

ABSTRACT
Models for the control of global cell-cycle transcription have advanced from a CDK-APC/C oscillator, a transcription factor (TF) network, to coupled CDK-APC/C and TF networks. Nonetheless, current models were challenged by a recent study that concluded that the cell-cycle transcriptional program is primarily controlled by a CDK-APC/C oscillator in budding yeast. Here we report an analysis of the transcriptome dynamics in cyclin mutant cells that were not queried in the previous study. We find that B-cyclin oscillation is not essential for control of phase-specific transcription. Using a mathematical model, we demonstrate that the function of network TFs can be retained in the face of significant reductions in transcript levels. Finally, we show that cells arrested at mitotic exit with non-oscillating levels of B-cyclins continue to cycle transcriptionally. Taken together, these findings support a critical role of a TF network and a requirement for CDK activities that need not be periodic.
KEYWORD: Cell-cycle transcription, mathematical modeling, time-series transcriptomics, transcriptional network
Introduction
A temporal program of cell-cycle transcription is observed across multiple species.1-5 The cell-cycle transcriptional program is characterized by the phase-specific transcription of a large number of genes (∼1000 in budding yeast), which can be organized into clusters based on their timing of expression and regulating transcription factors (TFs).6 The entire program is repeated in each new cell cycle, so that each of the genes oscillates in concert with successive cell-cycle progression.
Historically, the cell-cycle transcriptional program was thought to be controlled by a biochemical oscillator based on the antagonistic interactions between cyclin-dependent kinases (CDKs) and the anaphase-promoting complex/cyclosome (APC/C) (Fig. 1A).7-10 Because CDKs can trigger many phase-specific cell-cycle events, it was easy to imagine that they could also regulate phase-specific transcription by direct phosphorylation of TFs that control clusters of downstream genes (Fig. 1A).7,8,11-19 This model also assumes that the CDK-APC/C network functions as a post-transcriptional biochemical oscillator. While this is certainly true in early embryonic cells, where constitutive input from maternal stores of cyclin RNA is sufficient to drive rapid CDK oscillations,20,21 it is not clear that the CDK-APC/C network in somatic cells or yeast would similarly produce autonomous oscillations at the relevant (much longer) time scale without periodic input from the transcriptional program.
Figure 1.
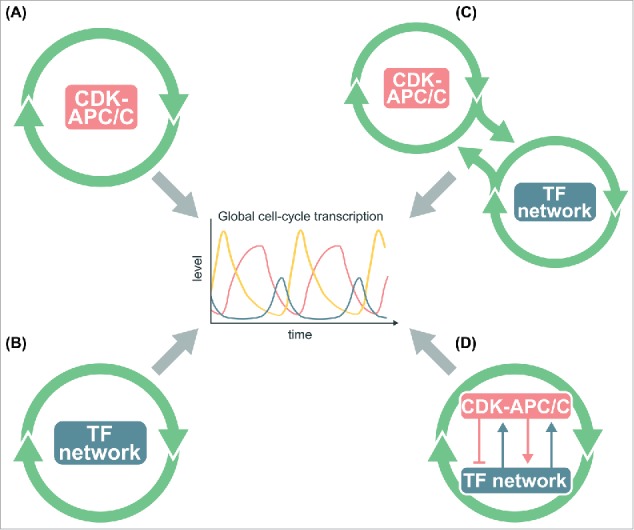
Models for the global control of cell-cycle transcription. (A) The CDK-APC/C network functions as an autonomous oscillator and drives the cell-cycle transcriptional program. (B) The network TFs drive the cell-cycle transcriptional program without CDK-APC/C input. (C) The TF network and CDK-APC/C network can function independently, but are coupled to drive the cell-cycle transcriptional program. (D) CDK-APC/C and TF networks are highly connected and act as a single network to control the cell-cycle transcriptional program. In models (B)-(D), periodic input from CDK-APC/C is not required for oscillations of the transcriptional program.
With the advent of systems-level analyses, it became evident that budding yeast has a highly interconnected network of TFs that can activate/repress each other as well as other cell-cycle genes.22-24 A second model thus suggested that the cell-cycle transcriptional program arose as an emergent property of a TF network, in which sequential waves of expression of TFs trigger phase-specific transcription with connections between M-phase TFs and G1 TFs restarting the cycle (Fig. 1B).23 With appropriate TF activity and stability, such networks could in principle produce phase-specific transcription without input from a CDK-APC/C oscillator.25,26 Support for this idea came from the finding that a large subset of the cell-cycle transcriptional program continued in cells lacking S-phase and mitotic cyclins, as well as in cells with constitutively high mitotic cyclins.27,28 As cyclins and other CDK regulators are expressed periodically as part of the transcriptional program, the finding that a TF network may be able to produce oscillations opened the door for a model in which CDK oscillations were driven by a TF network oscillator.29,30
In the experiments by Orlando et al.,27 about 30% of phase-specific genes were no longer periodically expressed in cells lacking all S-phase and mitotic cyclins, suggesting a third model in which the full program of phase-specific transcription requires some aspect of the CDK-APC/C network and TF network oscillators (Fig. 1C). Subsequent work proposed that the CDK-APC/C oscillator serves as a master oscillator that entrains other autonomous cell-cycle oscillators via a phase-locking mechanism.31,32 In aggregate, the studies described above suggested that the CDK-APC/C and the TF network might represent semi-independent oscillatory systems that were coupled by the fact that CDK activities regulate the TFs and the TFs regulate transcription of several CDK regulators.
When global transcript dynamics were examined in the cdc28–4/cdk1 cells lacking CDK activities, reproducible transcript oscillations were observed for only a fraction of cell-cycle genes.29 Even for these genes, transcript levels were substantially reduced, and the period of the oscillations was extended. Thus, while CDK oscillations were apparently not critical for phase-specific transcription, some level of CDK activity was required for high-amplitude transcriptional oscillations. These findings thus point to a fourth model in which CDK-APC/C and TFs exist in a highly interconnected network (Fig. 1D). This model accommodates data from wild-type cells where the entire network oscillates in concert with cell-cycle progression. In various cyclin or APC/C mutants where CDK-APC/C oscillations and cell-cycle progression are halted, the TF network continues to drive oscillations of portions of the cell-cycle transcriptional program.
