was extracted using the SV total RNA isolation kit (Promega) according to the manufacturer’s protocol. The contamination and degradation of the extracted total RNA were preliminarily assessed on 1% agarose gels. The RNA integrity was checked with an Agilent Bioanalyzer 2100 system, and the purity and amount determined using an IMPLEN NanoPhotometer® spectrophotometer. Total RNA was finally quantified using the Qubit RNA Assay Kit in a Qubit 2.0 Flurometer according to the manufacture protocol (Life Technologies, CA, USA).
Equimolar quantities of the total RNA from the nine samples (1 μg from each sample) were combined into one pool as the input material for the transcriptome library, which acts as a reference library. To build DGE libraries for the nine samples (three replicates in each of the three treatments), 3 μg of extracted RNA were used. The cDNA library was constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) and the final cDNA library was selectively enriched by PCR and purified with the AMPure XP system (Beckman Coulter, Beverly, USA). The cDNA library was then sequenced on the Illumina HiSeq 2000 platform by the Novogene Bioinformatics Technology Co. Ltd (Beijing, China). Finally, 100-bp paired-end raw reads for transcriptome and 100-bp single-end raw reads for DGE were generated. All raw read sequences were deposited in the NCBI Short Read Archive (SRA) database under the accession number of SRP073968.
De novo assembly and annotation
Prior to transcriptome assembly, raw sequencing reads were filtered through in-house Perl scripts to discard dirty reads, which included adaptors, unknown ‘N’ nucleotides (where the ‘N’ ratio was more than 10%), and low quality sequences (where the quality score was less than 5). Based on the clean reads, de novo transcriptome assembly into transcripts without a reference genome was carried out using short reads assembling program Trinity with min_kmer_cov set to 2 and all other parameters set by default [32]. For removing redundancy, if a component contained more than one assembled transcript, only the longest one was regarded as a unigene. To annotate the unigenes, we used NCBI BLAST 2.2.28+ with an E-value threshold of 10−3 in the database of eukaryotic orthologous groups (KOG), and with an E-value threshold of 10−5 in the NCBI non-redundant protein sequences (Nr) database, NCBI nucleotide collection (Nt) database and the Swiss-Prot protein database. Protein family (Pfam) was assigned using the HMMER 3.0 package. The database categories of Kyoto Encyclopedia of Genes and Genomes (KEGG) were assigned to the unigenes according to the KEGG Automatic Annotation Server (KAAS) online [33]. Based on BLASTX hits against the Nr and Swiss-Prot protein database (E value≤10−5), the gene ontology (GO) annotation of unigenes was obtained using Blast2GO v2.5 [34] and GO functional classification was performed using WEGO software [35]. The aligning results were compared against the Nr and Swiss-Prot databases with a priority order of Nr > Swiss-Prot to decide the unigenes’ direction and coding sequences (CDSs). When unigenes were unaligned to any of the above databases, ESTScan software [36] was used to predict the coding regions and sequence orientation.
Analysis of DGE tags
Raw reads of DGE sequences were deposited in the NCBI Short Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra). Raw sequence data of the libraries for DGE profiling analyses were filtered to remove reads containing adaptors and reads with more than 10% unknown nucleotides, and reads with more than 50% of low-quality base (value≤5). Clean reads were mapped into the assembled transcriptome reference database and the read counts for each gene derived from the mapping results were achieved by RSEM [37]. All read counts were normalized using expected number of fragments per kilobase of transcript sequence per millions base pairs (FPKM), as described by Trapnell et al. [38]. The DESeq R package (1.10.1) was used for differential expression analysis of unigenes between the control (26°C) and the treatments (4°C or 39°C). To evaluate the reproducibility of DGE library sequencing, a Pearson correlation analysis was performed using the replicates between the various treatments. A corrected P-value of <0.05 (P values adjusted using the Benjamini & Hochberg method, Padj) was used as the threshold to judge the significant differences in gene expression. The heatmap of the differentially expressed genes (DEGs) was constructed using the R packages of ggplot2 and pheatmap.
In order to analyze the biological functions and metabolic pathways of the DEGs, DEGs were subject to GO functional analysis using Blast2GO v2.5 [34] and KEGG enrichment analysis using the KOBAS software [39]. Pathways with P value cutoff of ≤0.05 were considered significantly enriched by DEGs.
Quantitative real-time PCR analysis
To confirm the DGE results, 18 genes were randomly selected for qRT-PCR verification. Specific primers for qRT-PCR were designed using the Primer Premier 5.0 software (Premier, Canada) and listed in the S1 Table. RNA for qRT-PCR analysis was extracted from the mycelia under the extreme temperature, as described above, using the SV total RNA isolation kit (Promega, USA). Reverse transcription was carried out according to the manufacturer’s protocol of GoScript™ Reverse Transcription System (Promega, USA). The qRT-PCR was run in the CFX96 TouchTM Real-Time PCR Detection Systems (Bio-Rad, USA) using BRYT-Green-based PCR reaction. PCR amplification was performed in a total reaction mixture of 20 μL, containing 20 ng cDNA, 10 μL 2 × GoTaq® qPCR Master Mix (Promega, USA) and 0.2 μM of each primer. PCR was run with the standard thermal cycle conditions using the two-step qRT-PCR method: an initial denaturation at 95°C for 30s, followed by 40 cycles of 3 s at 95°C and 30 s at 60°C. Each reaction was run in triplicate, and the average threshold cycle (Ct) was calculated for each replicate. The assembled glyceraldehyde-3-phosphate dehydrogenase unigene (GenBank accession no. KT944290) was employed as the internal gene and the relative expression of gene was calculated using the 2-ΔΔCt method. The correlation of the fold change of the gene expression ratios between qRT-PCR and RNA-seq was checked by linear regression analysis in SPSS 17.0 software.
Results
Assembled transcriptome
As shown in Table 1, a total of 2.92 Gb nucleotides were obtained with a Q20 percentage of 95.5%. The percentage of unassigned base “N” was 5.95% and the average GC content, 55.17% (Table 1). By overlapping information in high-quality reads, 24,707 transcripts were generated, with an average length of 1,581 bp and an N50 of 2,664 bp. From these transcripts, 17,514 unigenes were obtained with a mean size of 1,197 bp (Table 2).
Table 1. Throughput and quality of RNA-seq of the reference library and the DGE libraries.
| Library | Raw reads | Clean reads | Nucleotide (nt) | Q20 percentage (%) | N percentage (%) | GC percentage (%) |
|---|---|---|---|---|---|---|
| Reference library | 33,741,894 | 29,230,890 | 2,923,089,000 | 95.50 | 5.95 | 55.17 |
| NT1 | 10,935,341 | 10,568,185 | 1,056,818,500 | 95.00 | 0.00 | 55.12 |
| NT2 | 7,873,444 | 7,653,904 | 765,390,400 | 95.00 | 0.00 | 55.07 |
| NT3 | 7,655,454 | 7,435,760 | 743,576,000 | 95.00 | 0.00 | 56.30 |
| LT1 | 9,569,387 | 9,351,945 | 935,194,500 | 96.00 | 0.01 | 55.47 |
| LT2 | 10,030,572 | 9,782,547 | 978,254,700 | 96.00 | 0.01 | 55.01 |
| LT3 | 11,786,747 | 11,521,026 | 1,152,102,600 | 96.00 | 0.01 | 55.17 |
| HT1 | 9,328,176 | 9,094,778 | 909,477,800 | 96.00 | 0.01 | 55.61 |
| HT2 | 14,377,463 | 14,052,602 | 1,405,260,200 | 96.00 | 0.01 | 55.22 |
| HT3 | 11,799,489 | 11,524,052 | 1,152,405,200 | 96.00 | 0.01 | 56.11 |
Q20 percentage indicates the percentage of sequences with sequencing error rate lower than 1%. N percentage is the percentage of nucleotides which could not be sequenced.
