

Myc oncoproteins, including N-Myc, c-Myc and L-Myc, are essential for regulating cell proliferation and differentiation. Control of cell proliferation and differentiation is highly orchestrated, therefore, aberrant expression of the Myc oncoproteins due to chromosomal translocation, gene amplification, increased transcription, mRNA translation or protein stabilization can result in incessant cell proliferation, differentiation block, tumour initiation and progression. While c-Myc is over-expressed in hematological malignancies and solid tumours of various organ origins, N-Myc over-expression is restricted to ovarian cancer, rhabdomyosarcoma and tumours of neuroectodermal origins, for example neuroblastoma, retinoblastoma, medulloblastoma, glioblastoma and peripheral neuroectodermal tumours. Paradoxically, Myc oncoproteins induce apoptosis by promoting the transcription of pro-apoptotic genes, such as p53, and sensitizing cells to pro-apoptotic stimuli.1,2 Myc over-expressing tumour cells can evade apoptosis due to p53 mutations or deletions. While p53 is the most commonly mutated tumour suppressor gene in human cancers, p53 is not commonly mutated in c-Myc over-expressing lymphoma tissues, and is rarely mutated in neuroblastoma tissues harbouring N-Myc gene amplification. Although transcriptional activation of MDM2, which induces wild type p53 protein degradation, has been suggested to be important for protecting Myc over-expressing cancer cells against apoptosis, Myc oncoproteins can induce apoptosis through p53-independnet mechanisms such as cell cycle progression, DNA damage and generation of reactive oxygen species (ROS).3 It is not clear how Myc over-expressing cancer cells evade Myc-mediated apoptosis due to ROS production.
Myc oncoproteins are well-known to activate gene transcription by direct binding to Myc responsive element E-Boxes at target gene promoters. In an attempt to identify Myc target genes potentially important for the survival of Myc over-expressing cancer cells, we have identified a canonical E-Box at the gene core promoter of LYAR, and confirmed that N-Myc up-regulates LYAR expression by binding to its gene core promoter.4 As a nucleolar and nuclear protein with zinc finger DNA-binding motifs, LYAR is known to suppress gene transcription by forming a protein complex with the protein arginine methyltransferase PRMT5 at target gene promoters.5 We have confirmed through Affymetrix microarray gene expression study, gene set enrichment analysis and chromatin immunoprecipitation assay that LYAR considerably suppresses the transcription of oxidative stress genes, including SLC7A11, ULBP1, HMOX1 and CHAC1.4 In literature, CHAC1 acts as a γ-glutamyl cyclotransferase and specifically targets and degrades glutathione, resulting in glutathione depletion, oxidative stress and apoptosis. We have further found that knocking down LYAR induces CHAC1 gene expression and ROS production, and reduces cancer cell proliferation and survival, and that treatment with the glutathione supplement N-acetylcysteine blocks cell death due to LYAR knockdown. Based on our own data and the observations highlighted below, we propose a Working Model (Figure 1) suggesting that up-regulation of LYAR by Myc protects cancer cells against oxidative stress-mediated apoptosis through reducing CHAC1 gene expression.
Figure 1.
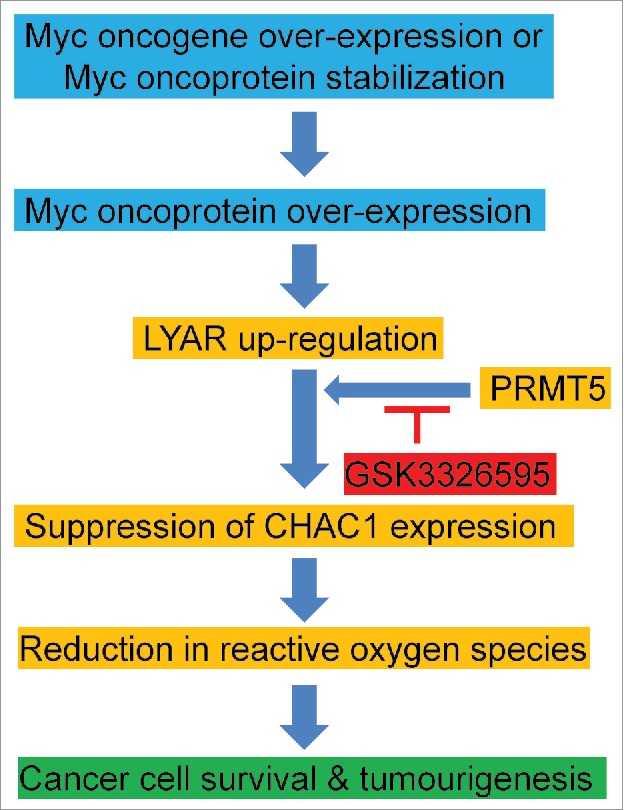
Up-regulation of LYAR by Myc protects cancer cells against apoptosis through reducing CHAC1 gene expression. Myc oncoproteins directly up-regulate LYAR expression by activating its gene transcription. LYAR interacts with PRMT5 and thereby suppresses CHAC1 gene transcription, leading to reactive oxygen species reduction, cancer cell survival and tumourigenesis. The PRMT5 inhibitor GSK3326595 is likely to block LYAR-mediated cell survival.