While the early CDK-APC/C models arose largely from classical genetic approaches that interrogate small sets of cyclin genes and targets,7,8 the TF network models were identified using systems-level analyses.22-24,27-29 Despite the accumulating evidence that supports the roles of a TF network, it was concluded in a recent publication that the cell-cycle transcriptional program was largely driven by a CDK-APC/C oscillator (Fig. 1A).33 Rahi et al.33 collected time-series transcriptome data of cells depleted of B-cyclins, but the analysis was focused on a very limited set of genes. Furthermore, the transcript dynamics in cells arrested with high levels of B-cyclins were only examined by single-cell fluorescent microscopy for a handful of genes. Thus, there is currently a measure of uncertainty regarding the mechanisms that drive global cell-cycle transcription.
Here we ask whether a more global analysis of the time-series transcriptome data sets of B-cyclin mutant cells27,33 would support a CDK-APC/C model (Fig. 1A) or an integrated network model (Fig. 1D). Of course it can never be ruled out that some undetectable level of residual cyclin-CDK activity is still functional and driving the periodic expression programs observed in cyclin mutant cells.27,29,33 To directly test whether periodic CDK inputs are required for transcriptional oscillations as described in the CDK-APC/C model (Fig. 1A), we block the CDK oscillations in the “on” state using mutants that arrest the cell cycle at mitotic exit and ask whether transcriptional oscillations could be observed. Here we present systems-level evidence from multiple experiments that the cell-cycle transcriptional program emerges from the function of a TF network tightly integrated with CDKs (Fig. 1D),25,27-29 rather than from the entrainment of individual TFs by periodic CDK activities produced by an autonomous CDK-APC/C oscillator (Fig. 1A).33
Results
Phase-specific transcription in cells lacking B-cyclins
In the B-cyclin mutant cells (clb1–6Δ, denoted as clbΔ below) from Orlando et al.,27 no DNA replication, SPB duplication, mitotic events, or inhibition of bud polarity were observed.27,34,35 Nevertheless, it is certainly possible that very low levels of Clb-CDK could drive the transcriptional program while remaining incapable of regulating other cell-cycle events. To test this hypothesis, Rahi et al.33 used a protocol to remove residual mitotic cyclin and then monitored the transcript dynamics in their cyclin mutant cells following a 90-minute pulse of CLN2 expression (cln1–3Δ clb1–6Δ MET-CLN2 0’-90’, denoted as CLN-pulse clbΔ below) (Fig. 2A). They reported that only 3 genes continued to oscillate, although no mechanism driving the oscillations of these 3 genes could be identified.33 However, there were differences in both the experimental approaches (Fig. 2A) and the analysis (Fig. 2B) in these 2 studies, so we set out to determine the source of the conflicting reports.
Figure 2.
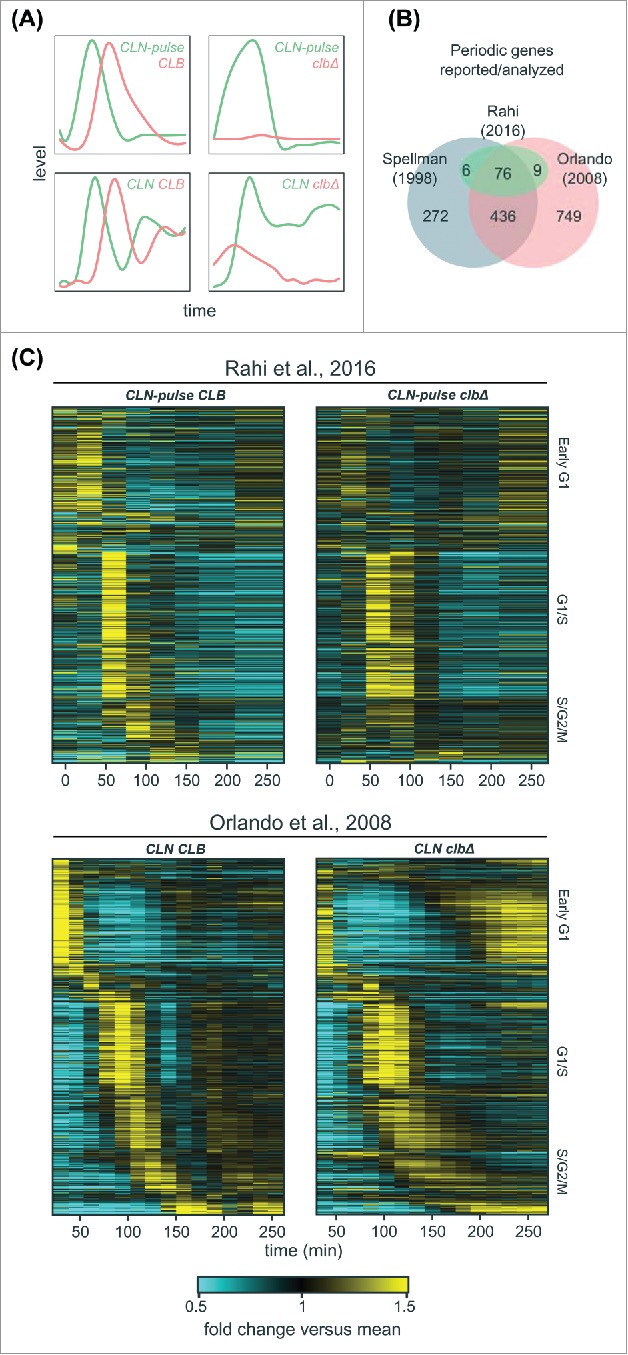
A large program of cell-cycle transcription persists in cells lacking B-cyclins. (A) Cartoon line graphs illustrating the levels of G1 cyclin-CDKs (green) and B-cyclin-CDKs (red) in the time-course experiments from Rahi et al.33 (top panels) and Orlando et al.27 (bottom panels). (B) Venn diagrams showing the relationships of sets of cell-cycle genes reported previously1,27 and those examined by Rahi et al.33 (C) Heat maps showing transcript dynamics of 881 cell-cycle genes (in the same order) in CLB control and clb△ mutant datasets from Orlando et al.27 (bottom panels) and Rahi et al.33 (top panels). In all experiments, early G1 cells were released into the cell cycle (with the CLB expression shut-off for B-cyclin mutants) for time-series gene expression profiling. Transcript levels are depicted as fold change versus mean in each individual dataset. Gene lists and corresponding microarray probes can be found in Table S2. See also Figures S1 and S2.