Table 2. Summary of the I. cateniannulata transcriptome.
| Category | Number | Total number |
Mean length (bp) |
N50 (bp) |
Total nucleotide | |||
|---|---|---|---|---|---|---|---|---|
| 200–500 bp | 500–1000 bp | 1–2 kb | >2kb | |||||
| Transcript | 7,633 | 4,250 | 5,718 | 7,106 | 24,707 | 1,581 | 2,664 | 39,071,015 |
| Unigene | 7,112 | 3,314 | 3,696 | 3,392 | 17,514 | 1,197 | 2,110 | 20,962,349 |
Functional annotation and classification
The 17,514 unigenes were accurately annotated by interrogating seven databases (Table 3): 65.34% (11,445) of unigenes could be annotated by BLASTx using the Nr database, 17.62% (3,087) by the Nt database, 16.58% (2,905) using the KEGG database, 37.41% (6,552) by the Swiss-Prot protein database, 45.22% (7,920) by the Pfam databases, 49.94% (8,748) according to the GO database, and 26.18% (4,586) according to the KOG database. In addition, 69.05% (12,095) of unigenes were annotated in at least one database, while only 8.81% (1,544) of unigenes were assigned to a homolog in all seven databases. A total of 17,204 coding sequences (12,174 predicted by BLASTX and 5,030 by ESTScan): 2,682 (15.59%) were smaller 200 bp, 12,939 (75.21%) were between 200 bp and 2,000 bp, and 1,583 (9.20%) were over 2,000 bp (S1 Fig). According to the BLASTX results of the Nr database, the sequence with the highest percentage of matching base (46.30%) was the C. militaris sequence, followed by the sequences of B. bassiana (42.00%) and M. anisopliae (1.10%) (Fig 1).
Table 3. Summary of the functional annotation of assembled unigenes.
| Public database | No. of unigene hit | Percentage (%) |
|---|---|---|
| Annotated in NR | 11,445 | 65.34 |
| Annotated in NT | 3,087 | 17.62 |
| Annotated in KEGG | 2,905 | 16.58 |
| Annotated in SwissProt | 6,552 | 37.41 |
| Annotated in PFAM | 7,920 | 45.22 |
| Annotated in GO | 8,748 | 49.94 |
| Annotated in KOG | 4,586 | 26.18 |
| Annotated in all databases | 1,544 | 8.81 |
| Annotated in at least one database | 12,095 | 69.05 |
| Total unigene | 17,514 | 100.00 |
Fig 1. Species distribution of the first BLAST hits against the Nr database.
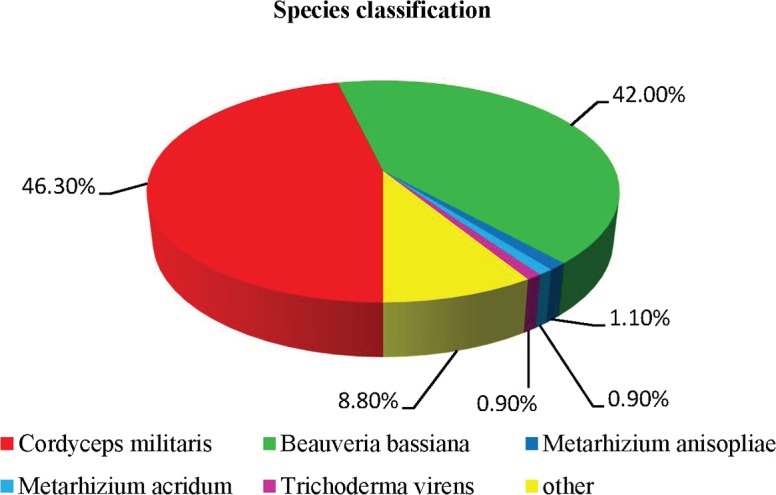
Gene ontology (GO) was used to classify the functions of the predicted I. cateniannulata unigenes. In total, 8,748 unigenes were classified into three different GO trees: biological process, cellular component, and molecular function (Table 3, Fig 2). The three different GO trees were divided into 47 functional groups, and further 1,179 functional terms (S2 Table). Seven groups were dominant clusters in GO classification: cellular process, metabolic process, single-organism process, cell, cell part, binding, and catalytic activity (Fig 2).
Fig 2. Histogram of gene ontology (GO) classification.

A total of 4,586 genes were assigned to KOG classification (Table 3, Fig 3) and grouped into 26 KOG categories (S3 Table). The largest category was “general function prediction only” (15.83%, 726), followed by “posttranslational modification, protein turnover, chaperones” (10.31%, 473), “signal transduction mechanisms” (8.16%, 374), “secondary metabolites biosynthesis, transport and catabolism” (7.26%, 333), and “translation, ribosomal structure and biogenesis” (6.69%, 307).
Fig 3. Histogram of KOG classification.

To further analyze the transcriptome of I. cateniannulata, the unigenes were searched against the KEGG pathway database. A total of 2,905 unigenes were annotated (Table 3) and assigned to 252 KEGG pathways. The most dominant pathways were “biosynthesis of amino acids” (ko01230, 115, 3.96%), “carbon metabolism” (ko01200, 95, 3.27%), and “ribosome” (ko03010, 91, 3.13%) (S4 Table).
DGE library sequencing and mapping to the reference transcriptome
Nucleotides of 0.74–1.41 Gb were obtained in each of the nine DGE libraries with a Q20 percentage of over 95%. The percentage of unassigned base “N” was below 0.01% and the average GC content was 55.01%-56.30% (Table 1). The clean reads of the nine DGE libraries were mapped to the above-constructed transcriptome reference database for each sample and the total mapped reads were above 96% for each replicate (S5 Table), which suggested the transcriptome was a reliable reference. The square of the Pearson correlation coefficient (R2) varied during 0.765–0.918 in HT treatment, 0.827–0.923 in LT treatment, and 0.882–0.933 in NT, indicating good operational stability and reliability of DEG library sequencing (Fig 4, S2 Fig).
Fig 4. Correlation tests for the replicates.

The abscissa represents the value log10 (FPKM + 1) of one duplicate; the ordinate represents the value log10 (FPKM + 1) of the other duplicate. R2 is the square of Pearson Correlation Coefficient.
Differentially expressed genes and qRT-PCR verification
There were a total of 5,686 DEGs in response to extreme temperature stresses, and the expression profile in the heatmap (Fig 5) showed significant differences among the three temperature treatments. In HT treatment, the expression of 4,483 genes was significantly changed when compared with the control (NT), with 2,905 genes being up-regulated and 1,578 genes down-regulated. In LT treatment, the expression of 1,245 genes was up-regulated and 682 genes down-regulated when compared with the control (Fig 6, S6 Table). Overlapped transcripts between heat and cold stresses showed that there were 724 DEGs responding to both heat and cold stresses, 3,759 DEGs only responding to heat stress, and 1,203 DEGs only responding to LT treatment (Fig 7).
Fig 5. Heatmap of the expression profile of DEGs in the three temperature treatments.
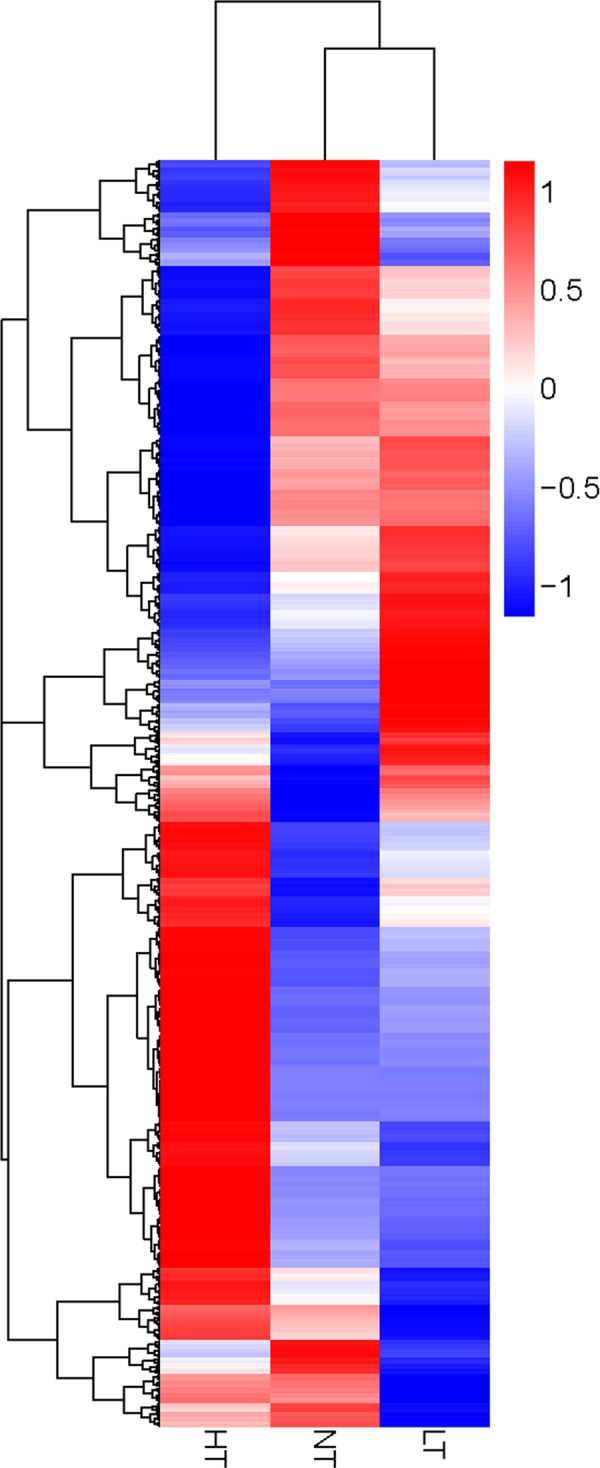
Each column represents a treatment, and each row represents a unigene. Differences in expression were shown in different colors. Data for gene expression level were normalized to z-score. Red represents up-regulated expression and blue represents down-regulated expression.