First, Myc up-regulation is well-known to induce the production of ROS, DNA damage and apoptosis, and antioxidants block Myc-mediated ROS production, DNA damage and apoptosis [reviewed in3]. Second, LYAR is highly expressed in undifferentiated embryonic stem cells, plays a critical role in maintaining embryonic stem cell identity and survival, and its downregulation significantly reduces embryonic stem cell growth and increases their apoptosis.6 The microRNA miR-34a is suppressed in human cancer tissues, inhibition of miR-34a impairs ROS production and apoptosis upon DNA damage, and LYAR has been identified as one of the key miR-34a targets. Third, CHAC1 has been found to be the gene most considerably up-regulated by treatment with the chemotherapy agent temozolomide in glioblastoma cells in a microarray differential gene expression study, and CHAC1 induces glioblastoma cell apoptosis via ROS generation and consequent loss of the mitochondria membrane potential.7
Taken together, the above findings in the literature and our own findings support the original hypothesis that Myc oncoproteins up-regulate LYAR expression by activating its gene transcription, and that up-regulation of LYAR, in turn, protects cancer cells against oxidative stress-mediated apoptosis through reducing CHAC1 gene expression. Consistent with our hypothesis, we have found that high levels of LYAR gene expression in human neuroblastoma tissues correlates with high levels of N-Myc gene expression and poorer patient survival, independent of traditional prognostic markers.4 We propose that up-regulation of LYAR by Myc oncoproteins in human tumour tissues lead to CHAC1 suppression, reduction in ROS production and resistance to apoptosis due to other Myc-dependent pathways. As LYAR suppresses target gene transcription through binding to the protein arginine methyltransferase PRMT5, and the PRMT5 inhibitor GSK3326595 is currently tested in clinical trials in patients with solid tumors and non-Hodgkin's lymphoma, we propose that GSK3326595 should be examined of its efficacy in blocking PRMT5 and LYAR interaction, enhancing ROS production, and inducing apoptosis in Myc over-expressing cancers.
Disclosure of interest
The authors declare no potential conflicts of interest.
References
- [1].Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. PMID:8091232 [DOI] [PubMed] [Google Scholar]
- [2].Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, Lunec J, Tweddle DA. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010;70:1377–1388. doi: 10.1158/0008-5472.CAN-09-2598. PMID:20145147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dang CV. MYC on the path to cancer. Cell. 2013;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sun Y, Atmadibrata B, Yu D, Wong M, Liu B, Ho N, Ling D, Tee AE, Wang J, Mungrue IN, et al. Upregulation of LYAR induces neuroblastoma cell proliferation and survival. Cell Death Differ. 2017;24:1645–1654. doi: 10.1038/cdd.2017.98. PMID:28686580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ju J, Wang Y, Liu R, Zhang Y, Xu Z, Wang Y, Wu Y, Liu M, Cerruti L, Zou F, et al. Human fetal globin gene expression is regulated by LYAR. Nucleic Acids Res. 2014;42:9740–9752. doi: 10.1093/nar/gku718. PMID:25092918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li H, Wang B, Yang A, Lu R, Wang W, Zhou Y, Shi G, Kwon SW, Zhao Y, Jin Y. Ly-1 antibody reactive clone is an important nucleolar protein for control of self-renewal and differentiation in embryonic stem cells. Stem Cells. 2009;27:1244–1254. doi: 10.1002/stem.55. PMID:19489080 [DOI] [PubMed] [Google Scholar]
- [7].Chen PH, Shen WL, Shih CM, Ho KH, Cheng CH, Lin CW, Lee CC, Liu AJ, Chen KC. The CHAC1-inhibited Notch3 pathway is involved in temozolomide-induced glioma cytotoxicity. Neuropharmacology. 2017;116:300–314. doi: 10.1016/j.neuropharm.2016.12.011. PMID:27986595 [DOI] [PubMed] [Google Scholar]