Several studies indicated that a large number of genes (∼1000) are periodically transcribed in wild-type cells (Fig. 2B).1,5,24,27,28,36-39 Rahi et al.33 focused on the analysis of a small set of 91 genes, so we first examined whether the conclusions reached by the 2 studies was affected by the gene sets chosen for analysis (Fig. 2).
In a previous study, Orlando et al.27 identified 881 genes (70% of wild-type periodic genes) whose phase-specific transcription remained “on schedule” in the clbΔ mutant cells. We directly examined the behaviors of these 881 genes in the RNA-seq data produced by Rahi et al.33 and compared them to data from Orlando et al.27 (Fig. 2C and Table S2). In the CLN-pulse CLB control (“wild-type-like”), the global dynamics during the first cycle were qualitatively similar to that in the wild-type (CLN CLB) cells from Orlando et al.27 (Fig. 2C, left panels), although distinct dynamical differences appear after the second cycle. Similarly, the transcript dynamics of the first cycle in the CLN-pulse clbΔ cells also look similar to the CLN clbΔ cells from Orlando et al.27 (Fig. 2C, right panels), including the partial activation of S/G2/M genes. Strikingly, despite the shut-off of CLN2 expression after 90 minutes in the experiments from Rahi et al.,33 a second peak of expression for early G1 genes was observed in both CLN-pulse CLB control and CLN-pulse clbΔ mutant cells (Fig. 2C, top panels), reminiscent of the results in the wild type and clbΔ mutant cells from Orlando et al.27 (Fig. 2C, bottom panels). Finally, a partial reduction in the peak-to-trough ratios (PTR, max/min) or amplitudes (max-min) of transcript dynamics for these 881 genes were observed in the CLN-pulse clbΔ mutant cells compared with the CLB control, whereas the clbΔ mutant cells from Orlando et al.27 exhibited transcript dynamics with similar PTR and amplitudes (Figure S1). These differences potentially resulted from the cyclin-depletion protocol or the shut-off of CLN2.
In both previous studies, the experimental protocols included a media shift at the beginning of the time course.27,33 To ask whether environmental stress response (ESR)40 or growth rate response (GRR)41 could be contributing to the transcript dynamics shown in Fig. 2C, we eliminated genes that are also in the ESR and GRR program from the 811 genes (Figure S2A). In the remaining 605 genes, similar results were obtained (Figure S2B). Taken together, these analyses suggest that phase-specific transcription can be observed in both data sets when a larger set of genes is analyzed.
Although global dynamics of all 4 data sets look similar in the first cycle, clear differences appear in the second cycle. Specifically, a second peak of G1/S transcription could be observed in both the wild type and clbΔ mutant cells from Orlando et al.27 (Fig. 2C, bottom panels); however, only one robust cycle of G1/S transcription was observed in the cells from Rahi et al. 33 (Fig. 2C, top panels). These differences are consistent with the experimental design in which CLN2 was shut-off 90 min into the experiment by Rahi et al.33 (Fig. 2A), as CLN1/2 are needed for full activation of G1/S transcription partly via inhibition of Whi5.13 In Orlando et al.,27 CLN1/2 were expressed from endogenous promoters at relatively high levels,27,35 likely due to the lack of repression of SBF transcription by Clb2.7,8 Presumably, persistent CLN expression promoted the re-initiation of G1/S transcription even in the clbΔ mutant cells in the experiments from Orlando et al.27 (Fig. 2C, bottom right). In support of the hypothesis that the shut-off of CLN2 rather than the depletion of residual Clb impaired the second cycle of transcription, the CLN-pulse CLB cells (Fig. 2C, top left panel) also have an impaired second cycle of transcription, despite expressing wild-type levels of Clbs. Moreover, it was reported that transcriptional oscillations could persist for some genes if CLN2 expression was maintained constitutively in the clbΔ mutant cells depleted for residual Clb.33 Thus, differences in Cln- rather than Clb-expression likely account for the lack of a measurable second peak of expression in the data from Rahi et al.33
Periodicity-ranking algorithms reveal that SIC1/CDC6/ CYK3 are not particularly periodic with respect to other cell-cycle genes in cells lacking B-cyclins (SIC1/CDC6/CYK3)
Using single-cell fluorescent microscopy, Rahi et al.33 reported that only the transcripts of 3 genes (SIC1/CDC6/CYK3) displayed persistent oscillations in cyclin mutant cells. Of course it is impractical to assay a large set of genes in single-cell assays, so we asked whether these 3 genes would rise to the top of an analysis of a larger set of genes interrogated by time-series RNA-seq in B-cyclin mutant cells. To evaluate the periodicity of all transcripts in the RNA-seq data of the CLN-pulse clbΔ cells, we used 2 distinct periodicity-ranking algorithms as described and implemented previously37,42-44 (Materials and Methods). We chose the de Lichtenberg algorithm because Rahi et al.33 applied a modified form of this algorithm to conclude that there are no clusters of periodic genes in the clbΔ mutant cells. We also used the Lomb-Scargle algorithm that was designed to analyze sparsely or unevenly sampled time-series data, such as those produced by Rahi et al.33
Due to differences in the sampling density, the periodicity measures returned by the algorithms are not directly comparable between the data sets generated by Rahi et al.33 and Orlando et al.27 Thus, we took the 3 genes (SIC1/CDC6/CYK3) that Rahi et al.33 determined were oscillating and asked where they appeared in the rank-ordered lists produced by the algorithms. Although the 2 algorithms differ in their quantitative criteria for periodicity,44 both consistently reported that these 3 genes did not stand out as particularly periodic and were ranked near the bottom of the 91 genes analyzed by Rahi et al.33 and at the bottom of the 881 periodic genes shown in Fig. 2C (Table 1).