Fig 6. DEGs of I. cateniannulata when exposed to heat and cold treatments.
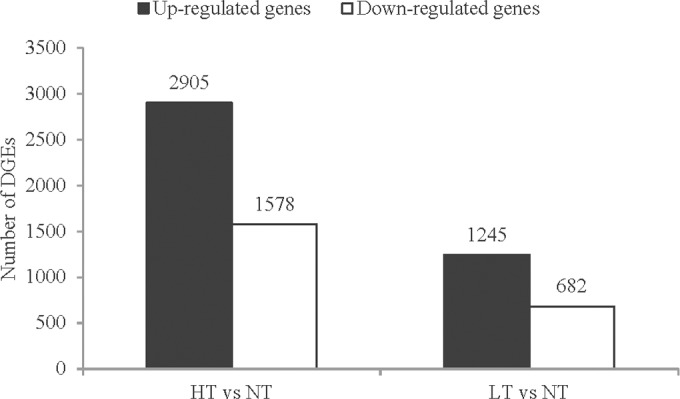
The number of DEGs was obtained from comparisons in NT versus HT and NT versus LT.
Fig 7. A Venn diagram describing the overlap of DEGs after treatments.

To verify the authenticity and reproducibility of the RNA-Seq results, 18 unigenes were randomly selected for qRT-PCR analysis and the expression profiles of the candidate unigenes by qRT-PCR were similar to the results of transcriptome analysis (Fig 8). The linear regression analysis showed significantly positive correlation of the relationship between gene expression ratios of qRT-PCR and RNA-seq was significantly positive (r = 0.75, P = 0.003) (Fig 9), confirming our transcriptomic data validity.
Fig 8. Expression profiles of eighteen unigenes uncovered by qRT-PCR (left side) and RNA-seq (right side).

Both the qRT-PCR data and the FPKM value of RNA-seq are means of three biological replicates and bars represent SE.
Fig 9. Correlation analysis of fold change data between qRT-PCR and RNA-seq.
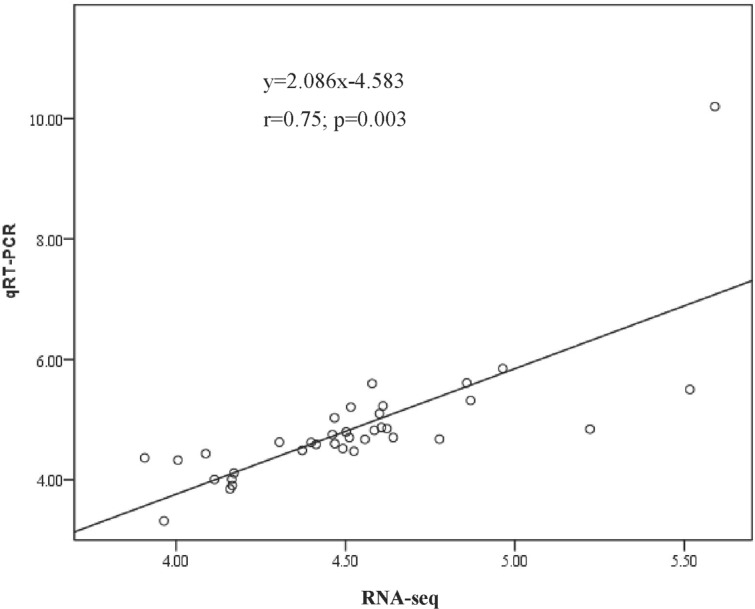
Data from qRT-PCR and RNA-seq are means of three replicates. Scatterplots were generated by the log2 expression ratios from RNA-seq (x-axis) and qRT-PCR (y-axis).
DEGs in response to heat treatment
A total of 4,483 DEGs including 2,905 up-regulated and 1,578 genes down-regulated were identified in HT. GO functional classification of 4,483 DEGs showed that they could be categorized into 50 functional groups, belonging to three main GO domains: biological processes (20), cellular components (16), and molecular functions (14). Among these groups, we found that cellular process, metabolic process, and single-organism process in the biological process ontology, cell and cell part in the cellular component ontology, and binding and catalytic activity in the molecular function ontology were the most common annotation terms (S3 Fig).
KEGG enrichment analysis showed that 8 pathways were significantly enriched (S7 Table). Among them, the ribosome biogenesis in eukaryotes was the most significantly enriched pathway, which contained 29 DEGs (28 down-regulated DEGs and 1 up-regulated DEG); the aminoacyl-tRNA biosynthesis was the second most significantly enriched pathway, which contained 18 DEGs (10 down-regulated DEGs and 8 up-regulated DEGs). However, we focused on the genes that were involved in endocytosis pathway, because of its ability to destroy misfolded and damaged proteins. After HT treatment, 12 DEGs related to clathrin-dependent endocytosis (CDE) were observed (Fig 10A). Among these 12 genes, 6 genes were up-regulated including PLD (comp7126_c0), dynamin (comp7305_c0), Hsc70 (comp7450_c0), VPS22 (comp2053_c0), CHMP4 (comp8162_c0) and CHMP3 (comp7520_c0); 6 genes were down-regulated including PLD (comp6558_c0), AP-2 (comp8686_c0 and comp6289_c0), Hsc70 (comp5575_c1), ArfGEF (comp4739_c0) and VPS45 (comp4862_c0).
Fig 10. Differential expression genes involved in the pathway of endocytosis in I. cateniannulata.
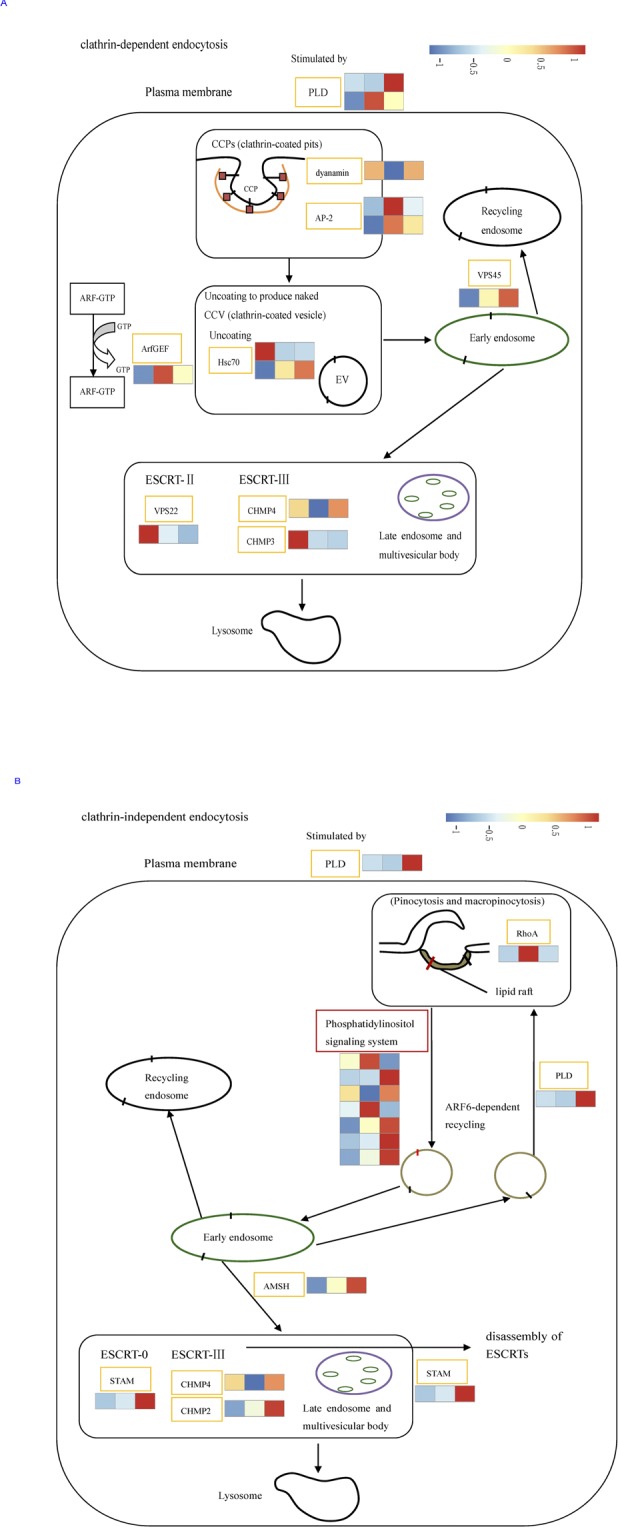
A. DEGs involved in clathrin-dependent endocytosis (CDE); B. DEGs involved in clathrin-independent endocytosis (CIE). Columns in each heatmap from left to right represented the value of FPKM: HT, NT and LT. And each row represents a gene. Expression differences are shown in different colors. Red means high expression and blue means low expression.