Table 1.
Ranks of periodicity scores in the RNA-seq data of CLN-pulse clbΔ experiments. Among the 91 genes analyzed by Rahi et al.33
| LSa |
DLa |
|||
|---|---|---|---|---|
| Gene | Rep. 1 | Rep. 2 | Rep. 1 | Rep. 2 |
| SIC1 | 83 | 66 | 83 | 86.5 |
| CDC6 | 71 | 55 | 79 | 81 |
|
CYK3 |
90 |
90 |
69 |
62 |
| Among the 881 genes analyzed by Orlando et al.27+ CDC6 | ||||
| LSa |
DLa |
|||
| Gene |
Rep. 1 |
Rep. 2 |
Rep. 1 |
Rep. 2 |
| SIC1 | 749 | 599.5 | 717 | 783 |
| CDC6 | 654 | 484.5 | 690 | 717 |
| CYK3 | 807 | 822 | 561 | 479 |
Ranks of periodicity scores by the Lomb-Scargle (LS) or the de Lichtenberg (DL) algorithms.
Whether these 3 specific genes should be considered periodic in this data set is certainly arguable. However, the conclusion that only 3 genes oscillated in the B-cyclin mutant cells is not well supported by the RNA-seq data, which in turn does not argue strongly against the TF network models (Figs. 1B and 1C).
Clusters of phase-specific gene expression are observed in cells lacking B-cyclins
Viewing global transcript dynamics of many genes by heat map can be visually misleading (Fig. 2). So we focused on comparing the behaviors of canonical gene clusters in the budding yeast cell cycle by line graphs (Fig. 3).1,6 These include the Mcm1 cluster (early G1 genes), SBF/MBF cluster (G1/S genes), histone cluster, Hcm1 cluster (S-phase genes), CLB2 cluster (G2/M genes), and Swi5/Ace2 cluster (M/G1 genes). For each cluster except the Swi5/Ace2 cluster, 2 genes whose periodicity was ranked above SIC1/CDC6/CYK3 in the CLN-pulse clbΔ mutant are chosen for illustrations.
Figure 3.

Phase-specific transcription of cell-cycle gene clusters in cells lacking B-cyclins. Line graphs showing transcript dynamics of selected genes in the indicated gene clusters for the CLB control and clbΔ mutant data sets from Orlando et al.27 and Rahi et al.33 Early G1 cells were released into the cell cycle (with the CLB expression shut-off for B-cyclin mutants) for time-series gene expression profiling. CLN and CLB levels are as shown in Figure 2A. Transcript dynamics are plotted as fold change vs. mean as used in Figure 2C. See also Figure S3 and S4.
As shown in Fig. 3, the dynamical behaviors (fold change versus mean) of genes from all clusters (except for the Mcm1 cluster) were remarkably reproducible in the cyclin-depleted cells from both studies (Fig. 3; blue vs. green lines and orange vs. red lines), suggesting that the sequential activation of gene clusters was not driven by residual Clb in the mutant from Orlando et al.27 The conclusion that the CLB2 cluster is not activated in the CLN-pulse clbΔ cells from Rahi et al.33 likely reflects the small number of CLB2 cluster genes used in the analysis. As shown in Figures S3 and S4, among the 30 canonical CLB2 cluster genes,1 many were still expressed similarly in both CLN-pulse CLB and CLN-pulse clbΔ cells. Again, the lack of a strong second pulse for SBF/MBF cluster in the CLN-pulse clbΔ cells is consistent with the loss of positive feedback mediated by G1 cyclin-CDKs as discussed above.13
Phase-specific transcription of the network TFs at lower amplitudes in cells lacking B-cyclins
To ask if these canonical gene clusters could be regulated in a phase-specific manner by a chain of serially activating TFs as previously proposed,23,24,27 we examined the transcript behaviors of the core TFs in the network model (Fig. 4A and Table S3). Consensus TF genes include SWI4 (SBF), HCM1, YHP1, NDD1 (SFF), SWI5, and an output SIC1. As shown in Figs. 4B and 4C, these network TFs components exhibited qualitatively similar dynamics with identical temporal order of phase-specific transcription in all 4 CLB control and clbΔ mutant data sets.27,33 However, a significant reduction in the amplitude of SWI5 transcripts was observed in the CLN-pulse clbΔ mutant cells, as compared with the CLN-pulse CLB cells from Rahi et al.33 This observation is consistent with previous studies showing that the activity of SFF is increased by Clb2-CDK, which phosphorylates the components of SFF.14,15 Thus, loss of the positive feedback between SFF and Clb2-CDK should decrease the amplitude of SFF targets. A similar but less severe reduction in transcript levels was also observed in the clbΔ mutant cells from Orlando et al.27 (Fig. 4B). It is unclear yet whether the higher level of SWI5 might have resulted from residual Clb1 or from the fact that CLN1/2 expression was not shut-off in these experiments.
Figure 4.

Evidence for serial activation of network TFs in cells lacking B-cyclins. (A) Diagram of the network TFs model proposed by Orlando et al.27 SIC1 is an output normally activated by Swi5 during mitotic exit. See Table S3 for edge evidence. (B)(C) Line graphs showing the absolute transcript levels (arbitrary units) of the network TFs components in the CLB control and clbΔ mutant data sets from Orlando et al.27 and Rahi et al.33
Regardless of the source, the significant drop in the transcript levels for CLB2 cluster genes (Fig. 4B, specifically the network TFs genes encoding Swi5 and Ace2) called into question whether they could still function to activate their downstream targets. The ability of these proteins to activate their targets is an important prediction of network models. Paradoxically, the Swi5/Ace2 targets SIC1/CDC6/CYK3, were shown to oscillate in the CLN-pulse clbΔ mutant cells without substantial reduction in amplitude even though the transcript levels of SWI5 and ACE2 peak at only 8% and 10% of wild-type levels (Fig. 4B).33 An explicit assumption was made by Rahi et al.33 that a drop of 10-fold or more in amplitude in the mutant cells would render Swi5 and Ace2 biologically inactive. The idea that a 10-fold drop in the expression of a TF gene should produce a substantial drop in the expression of its target genes seems like a reasonable assumption and would likely fit the intuition of many geneticists. However, systems-level analyses of networks have demonstrated that motifs in gene regulatory networks can buffer substantial perturbations in expression levels.45 The ability to buffer large drops has also been observed in the circadian rhythm network in mouse by a mechanism called paralog compensation.46
Given that these transcription factors are interacting with Clb2-CDK in complex feedback loops, we investigated whether the network could be capable of buffering perturbations in amplitude, specifically asking whether it is reasonable to believe that Swi5/Ace2 could function to activate their targets as predicted by the network models (Figs. 1B-D).
A mathematical model demonstrates that the low-amplitude transcriptional oscillations can be biologically relevant in the context of the global network
As discussed above, it is a common assumption that TFs must achieve some threshold level of expression before they can efficiently activate (or repress) their target genes. This assumption is often made precise with the use of a Hill function nonlinearity in ODE models of transcriptional regulation. Explicitly, if TF A activates transcription of gene B, it is common to model the transcriptional rate of gene B with an ODE model of the form:
where represents a (soft) threshold of activation, below which transcription of gene B may be mostly unaffected by levels of TF A, but above which the contribution of TF A to the transcription of gene B is dramatically enhanced.
Using the above equation to consider the activation of the SIC1 gene by Swi5 in isolation, it is reasonable to assume that the dramatic reduction in the expression of SWI5 in the clbΔ mutant cells would reduce the abundance of Swi5 protein well below the SIC1 activation threshold. Alternatively, if a very low SIC1 activation threshold explains the observation of SIC1 pulses in the clbΔ cells, then the equation would predict SIC1 to be always highly activated in CLB cells.
The logic above assumes that there is no additional input to either gene A or B. However, the regulatory interaction between Swi5 and SIC1 is part of a network with additional inputs including Clb2-CDK and the opposing phosphatase Cdc14 (Fig. 5A). The diminished level of SWI5 transcripts in the clbΔ cells is predicted by the network interactions, as Clb2-CDK increases the transcript level of SWI5 by fully activating SFF. Moreover, Clb2-CDK phosphorylation of Swi5 inhibits its activity by sequestering it in the cytosol (Fig. 5A).15,16 Thus, the timing of SIC1 activation is not closely tied to the accumulation of Swi5, but rather is delayed until Cdc14 dephosphorylates and activates Swi5 during mitotic exit (Fig. 5A).47 In the clbΔ cells, the inhibition of Swi5 by Clb2-CDK is relieved, potentially leading to the robust activation of SIC1 by low-amplitude expression of SWI5. In support of this hypothesis, SIC1 transcripts accumulated earlier in the clbΔ cells than they did in wild-type cells (Fig. 4B).27
Figure 5.
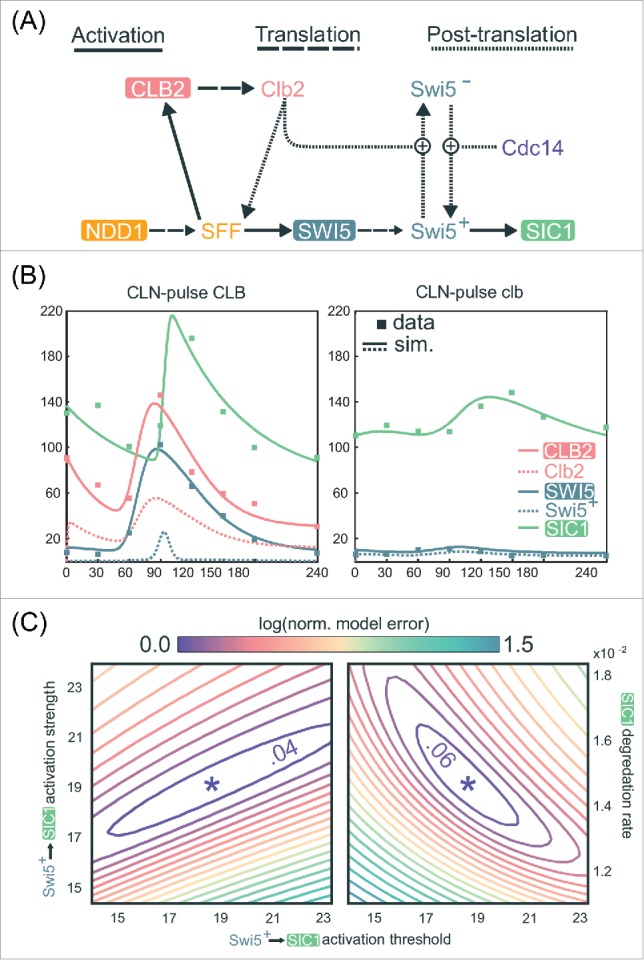
A quantitative model demonstrates the robust activation of SIC1 by low-amplitude SWI5 oscillation. (A) Network topology used for quantitative modeling of transcriptional regulation of SIC1 (see Document S1 for explicit description of equations). (B) Line graphs of selected variables generated by numerical simulation of mathematical model in (A) for a specific choice of parameters, , along with scatter plots of CLB2, SWI5, and SIC1 levels in the RNA-seq data (in FPKM values) from Rahi et al.33 See Document S1 for the parameter values in . (C) Contour plots of the logarithm of the local-minimum-normalized model error over 2 2-dimensional regions of parameters space, centered at . In particular we plot , where is the objective function defined in Document S1, as we independently vary several parameters in a neighborhood of .
See also Figure S5.