In this study, four categories of DEGs functioning as temperature sensors were observed, including CWMCMR genes, Hsps, ICSMR genes and glutathione S-transferase (GST) genes. DEG analysis showed that there were 14 CWMCMR unigenes in the up-regulated unigenes, but no CWMCMR unigene was observed in the down-regulated unigenes (Table 4). In the Hsp superfamily, 25 unigenes from 6 Hsp families (Hsp100, Hsp90, Hsp70, Hsp60, Hsp40 and sHsp) were differentially regulated under heat treatments, including 24 up-regulated unigenes and 1 down-regulated unigene. Hsp40 family was the largest group with 8 family members and Hsp70 family was the second most abundant group with 6 family members (Table 5). Eight ICSMR genes including trehalose metabolism-related genes, mannitol metabolism-related genes, glycerol metabolism-related genes and arabinitol metabolism-related genes were differentially regulated (Table 6). Six GST genes were found to be up-regulated, and no GST gene was observed to be down-regulated (Table 7).
Table 4. Variation in the expression of CWMCMR genes when exposed to heat and cold treatments in I. cateniannulata.
| Unigene ID | Annotation | HT/NT | LT/NT | Category |
|---|---|---|---|---|
| comp34522_c0 | β-1,3-glucan synthase | ☆ | ↑ | Glucan metabolism-related genes |
| comp8138_c0 | 1,3-β-glucanosyltransferase | ↑ | ↑ | |
| comp2635_c0 | exo-β-1,3-glucanase | ↑ | ☆ | |
| comp7516_c0 | GPI-anchored cell wall β-1,3-endoglucanase | ↑ | ↑ | |
| comp4353_c0 | endoglucanase EG-II | ☆ | ↑ | |
| comp3551_c0 | endo-1,3(4)- β-glucanase | ↑ | ☆ | |
| comp49089_c0 | endo-1,3(4)- β-glucanase 1 | ↑ | ↑ | |
| comp48493_c0 | glucan endo-1,4-β-glucanase activity | ↑ | ↑ | |
| comp4816_c0 | cell wall glucanase | ↑ | ☆ | |
| comp4822_c0 | xyloglucan endo-transglycosylase | ↑ | ☆ | |
| comp118120_c0 | class 5 chitinase 1 | ↑ | ☆ | Chitin/ chitosan metabolism-related genes |
| comp1058_c0 | class III chitinase ChiA1 | ↑ | ☆ | |
| comp129434_c0 | chitinase II | ↑ | ☆ | |
| comp105600_c0 | class V chitinase | ↑ | ☆ | |
| comp37786_c0 | chitinase activity | ☆ | ↑ | |
| comp8305_c0 | exochitinase | ↑ | ↑ | |
| comp1759_c0 | cell wall mannoprotein CIS3 | ☆ | ↑ | Mannoprotein metabolism-related genes |
| comp29349_c0 | cell wall serine-threonine-rich galactomannoprotein Mp1 | ↑ | ☆ | |
| comp7200_c0 | mannan endo-1,6-alpha-mannosidase | ☆ | ↑ | |
| comp2210_c0 | GPI anchored cell wall protein | ☆ | ↑ | GPI-anchored protein |
“↑” indicates up-regulated-genes and “☆” indicates unigenes no differentially expressed.
Table 5. Variation of Hsp when exposed to heat and cold treatments in I. cateniannulata.
| Unigene ID | Annotation | HT/NT | LT/NT | Category |
|---|---|---|---|---|
| comp7457_c0 | heat shock protein Hsp98 | ↑ | ↑ | Hsp100 family |
| comp7486_c0 | heat shock protein 78 | ↑ | ☆ | |
| comp5388_c0 | heat shock protein 90 | ↑ | ☆ | Hsp90 family |
| comp4106_c0 | heat shock protein 70 | ↑ | ☆ | Hsp70 family |
| comp4914_c0 | hsp70-like protein | ↑ | ☆ | |
| comp7450_c0 | heat shock protein 70 | ↑ | ☆ | |
| comp6682_c0 | heat shock protein 70 | ↑ | ☆ | |
| comp5575_c1 | heat shock protein 70 | ↓ | ☆ | |
| comp6246_c0 | chaperone protein dnaK | ↑ | ☆ | |
| comp7656_c0 | heat shock protein 60 | ↑ | ☆ | Hsp60 family |
| comp2707_c0 | heat shock protein 60 | ↑ | ☆ | |
| comp4697_c0 | heat shock protein 60 | ↑ | ☆ | |
| comp5545_c0 | DnaJ domain containing protein | ↑ | ☆ | Hsp40/DnaJ family |
| comp8061_c0 | DnaJ domain protein | ↑ | ☆ | |
| comp6354_c0 | DnaJ domain protein | ☆ | ↑ | |
| comp2405_c1 | Chaperone protein DnaJ | ☆ | ↓ | |
| comp7860_c0 | DnaJ domain protein | ☆ | ↑ | |
| comp7085_c0 | chaperone DnaJ | ↑ | ☆ | |
| comp6804_c0 | DnaJ domain protein | ↑ | ☆ | |
| comp201_c0 | DnaJ central domain | ↑ | ☆ | |
| comp7527_c0 | DnaJ domain-containing protein | ↑ | ☆ | |
| comp2189_c0 | DnaJ homolog subfamily A member 2 | ↑ | ☆ | |
| comp27058_c0 | DnaJ central domain | ↑ | ↑ | |
| comp2221_c0 | 30 kD heat shock protein | ↑ | ↑ | sHsp family |
| comp6572_c0 | 30 kD heat shock protein | ↑ | ↑ | |
| comp1380_c0 | hsp20-like protein | ↑ | ☆ | |
| comp1717_c1 | chaperonin 10 kD subunit | ↑ | ☆ |
“↑” indicates up-regulated-genes, “↓” indicates down-regulated-genes and “☆”indicates unigenes no differentially expressed.
Table 6. Variation in the expression of ICSMR genes when exposed to heat and cold treatments in I. cateniannulata.
| Unigene ID | Annotation | HT/NT | LT/NT | Category |
|---|---|---|---|---|
| comp6260_c0 | trehalase precursor | ☆ | ↑ | Trehalose metabolism-related genes |
| comp55474_c0 | trehalose-phosphatase | ↑ | ↑ | |
| comp1429_c0 | trehalose synthase | ☆ | ↑ | |
| comp2294_c0 | trehalose-6-phosphate synthase (tps1) | ↓ | ☆ | |
| comp7547_c0 | mannitol-1-phosphate dehydrogenase | ↑ | ☆ | Mannitol metabolism-related genes |
| comp13549_c0 | mannitol-1-phosphate dehydrogenase | ↑ | ☆ | |
| comp2518_c0 | mannitol dehydrogenase | ↓ | ☆ | |
| comp6842_c1 | glycerol dehydrogenase Gcy1 | ↑ | ↑ | Glycerol metabolism-related genes |
| comp6167_c0 | glycerophosphoryl diester phosphodiesterase | ☆ | ↑ | |
| comp10601_c0 | glycerophosphoryl diester phosphodiesterase | ☆ | ↑ | |
| comp7775_c0 | triacylglycerol lipase | ↑ | ↑ | |
| comp5307_c0 | D-arabinitol 2-dehydrogenase | ↑ | ☆ | Arabinitol metabolism-related genes |
“↑” indicates up-regulated-genes, “↓” indicates down-regulated-genes and “☆”indicates unigenes no differentially expressed.