To test this hypothesis quantitatively, we constructed a minimal ODE model based only on well-established regulatory interactions (Fig. 5A, Materials and Methods; Document S1).10,48 Specifically, we aimed to determine whether this model could explain the observation that the roughly 10-fold reduction in SWI5 peak expression in the clbΔ cells does not correspondingly reduce SIC1 expression. Remarkably, after parameter optimization, this simple model generates dynamical behaviors of SWI5 and SIC1 that closely match the experimental data produced by Rahi et al.33 (Materials and Methods and Document S1). For the CLN-pulse CLB control cells (Fig. 5B, left), the simulations indeed recapitulate the transient burst of SIC1 expression in late mitosis due to the opposing regulations of Swi5 by Clb2 and Cdc14. Using the same parameters to simulate the CLN-pulse clbΔ mutant cells (Fig. 5B, right), the model also successfully recapitulates the intermediate activation of SIC1 by the low-amplitude oscillation of SWI5. These dynamical behaviors are achievable by a wide variety of parameter choices, including a large range of activation thresholds for Swi5 activation of SIC1 (Figs. 5C, S5A, and S5B). Finally, similar fits to data can also be observed elsewhere in parameter space (Fig. 5C), further supporting the biologic plausibility of a model in which substantially reduced levels of Swi5 could still regulate SIC1 oscillations in clb∆ mutants.
While the above model and parameters may not fully represent the physiology of budding yeast cells, these results clearly demonstrate that in the context of a network, a 10-fold drop in expression of a regulator does not necessarily cripple its ability to regulate downstream targets. Given these findings, it is certainly plausible that a pulse of transcription is indeed passed through the TF network (Fig. 4A). The direct experimental test of this hypothesis is to assess the impact of the deletion of the SWI5 gene on the expression of its oscillating targets. Rahi et al.33 performed this experiment and found that SIC1 expression was eliminated. Both the mathematical modeling and genetic experiments support a model in which the network is robust to amplitude changes in SWI5 and activates downstream targets such as SIC1/CDC6/CYK3. Thus, the parsimonious explanation for the SIC1 oscillations during CDK-APC/C arrests observed by Rahi et al.33 is that they are produced by a TF network that can function in the absence of oscillating CDK activity (Fig. 1D).
The cell-cycle transcriptional program in mitotically arrested cells
The experiments described thus far were aimed at investigating the degree to which CDK-APC/C oscillations are driving oscillations in the cell-cycle transcriptional program by depleting cyclins and thus inhibiting CDK activity. The caveat with all experiments based on conditional mutants is that one can never “prove” the activity is completely eliminated, only that it is undetectable. Another approach to test the same question is to block CDK oscillations but to block them in the “on” state, and then ask how it affects oscillations of the cell-cycle transcriptional program. We chose to analyze transcriptome dynamics in cdc14–3 and cdc15–2 mutant cells where the cell cycle arrests at mitotic exit at restrictive temperature, and the cells contain non-oscillating levels of mitotic cyclins.47,49,50
In budding yeast, Cdc14 phosphatase is a CDK-counteracting phosphatase and the key effector for anaphase progression and mitotic exit.47,51,52 The release of Cdc14 from its sequestration in the nucleolus is promoted by mitotic exit pathways upon anaphase entry (Fig. 6A). Particularly, APC-Cdc20 initiates the FEAR pathway to trigger the early-anaphase Cdc14 release into the nucleus,53-55 while Cdc15 is a component in the MEN pathway that promotes the late-anaphase Cdc14 release into the cytosol.56,57 In arrested cdc14–3 and cdc15–2 mutants, only APC-Cdc20 but not APC-Cdh1 is thought to be active, and thus Clb2 is maintained constitutively at moderate level as compared with the cdc20Δ mutant.47,49,50
Figure 6.
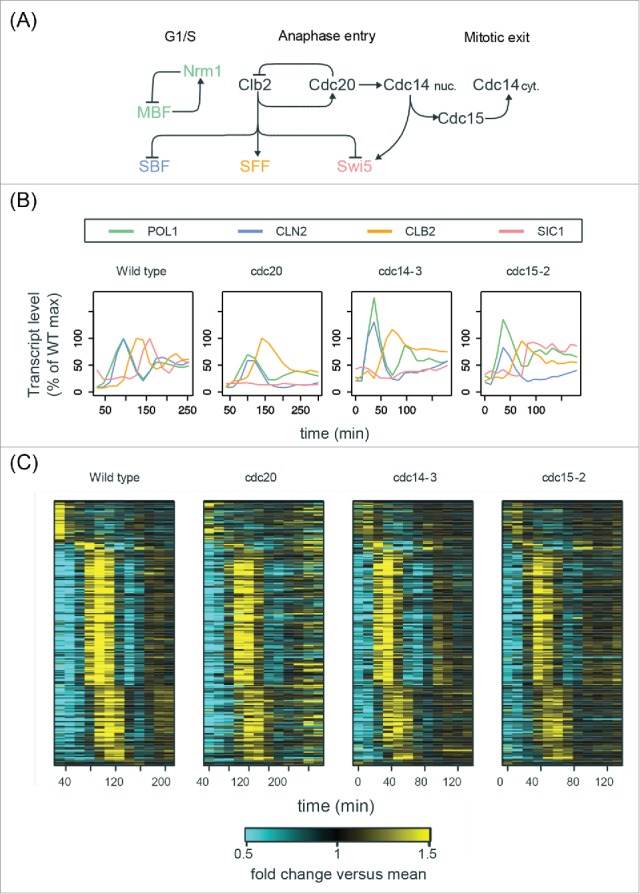
The cell-cycle transcriptional program in mutants defective in mitotic exit. (A) Simplified diagram of part of the CDK-APC/C network (black) controlling progression through mitosis and their established input into network TFs (colored). See Table S3 for edge evidence. (B) Line graphs showing transcript dynamics of canonical targets of network TFs in indicated strains. In all experiments, early G1 cells were released for time-series gene expression profiling by microarray. Transcript levels are depicted as percentage of maximal level in corresponding wild-type controls at the same temperature from previous studies.27,29 Results of the wild-type control from Orlando et al.27 are shown. (C) Heat maps showing transcript dynamics of 249 periodic genes in indicated strains. The wild-type data from Orlando et al.27 are shown. Transcript levels were measured by microarray analysis and are depicted as fold change vs. mean in individual data sets. See also Figures S6 and S7.
Early G1 cells of the cdc14–3 and cdc15–2 mutants were obtained by α-factor arrest at permissive temperature (25°C) and then released into YEP-dextrose medium at restrictive temperature (37°C). Time-series samples were taken and subjected to microarray analysis (Fig. 6). The mitotic arrests of the bulk of populations were confirmed by budding indices, DNA content, and the absence of nuclear division (Figure S6).
First, we confirmed that the transcript behaviors of cell-cycle genes were consistent with well-established regulations by Clb-CDKs (Figs. 6A and 6B). For example, the SBF-regulated gene (CLN2) was indeed expressed at constitutively low level after the first peak during 3 different mitotic arrests (Fig. 6B), indicating the inhibition of SBF by Clb2-CDK.7,8 The SFF-regulated gene (CLB2) was also impaired in its transcriptional downregulation, likely due to constitutive Clb-CDK activity. Interestingly, the Swi5-regulated gene (SIC1) was weakly expressed in both cdc20Δ and cdc14–3 mutants but was robustly activated in the cdc15–2 mutant cells (Fig. 6B), suggesting that nuclear Cdc14 can partially activate Swi5 to trigger M-G1 transcription. Importantly, we observed strong re-initiation of the MBF/Nrm1-regulated gene (POL1) during these mitotic arrests (Fig. 6B),58 consistent with the previous observations in Cdc20-depleted cells.28
To further identify the periodic transcripts in arrested cdc14–3 mutant cells, we ran the de Lichtenberg and Lomb-Scargle algorithms on the microarray data (see Materials and Methods for details). Among the intersection of top periodic genes reported by the 2 algorithms, we examined 249 genes that were also high-confidence periodic genes in wild-type cells reported previously (Figure S7A).28 As shown in Fig. 6C, these 249 genes oscillated for 2 cycles with the same temporal order and similar amplitudes (Figure S7C) in both wild-type cycling cells and arrested cdc14–3 mutant cells. Consistent with the observations in Fig. 6B, a portion of the wild-type transcriptional program was impaired during the mitotic arrests in the cdc14–3 mutant cells, likely due to the constitutive level of Clb-CDKs. Similar results could also be observed in the cdc20Δ and cdc15–2 mutants (Figs. 6C, S7C, and S7D).
We argue that the transcriptional oscillations observed in these mitotically arrested cells (and the Cdc20-depleted cells from Bristow et al.28) did not result from cells leaking through the arrest for the following reasons. First, as indicated by budding indices and the Clb2 level in western blots (Figure S6A; Bristow et al.28: Figure S1), the bulk of the population remained mitotically arrested throughout the experiments, and thus very few cells leaked through the arrest. Second, the amplitudes of transcriptional oscillations generated by a small population of cells leaking through the arrest would be small and should be directly proportional to the size of the leaking population, assuming that the leaking population was perfectly synchronous. Nonetheless, the second cycle of transcription observed in the mutants exhibited amplitudes very similar to wild-type cells where nearly the entire population is cycling (Figs. 6C and S7B; Bristow et al.28: Fig. 3). For instance, the 249 genes shown in Fig. 6C exhibited a second transcriptional cycle with an average of 54.7% and 59.7% amplitudes relative to the first cycle in wild-type and cdc14–3 mutant cells, respectively (Figure S7B). These results are not consistent with a small fraction of cells leaking through the arrests. Finally, consistent with the maintenance of Clb2 levels, the transcript behaviors of established Clb2-CDK transcriptional targets were indeed impaired during the arrest, including the SBF-regulated genes (inhibited by Clb27,8) and the SFF-regulated genes (upregulated by Clb214,15) (Fig. 6B; Bristow et al.28: Fig. 1). These findings are consistent with the results of the single-cell assays.33
Taken together, these data demonstrate that a large subset of the cell-cycle transcriptional program continues to oscillate in a variety of mutant cells arrested with constitutive Clb-CDK activity. These results are not consistent with a model in which oscillations of the CDK-APC/C network predominantly control periodic cell-cycle transcription (Fig. 1A).
Discussion
Determining how the cell-cycle transcriptional program is generated is important for understanding principles of somatic cell-cycle control. Multiple studies have sought to address this question by monitoring transcript dynamics during a variety of CDK-APC/C arrests,27-29,33 and currently there are 2 conflicting models proposed for the global control of cell-cycle transcription. In the first, a CDK-APC/C oscillator drives periodic transcription (Fig. 1A);33 in the other, periodic transcription is driven by a TF network coupled with CDK activities (Fig. 1C).23,27,28 Our findings herein indicate that phase-specific transcription can occur in the absence of periodic input from CDK-APC/C, and are inconsistent with a model in which an autonomous CDK-APC/C oscillator drives cell-cycle transcription. While periodic CDK activities are not required for transcriptional oscillations, it has been shown previously that CDKs contribute to robust, high-amplitude oscillations, likely through various feedback loops (Fig. 7A). We propose a network model in which TFs and CDKs are interconnected (Figs. 1D and 7A). The high degree of interconnection between network TFs and CDKs argues against models where one autonomous oscillator is entraining another during normal cell cycles. Such an integrated network would couple transcriptional oscillations with normal cell-cycle progression as well as promote robustness of cell-cycle oscillations to a variety of perturbations.
Figure 7.
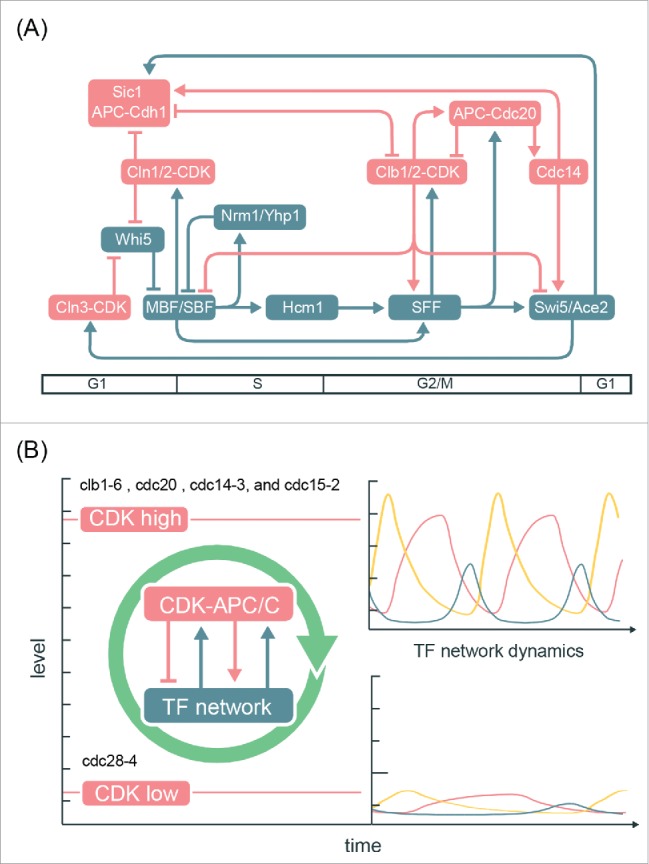
Integrated network model for the control of the cell-cycle transcriptional program in budding yeast. (A) Network diagram incorporating components of the CDK-APC/C model proposed by Rahi et al.33 and the TF network model proposed by Orlando et al.27 Nodes are ordered horizontally by their approximate time of activation during the cell cycle. See Table S3 for edge evidence. (B) Functional outcomes of the cell-cycle-transcriptional program during different CDK-APC/C perturbations with either high or low CDK activities.