Table 7. Variation in the expression of GSTs when exposed to heat and cold treatments in I. cateniannulata.
| Unigene ID | Annotation | HT/NT | LT/NT | Category |
|---|---|---|---|---|
| comp72432_c0 | glutathione S-transferase kappa 1 | ↑ | ☆ | glutathione S-transferase |
| comp5315_c0 | glutathione S-transferase | ↑ | ☆ | |
| comp3427_c0 | glutathione S-transferase | ↑ | ☆ | |
| comp7962_c0 | glutathione S-transferase domain-containing protein | ↑ | ☆ | |
| comp9890_c0 | glutathione S-transferase | ↑ | ☆ | |
| comp7963_c0 | glutathione S-transferase domain-containing protein | ↑ | ↑ |
“↑” indicates up-regulated-genes and “☆”indicates unigenes no differentially expressed.
DEGs in response to cold treatment
In comparison with NT, 1,927 DEGs were found in LT including 1,245 up-regulated genes and 682 down-regulated genes. All DEGs were categorized into 49 functional groups by GO functional classification, including biological processes (20), cellular components (14), and molecular functions (15). Among these groups, we found cellular process, metabolic process, and single-organism process in the biological process ontology, cell, cell part, and membrane in the cellular component ontology, and binding and catalytic activity in the molecular function ontology were the dominant clusters (S4 Fig).
The DEGs were highly enriched in polycyclic aromatic hydrocarbon degradation, phosphatidylinositol signaling system, tyrosine metabolism, inositol phosphate metabolism and drug metabolism—other enzymes by KEGG analysis (S7 Table). We also focused on the genes that were related to endocytosis pathway. After LT treatment, 10 DEGs related to clathrin-independent endocytosis (CIE) were up-regulated including PLD (comp7126_c0), PTEN (comp5571_c0),PLC (comp7099_c2), E3.1.3.56 (comp7092_c0 and comp4008_c0), PKC (comp5597_c0), AMSH (comp3635_c0), STAM(comp7300_c0), CHMP4 (comp8162_c0), CHMP2 (comp4090_c0), PTEN (comp5571_c0), PLC (comp7099_c2), E3.1.3.56 (comp7092_c0 and comp4008_c0) and PKC (comp5597_c0), while 3 of them were down-regulated including RhoA (comp6998_c0), PI4K(comp5407_c0) and PLC (comp7040_c0) (Fig 10B).
DEG analysis revealed that only CWMCMR (11) (Table 4), ICSMR (7) (Table 6), and GST unigenes (1) (Table 7) were up-regulated, and Hsp unigene (7) were differentially regulated including 6 members up-regulation and 1 member down-regulation (Table 5). In addition, one eubacteria-like cold shock protein (CSP) homologue (comp1755_c0) and one glycine-rich RNA binding protein (GRP) homologue (comp7081_c0) were found to be up-regulated under cold stress.
DEGs in response to both heat and cold treatments
A total of 724 co-regulated DEGs (Fig 7) including 496 up-regulated and 228 down-regulated genes were identified in both heat and cold treatment. All DEGs were categorized into 45 functional groups by GO functional classification, including biological processes (21), cellular components (15), and molecular functions (9). Among these groups, we found cellular process, metabolic process, and single-organism process in the biological process ontology, cell, cell part, and organelle in the cellular component ontology, and binding and catalytic activity in the molecular function ontology were the dominant clusters (S5 Fig).
The co-regulated DEGs were highly enriched in glycolysis / gluconeogenesis, regulation of mitophagy–yeast, sulfur metabolism, glycerolipid metabolism and ABC transporters by KEGG analysis (S7 Table). Besides, there were two co-regulated DEGs, PLD (comp7126_c0) and CHMP4 (comp8162_c0), related to endocytosis pathway. DEG analysis revealed that 5 CWMCMR genes (Table 4), 4 Hsp genes (Table 5), 3 ICSMR genes (Table 6) and 1GST gene (Table 7) were both up-regulated under heat and cold treatments.
Discussion
As the genomic information of I. cateniannulata was unavailable, and the molecular response mechanisms of the species to environmental variation not well understood, we used next-generation sequencing technology and transcriptome analysis as an alternative for in-depth analysis of molecular responses of I. cateniannulata to high and cold temperatures. In the present study, the transcriptome characterization of I. cateniannulata generated 29.2 million clean reads and assembled into 17,514 unigenes with a mean size of 1,197 bp. While 12,095 (69%) unigenes were successfully annotated using the public databases, 5,419 (31%) unigenes had no homologous sequences to public databases. This indicates that I. cateniannulata may contain several species-specific unigenes. The variation in number of unigenes and the expression profiles of DEGs under heat and cold temperature stresses suggests that the molecular responses of I. cateniannulata to high and low temperatures may greatly vary as reported in Pyropia yezoensis [40]. Though the cold temperature slows down the metabolic rates and results in the dormancy in organism, we still identified many DEGs in metabolic pathways under cold temperature treatment in our study. It maybe the cold temperature treatment (4°C for 4h) was not devastating to I. cateniannulata, and there were many DEGs have been activated. Our result is well in accordance with the study of Pyropia yezoensis in response to chilling and freezing stress [40], Chrysanthemum nankingense under low temperature [41], and Anthurium andraeanum under cold stress [42].
Endocytosis involves cells take up molecules (such as proteins) via vesicles, which is essential for cell-to-cell communication and cellular responses to external stimuli in all eukaryotic cells [43, 44]. Endocytosis pathway is significantly enriched in lotus (Nelumbo Adans) [45] and filamentous fungus (M. anisopliae) [16] under conditions of heat shock stress. And this pathway is also induced by cold stress in amur carp (Cyprinus carpio haematopterus) [46]. Endocytosis pathway contains two major categories: CDE and CIE [47]. CDE is well characterized and highly conserved cellular process from humans to fungi [48]. After heat treatment, the pathway of CDE was activated in I. cateniannulata as reported in M. anisopliae [16]. Dynamin is one of critical factors for different stages of CDE [49]. It was significantly up-regulated in I. cateniannulata under heat stress. Here, one Hsc70 gene (comp7450_c0) up-regulation and the down-regulation of comp5575_c1 were observed after heat stress. Hsc70 play a very important role in the ATP-dependent dissociation of clathrin from clathrin coated vesicles (CCVs) and other key processes in CDE [50]. Late endosomes (also known as multivesicular bodies, MVBs) is one of the major trafficking hub of the endocytic pathway, which formation is essential for cells to destroy misfolded and damaged proteins [47]. The endosomal sorting complex required for transport (ESCRT) plays a very important role in the MVB sorting pathway [47]. In this study, three genes involved in the ESCRT pathway, including ESCRT-Ⅱ (VPS22) and ESCRT-Ⅲ (CHMP3 and CHMP4) were up-regulated under heat stress. It was speculated that the activation of CDE would facilitate the elimination of misfolded and damaged proteins, which caused by heat shock in I. cateniannulata. Apart from CDE, CIE pathway have been found in eukaryotic cells, which mediates many biological processes [51]. In this study, CIE was activated when I. cateniannulata was exposed to cold treatment. The gene encoding AMSH (associated molecule with a Src homology 3 domain), a deubiquitinating enzyme, was up-regulated as well as, three genes involved in the ESCRT pathway, ESCRT-0 (STAM (signal transducing adaptor molecule)) and ESCRT-Ⅲ (CHMP (charged multivesicular body proteins) 2 and 4). These results suggest that the activation of CIE pathway would accelerate the removal of denatured proteins caused by low temperature stress in I. cateniannulata.
A total of 20 CWMCMR genes were up-regulated in response to high and/or low temperatures. Cell wall plays a very important role in protecting fungi against a variety of harsh environments such as heat, cold, desiccation and osmotic stress [52]. In this study, 1 β-1,3-glucan synthase orthologous gene (comp34522_c0) was up-regulated under cold temperature. We suggest that the cell may synthesize more β-1,3-glucan to enhance cell wall tensile strength against cold temperature. β-1,3-glucan has a coiled spring-like structure that confers a degree of elasticity and tensile strength to the cell wall [11]. β-1,3-glucan synthase (MaFKS)-RNAi transformants of entomopathogenic fungus M. acridum are more sensitive to agents that disturb the cell wall or cell membrane and to hyperosmotic stress in comparison with the wild type [12].
GPI-anchored protein orthologous gene (comp2210_c0) was up-regulated in response to cold temperature. GPI-anchored protein may not only play a role in maintaining the fungal cell wall integrity [13, 53], but also contribute to their multi-stress tolerance [13]. So the up-regulation of orthologous genes (comp2210_c0) of GPI-anchored protein may enhance the cell wall integrity and offer more resistance to cold temperature. Other up-regulated genes such as glucan-metabolism-related genes, chitin/chitosan-metabolism-related genes, and mannoprotein-metabolism-related genes, might have also been involved in maintaining cell wall integrity and increasing tensile strength against heat and cold. However, the exact functions of these genes are elusive.
Twenty-seven Hsp genes from 6 major families (Hsp100, Hsp90, Hsp70, Hsp60, Hsp40 and sHsp) were differentially regulated when exposed to heat and/or cold in I. cateniannulata. Hsps are ubiquitous in all prokaryotes and eukaryotes and can be induced by several kinds of stresses, including extremes temperatures, desiccation, and toxic substances [54–56]. In the filamentous fungus M. anisopliae, 10 Hsp genes are up-regulated in heat-treated conidia [16]. Here, one hsp98 homolog (comp7457_c0) was up-regulated in response to both heat and cold and one hsp78 homolog (comp7486_c0) was up-regulated in response to cold treatment. Hsp100 family is a major heat-regulated protein family in several species [57]. The Neurospora crassa Hsp98 protein, a member of hsp100 family, is highly expressed in response to heat shock [58]. Hsp78, a member of the Clp/Hsp100 family in S. cerevisiae, is required for the maintenance of mitochondrial function under heat stress [59]. Hsp90 homolog (comp5388_c0) was up-regulated upon exposure to heat. All eukaryotic cells produce prominent heat-shock protein with the molecular weight ranging from 80 to 90 kD, which are classified as the Hsp90 family [14, 60]. Hsp90 transcription is significantly induced when exposed to various abiotic stresses such as heat, cold, and oxygen deprivation [61, 62].
Interestingly, 5 members of Hsp70 family in I. cateniannulata were up-regulated in response to heat and might be invovled with the removal of denatured proteins. This is in agreement with other studies. For example, Hsp70 can prevent the aggregation of unfolding proteins and even refold aggregated proteins under heat [63]. The fungus B. emersonii has 10 putative Hsp70 homologs, and all Hsp70 genes (except for Hsp70-4 and Hsp70-6) are induced to different degrees upon exposure to heat [64]. The yeast S. cerevisiae has 14 Hsp70 genes, in which cytosolic Hsp70-Ssa (Ssa1, Ssa2, Ssa3 and Ssa4) is heat-inducible [65]. The transcriptional level of Hsp70 is up-regulated in B. bassiana when exposed to heat (38°C), cold (4°C), or UV stress [17].
Three Hsp60 homologs were up-regulated under heat in I. cateniannulata. The Hsp60 gene expression levels are also up-regulated in the Aspergillus fumigatus, A. terreus and Scedosporium apiospermum under heat [66]. In addition, 10 Hsp40/DnaJ homologs were up-regulated under heat and/or cold temperatures. Hsp40 family, also known as J-protein family, is the largest class of Hsp70 cofactors, which bind nonnative proteins and deliver them to Hsp70 [67, 68]. Both Mas5 and Mdj1, the members of Hsp40 in B. bassiana, have been shown to be indispensable for the environmental adaptation and virulence [19, 20].
In our study, 2 Hsp30 homologs (comp2221_c0 and comp6572_c0) were up-regulated under both heat and cold temperatures, and 1 Hsp20 (comp1380_c0) and 1 Hsp10 homologs (comp1717_c1) up-regulated when exposed to heat. The expression of Hsp23, a small heat-shock protein gene in Trichoderma virens is increased when the fungus is grown at extreme temperatures (4, 10 or 41°C) [69]. A small heat shock protein gene hsp25 is up-regulated in M. robertsii in response to extreme temperatures (4, 35, and 42°C), and overexpression of hsp25 improves the growth of M. robertsii when exposed to heat [18]. The sHsps of A. nidulans have been shown to take part in resisting adverse conditions, including heat and cold as well as oxidative/osmotic stresses [70].
Three trehalose-metabolism-related genes were up-regulated after heat and/or cold treatments in I. cateniannulata. Trehalose is present in a wide range of organisms, and could serve as a stabilizer and protectant of proteins and cellular membranes against a variety of stresses such as heat, cold, oxidation, and desiccation [71]. Under heat or chemical stress, the increasing of trehalose in the cell, which associated with up-regulation of the trehalose-6-P phosphatase transcript in arbuscular mycorrhizal (AM) fungi Glomus intraradices was observed [72]. Several trehalose accumulation-related genes are up-regulated in M. anisopliae in response to heat [16].
Here, two mannitol 1-phosphate dehydrogenase (MPD) orthologous genes (comp7547_c0 and comp13549_c0) showed up-regulation in response to heat. It was speculated that the up-regulation of MPD would increase the content of mannitol in I. cateniannulata. One D-arabinitol 2-dehydrogenase orthologous gene (comp5307_c0) was up-regulated under heat treatment. D-arabitol is one of the polyols found most frequently in fungi [73], which may act as adversity protectant [74]. In our study, 2 glycerol-metabolism-related genes were up-regulated when exposed to cold treatment, and 2 glycerol-metabolism-related genes were up-regulated in response to both heat and cold treatment in I. cateniannulata. The up-regulation of glycerol-metabolism-related genes may contribute to the increasing glycerol content against heat and cold temperatures.
GSTs play a very important role in response to oxidative stress by removing reactive oxygen species and regenerate S-thiolated proteins [75]. The GSTs involved in protecting Schizosaccharomyces pombe cells from damage causing by oxidative stress [76]. In our study, 6 GST genes were up-regulated under heat treatment, and 1 GST gene was up-regulated under cold treatment. We suggest that GST genes up-regulation might protect I. cateniannulata cells against damage resulting from oxidative stress induced by heat and cold treatments.
When I. cateniannulata was exposed to cold stress, the homologue of CSP (comp1755_c0) and GRP (comp7081_c0) were up-regulated. This is well in accordance with M. anisopliae’s CSP (CRP1) and GRP (CRP2) homologue, which play a key role in against cold stress [77].
Conclusions
The combination of RNA-seq and DGE analysis based on next generation sequencing technology provided comprehensive information on gene expression of I. cateniannulata, an entomopathogenic fungus for which little genomic information was available. Many DEGs of I. cateniannulata were identified under heat and cold temperatures with significant differences in molecular responses. In this study, we mainly focused on endocytosis pathway and identified several genes that were either up or down regulated when exposed to changing temperatures. Candidate stress-related genes may be useful tools for improvement of strain tolerance against extreme environmental temperatures in I. cateniannulata.
Supporting information
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(XLSX)
(XLS)
(XLS)
(XLS)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Mr. Yong Li for technical assistance during qRT-PCR analysis.
Data Availability
All raw read sequences were deposited in the NCBI Short Read Archive (SRA) database under the accession number of SRP073968. And all other relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was financially supported by the programs of Natural Science Foundation of Fujian Province, China (grant no. 2015J01099; http://www.fjkx.org/), Modern Agro-Industry Technology Research System (grant no. CARS-23; http://www.moa.gov.cn/), Key Project of Tea Research Institute of FAAS (grant no. 2014-cys-02; http://www.faas.cn/dept/cys/index.html) and Collaborative Innovation Center of Chinese Oolong Tea Industry-Collaborative Innovation Center (2011) of Fujian Province (http://www.fjedu.gov.cn/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Feng MG, Poprawski TJ, Khachatourians GG. Production, formulation and application of the entomopathogenic fungus Beauveria bassiana for insect control: current status. Biocontrol sci. Techn. 1994; 4: 3–34. [Google Scholar]
- 2.Goettel MS, Eilenberg J, Glare T. Entomopathogenic fungi and their role in regulation of insect populations In: Gilbert LI, Iatrou K, Gill S, editors. Comprehensive molecular insect science. Oxford: Elsevier Pergamon; 2005. pp. 361–406. [Google Scholar]
- 3.Roberts D, St Leger RJ. Metarhizium spp., cosmopolitan insect-pathogenic fungi: mycological aspects. Adv. Appl. Microbiol. 2004; 54: 1–70. doi: 10.1016/S0065-2164(04)54001-7 [DOI] [PubMed] [Google Scholar]
- 4.Rangel DE, Braga GU, Anderson AJ, Roberts DW. Variability in conidial thermotolerance of Metarhizium anisopliae isolates from different geographic origins. J. Invertebr. Pathol. 2005; 88: 116–125. doi: 10.1016/j.jip.2004.11.007 [DOI] [PubMed] [Google Scholar]
- 5.Fernandes EK, Rangel DE, Moraes AM, Bittencourt VR, Roberts DW. Cold activity of Beauveria and Metarhizium, and thermotolerance of Beauveria. J. Invertebr. Pathol. 2008; 98: 69–78. doi: 10.1016/j.jip.2007.10.011 [DOI] [PubMed] [Google Scholar]
- 6.Ouedraogo A, Fargues J, Goettel MS, Lomer CJ. Effect of temperature on vegetative growth among isolates of Metarhizium anisopliae and M. flavoviride. Mycopathologia. 1997; 137: 37–43. doi: 10.1023/A:1006882621776 [DOI] [PubMed] [Google Scholar]
- 7.Devi KU, Sridevi V, Mohan ChM, Padmavathi J. Effect of high temperature and water stress on in vitro germination and growth in isolates of the entomopathogenic fungus Beauveria bassiana (Bals.) Vuillemin. J. Invertebr. Pathol. 2005; 88: 181–189. doi: 10.1016/j.jip.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 8.Bugeme DM, Maniania NK, Knapp M, Boga HI. Effect of temperature on virulence of Beauveria bassiana and Metarhizium anisopliae isolates to Tetranychus evansi. Exp. Appl. Acarol. 2008; 46: 275–285. doi: 10.1007/s10493-008-9179-1 [DOI] [PubMed] [Google Scholar]
- 9.Hong TD, Gunn J, Ellis RH, Jenkins NE, Moore D. The effect of storage environment on the longevity of conidia of Beauveria bassiana. Mycological Research. 2001; 105: 597–602. [Google Scholar]
- 10.Cid VJ, Durán A, del Rey F, Snyder MP, Nombela C, Sánchez M. Molecular basis of cell integrity and morphogenesis in Saccharomyces cerevisiae. Microbiol. Rev. 1995; 59: 345–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesage G, Bussey H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006; 70: 317–343. doi: 10.1128/MMBR.00038-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang M, Jin K, Xia Y. MaFKS, a β-1,3-glucan synthase, is involved in cell wall integrity, hyperosmotic pressure tolerance and conidiation in Metarhizium acridum. Curr. Genet. 2011; 57: 253–260. doi: 10.1007/s00294-011-0344-4 [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Zhu J, Ying SH, Feng MG. The GPI-anchored protein Ecm33 is vital for conidiation, cell wall integrity, and multi-stress tolerance of two filamentous entomopathogens but not for virulence. Appl. Microbiol. Biotechnol. 2014; 98: 5517–29. doi: 10.1007/s00253-014-5577-y [DOI] [PubMed] [Google Scholar]
- 14.Lindquist S. The heat-shock response. Annu. Rev. Biochem. 1986; 55: 1151–1191. doi: 10.1146/annurev.bi.55.070186.005443 [DOI] [PubMed] [Google Scholar]
- 15.Wyatt TT, van Leeuwen MR, Golovina EA, Hoekstra FA, Kuenstner EJ, Palumbo EA, et al. Functionality and prevalence of trehalose-based oligosaccharides as novel compatible solutes in ascospores of Neosartorya fischeri (Aspergillus fischeri) and other fungi. Environ Microbiol. 2015; 17: 395–411. doi: 10.1111/1462-2920.12558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang ZX, Zhou XZ, Meng HM, Liu YJ, Zhou Q, Huang B. Comparative transcriptomic analysis of the heat stress response in the filamentous fungus Metarhizium anisopliae using RNA-Seq. Appl Microbiol Biotechnol. 2014; 98: 5589–97. doi: 10.1007/s00253-014-5763-y [DOI] [PubMed] [Google Scholar]
- 17.Xie L, Chen HM, Tang Q, Pu SC, Li ZZ, Huang B. Expression analysis of hsp70 gene from Beauveria bassiana under several stress conditions by Realtime-PCR. Mycosystema. 2009; 28: 806–812. Chinese. [Google Scholar]
- 18.Liao X, Lu HL, Fang W, St Leger RJ. Overexpression of a Metarhizium robertsii HSP25 gene increases thermotolerance and survival in soil. Appl. Microbiol. Biotechnol. 2014; 98: 777–783. doi: 10.1007/s00253-013-5360-5 [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Ying SH, Hu Y, Feng MG. Mas5, a homologue of bacterial DnaJ, is indispensable for the host infection and environmental adaptation of a filamentous fungal insect pathogen. Environ. Microbiol. 2016; 18: 1037–1047. doi: 10.1111/1462-2920.13197 [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Ying SH, Hu Y, Feng MG. Vital role for the J-domain protein Mdj1 in asexual development, multiple stress tolerance, and virulence of Beauveria bassiana. Appl. Microbiol. Biotechnol. 2017; 101: 185–195. doi: 10.1007/s00253-016-7757-4 [DOI] [PubMed] [Google Scholar]
- 21.Wang ZL, Lu JD, Feng MG. Primary roles of two dehydrogenases in the mannitol metabolism and multi-stress tolerance of entomopathogenic fungus Beauveria bassiana. Environ Microbiol. 2012; 14: 2139–2150. doi: 10.1111/j.1462-2920.2011.02654.x [DOI] [PubMed] [Google Scholar]
- 22.Luangsa-ard JJ, Hywel-Jones NL, Manoch L, Samson RA. On the relationships of Paecilomyces sect. Isarioidea species. Mycological Res. 2005; 109: 581–9. [DOI] [PubMed] [Google Scholar]
- 23.Liang ZQ. Two new species of Paecilomyces from insects. Acta Microbiologica Sinica. 1981; 21: 31–34. Chinese. [Google Scholar]
- 24.Wang DF, Yang G, Wang QS, Zeng MS, Wu GY. Identification of two Isaria isolates and bioassay of their pathogenicity against the Homona coffearia and Adoxophyes honmai. Acta Phytophylacica Sinica. 2014; 41: 531–539. Chinese. [Google Scholar]
- 25.Mitsuhashi W, Shimazu M, Hashimoto H. Control of Epinotia granitalis (Lepidoptera: Tortricidae) with Paecilomyces spp. on cotton bands wrapped on the trunks of Cryptomeria japonica. Appl. Entomol. Zool. 1992; 27: 295–6. [Google Scholar]
- 26.Rocha LF, Luz C. Activity of Metarhizium spp. and Isaria spp. from the central Brazilian cerrado against Triatoma infestans nymphs. Trans R Soc Trop Med Hyg. 2011; 105: 417–419. doi: 10.1016/j.trstmh.2011.04.012 [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Jin D, Zou X, Guo J. Laboratory and field evaluation of an entomopathogenic fungus, Isaria cateniannulata strain 08XS-1, against Tetranychus urticae (Koch). Pest Manag Sci. 2016; 72: 1059–1066. doi: 10.1002/ps.4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang ZQ, Han YF, Chu HL, Liu AY. Studies on the genus Paecilomyces in China I. Fungal Diversity. 2005; 20: 83–101. [Google Scholar]
- 29.Chen MJ, Huang B, Li ZZ. Niche comparison of dominant entomopathogenic fungi in three forest ecosystems. Chinese Journal of Applied Ecology. 2011; 22: 1275–1279. Chinese. [PubMed] [Google Scholar]
- 30.Wang P, Huang C, Li JL, Li ZZ, Wang B. The community structure and diversity of entomopathogenic fungi in the tea garden soil of Guangdong province. Journal of Tea Science. 2013; 33: 562–569. Chinese. [Google Scholar]
- 31.Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010; 20: 1432–1440. doi: 10.1101/gr.103846.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011; 29: 644–652. doi: 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008; 36: D480–4. doi: 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008; 36: 3420–35. doi: 10.1093/nar/gkn176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006; 34: W293–7. doi: 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB. 1999; 99: 138–148. [PubMed] [Google Scholar]
- 37.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12: 323 doi: 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech. 2010; 28: 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005; 21: 3787–93. doi: 10.1093/bioinformatics/bti430 [DOI] [PubMed] [Google Scholar]
- 40.Sun P, Mao Y, Li G, Cao M, Kong F, Wang L, et al. Comparative transcriptome profiling of Pyropia yezoensis (Ueda) M.S. Hwang & H.G. Choi in response to temperature stresses. BMC Genomics. 2015; 16: 463 doi: 10.1186/s12864-015-1586-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren L, Sun J, Chen S, Gao J, Dong B, Liu Y, et al. A transcriptomic analysis of Chrysanthemum nankingense provides insights into the basis of low temperature tolerance. BMC Genomics. 2014; 15: 844 doi: 10.1186/1471-2164-15-844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian DQ, Pan XY, Yu YM, Wang WY, Zhang F, Ge YY, et al. De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. BMC Genomics. 2013; 14: 827 doi: 10.1186/1471-2164-14-827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baggett JJ, Wendland B. Clathrin function in yeast endocytosis. Traffic. 2001; 2: 297–302. [DOI] [PubMed] [Google Scholar]
- 44.Fan L, Li R, Pan J, Ding Z, Lin J. Endocytosis and its regulation in plants. Trends Plant Sci. 2015; 20: 388–397. doi: 10.1016/j.tplants.2015.03.014 [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Du F, Li N, Chang Y, Yao D. Gene expression profile in the long-living lotus: insights into the heat stress response mechanism. Plos One. 2016; 11: e0152540 doi: 10.1371/journal.pone.0152540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang L, Chang Y, He X, Tang R. Transcriptome analysis to identify cold-responsive genes in amur carp (Cyprinus carpio haematopterus). Plos One 2015; 10: e0130526 doi: 10.1371/journal.pone.0130526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miaczynska M, Stenmark H. Mechanisms and functions of endocytosis. J Cell Biol. 2008; 180: 7–11. doi: 10.1083/jcb.200711073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011; 12: 517–533. doi: 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- 49.Liu YW, Su AI, Schmid SL. The evolution of dynamin to regulate clathrin-mediated endocytosis: speculations on the evolutionarily late appearance of dynamin relative to clathrin-mediated endocytosis. Bioessays. 2012; 34: 643–7. doi: 10.1002/bies.201200033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007; 8: 640–6. doi: 10.1111/j.1600-0854.2007.00568.x [DOI] [PubMed] [Google Scholar]
- 51.Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol. 2010; 22: 519–527. doi: 10.1016/j.ceb.2010.04.001 [DOI] [PubMed] [Google Scholar]
- 52.Free SJ. Fungal cell wall organization and biosynthesis. Adv. Genet. 2013; 81: 33–82. doi: 10.1016/B978-0-12-407677-8.00002-6 [DOI] [PubMed] [Google Scholar]
- 53.Levdansky E, Kashi O, Sharon H, Shadkchan Y, Osherov N. The Aspergillus fumigatus cspA gene encoding a repeat-rich cell wall protein is important for normal conidial cell wall architecture and interaction with host cells. Eukaryot Cell. 2010; 9: 1403–15. doi: 10.1128/EC.00126-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu. Rev. Physiol. 1999; 61: 243–282. doi: 10.1146/annurev.physiol.61.1.243 [DOI] [PubMed] [Google Scholar]
- 55.Sørensen JG, Kristensen TN, Loeschcke V. The evolutionary and ecological role of heat shock proteins. Ecol. Lett. 2003; 6: 1025–37. [Google Scholar]
- 56.Liu D, Zhang X, Cheng Y, Takano T, Liu S. rHsp90 gene expression in response to several environmental stresses in rice (Oryza sativa L.). Plant Physiology Biochemistry. 2006; 44: 380–386. doi: 10.1016/j.plaphy.2006.06.011 [DOI] [PubMed] [Google Scholar]
- 57.Ye SF, Yu SW, Shu LB, Wu JH, Wu AZ, Lou LJ. Expression profile analysis of 9 heat shock protein genes throughout the life cycle and under abiotic stress in rice. Chinese Science Bulletin. 2012; 57: 336–343. [Google Scholar]
- 58.Vassilev AO, Plesofsky-Vig N, Brambl R. Isolation, partial amino acid sequence, and cellular distribution of heat-shock protein hsp98 from Neurospora crassa. Biochim. Biophys. Acta. 1992; 1156: 1–6. [DOI] [PubMed] [Google Scholar]
- 59.Krzewska J, Langer T, Liberek K. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 2001; 489: 92–6. [DOI] [PubMed] [Google Scholar]
- 60.Roy SS, Wheatley RW, Kapoor M. Homology modeling, ligand docking and in silico mutagenesis of neurospora Hsp80 (90): insight into intrinsic ATPase activity. J Mol Graph Model. 2013; 44: 54–69. doi: 10.1016/j.jmgm.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 61.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010; 11: 515–528. doi: 10.1038/nrm2918 [DOI] [PubMed] [Google Scholar]
- 62.Dal Piaz F, Terracciano S, De Tommasi N, Braca A. Hsp90 activity modulation by plant secondary metabolites. Planta Medica, 2015; 81: 1223–39. doi: 10.1055/s-0035-1546251 [DOI] [PubMed] [Google Scholar]
- 63.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005; 62: 670–684. doi: 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Georg Rde C, Gomes SL. Comparative expression analysis of members of the Hsp70 family in the chytridiomycete Blastocladiella emersonii. Gene. 2007; 386: 24–34. doi: 10.1016/j.gene.2006.07.033 [DOI] [PubMed] [Google Scholar]
- 65.Hasin N, Cusack SA, Ali SS, Fitzpatrick DA, Jones GW. Global transcript and phenotypic analysis of yeast cells expressing Ssa1, Ssa2, Ssa3 or Ssa4 as sole source of cytosolic Hsp70-Ssa chaperone activity. BMC Genomics. 2014; 15: 194 doi: 10.1186/1471-2164-15-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raggam RB, Salzer HJ, Marth E, Heiling B, Paulitsch AH, Buzina W. Molecular detection and characterisation of fungal heat shock protein 60. Mycoses. 2011; 54: e394–9. doi: 10.1111/j.1439-0507.2010.01933.x [DOI] [PubMed] [Google Scholar]
- 67.Walsh P, Bursać D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO J. 2004; 5: 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010; 11: 579–592. doi: 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Montero-Barrientos M, Cardoza RE, Gutiérrez S, Monte E, Hermosa R. The heterologous overexpression of hsp23, a small heat shock protein gene from Trichoderma virens, confers thermotolerance to T. harzianum. Curr. Genet. 2007; 52: 45–53. doi: 10.1007/s00294-007-0140-3 [DOI] [PubMed] [Google Scholar]
- 70.Wu J, Wang M, Zhou L, Yu D. Small heat shock proteins, phylogeny in filamentous fungi and expression analyses in Aspergillus nidulans. Gene. 2016; 575: 675–9. doi: 10.1016/j.gene.2015.09.044 [DOI] [PubMed] [Google Scholar]
- 71.Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology, 2003; 13: 17R–27R. doi: 10.1093/glycob/cwg047 [DOI] [PubMed] [Google Scholar]
- 72.Ocón A, Hampp R, Requena N. Trehalose turnover during abiotic stress in arbuscular mycorrhizal fungi. New Phytol. 2007; 174: 879–891. doi: 10.1111/j.1469-8137.2007.02048.x [DOI] [PubMed] [Google Scholar]
- 73.Link T, Lohaus G, Heiser I, Mendgen K, Hahn M, Voegele RT. Characterization of a novel NADP(+)-dependent D-arabitol dehydrogenase from the plant pathogen Uromyces fabae. Biochem. J. 2005; 389 (Pt 2): 289–95. doi: 10.1042/BJ20050301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maclean DJ, Scott KJ. Identification of glucitol (sorbitol) and ribitol in a rust fungus, Puccinia graminis f. sp. tritici. J. Gen. Microbiol. 1976; 97: 83–89. doi: 10.1099/00221287-97-1-83 [DOI] [PubMed] [Google Scholar]
- 75.Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001; 360(Pt 1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Veal EA, Toone WM, Jones N, Morgan BA. Distinct roles for glutathione S-transferases in the oxidative stress response in Schizosaccharomyces pombe. J. Biol. Chem. 2002; 277: 35523–31. doi: 10.1074/jbc.M111548200 [DOI] [PubMed] [Google Scholar]
- 77.Fang W, St Leger RJ. RNA binding proteins mediate the ability of a fungus to adapt to the cold. Environ Microbiol. 2010; 12: 810–820. doi: 10.1111/j.1462-2920.2009.02127.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(XLSX)
(XLS)
(XLS)
(XLS)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All raw read sequences were deposited in the NCBI Short Read Archive (SRA) database under the accession number of SRP073968. And all other relevant data are within the paper and its Supporting Information files.