Moreover, this integrated model can explain the transcript dynamics observed in multiple mutant backgrounds (Figs. 2C and 6C). In the CDK-APC/C mutants, low levels of CDK activities are expected to impair the capability of the TF network to generate a robust cell-cycle transcriptional program,29 while constitutively high CDK activities (such as cdc14ts, cdc15ts, and cdc20Δ mutant cells) can promote the TF network to generate global cell-cycle transcription even without oscillating CDK activities (Figs. 6B and 6C). The CLN-pulse clbΔ experiments from Rahi et al.33 can then be viewed as a hybrid of high- and low-CDK arrests sequentially, and thus only one robust cycle of transcription could be observed. An interesting question to ask is whether the impaired dynamics of the TF network in low-CDK conditions could be genetically restored, such as by deleting the transcriptional corepressor (Whi5). Analogously, the lethality of cln1 cln2 cln3 triple mutant can be rescued by the sic1 mutation that restores B-cyclin-CDK activities.59,60
Our findings highlight the advantages of a systems-level analysis of the data. By directly comparing the transcriptomic dynamics of an expanded set of 881 genes, we demonstrate that, as observed previously by Orlando et al.,27 cells lacking B-cyclin genes exhibit a very similar set of dynamics to those with their full complement of B-cyclin genes (Fig. 2C). In the cdc20Δ mutant cells where mitotic cyclins are constitutively expressed, CLN2 and CLB2 were not observed to oscillate in single-cell assays from Rahi et al.33 However, a more expansive microarray study of transcriptome dynamics in Cdc20-depleted cells28 uncovered a large number of transcripts continue to oscillate robustly, despite the fact that CLN2 and CLB2 transcripts did not oscillate in these mutants (Figs. 6B and 6C).
Quantitative modeling also revealed that the integrated network could buffer the effects of substantial drops in amplitude for SWI5 and thus offers a simple explanation of the SIC1 transcript dynamics observed in the cyclin mutant cells. This finding underscores the fact that network context for TF-target interactions can have substantial impacts on regulatory outputs. If extracted from the influence of the network, it would be unlikely that a 10-fold drop in SWI5 levels could drive relatively normal levels of transcription of the SIC1 gene.
We have proposed that the ancestral oscillatory mechanism for the cell cycle was likely a TF network,30 while CDKs have been proposed to arise in evolution well after mechanisms of cell division had been established.61 In modern eukaryotes, CDKs and APC/C undoubtedly provide important feedback regulations onto the TF network to refine the control of the cell-cycle transcriptional program and to increase its robustness to perturbation (Fig. 7). Indeed, a recent study in fission yeast has shown that Cdk1 activity can modulate the progression of cell-cycle transcriptional program; however, the potential contribution of a TF network has yet to be determined by experiments analogous to the cdc20Δ, cdc14–3, or cdc15–2 mutants in budding yeast.62 Furthermore, dissecting and establishing the molecular mechanisms that couple the oscillations of CDK-APC/C and the TF network will be imperative for moving toward an integrated model of the eukaryotic cell cycle.
Materials and methods
Requests of further information may be directed to the corresponding author Steven B. Haase ( shaase@duke.edu).
Processing and analyses of RNA-seq data
Raw RNA-Sequencing data from Rahi et al.33 were downloaded from the SRA database (http://www.ncbi.nlm.nih.gov/sra/?term = SRP073907). FASTQ files were aligned using STAR.63 The S. cerevisiae S288C reference genome (Ensembl build R64–1–1) was downloaded from Illumina iGenomes on March 2, 2016 (https://support.illumina.com/sequencing/sequencing_software/igenome.html). Aligned reads were assembled into transcripts, quantified, and normalized using Cufflinks2.64 Samples from all time-series experiments were normalized together using the CuffNorm feature. Replicate time-series data were available on SRA, but not discussed in detail by Rahi et al.33 Therefore, we relied on the SRA annotation to organize them as summarized in Table S1.
The normalized FPKM gene expression outputs (“genes.fpkm_table”) were used in the analyses presented. To avoid fractional and zero values, 1 was added to every FPKM value in each data set. Fractions and zeros were found to interfere with the de Lichtenberg periodicity algorithm, which involves log-transformation of data points (data not shown).
Four time-series data sets corresponding to the CLB control and clbΔ mutant experiments (2 replicates each) described in Rahi et al.33 were run through 2 periodicity-ranking algorithms: Lomb-Scargle (LS) and de Lichtenberg (DL).37,42,43 Each algorithm was implemented as described previously.44 For all time-series data, we first tested a large range of periods from 50–130 minutes as described previously.33 We examined the top-scoring genes in the LS output for each replicate data set. At a p-value cutoff of 0.2, about 600–900 genes were reported as periodic by LS. This numerical p-value cutoff is arbitrary, but the size of the output periodic gene lists matches previous literature.1,5,24,27,28,36-39 For those top periodic genes, we then examined their period length reported by LS in the following table.
| Experiment | Number_genes_LS<0.2 | Average_period_LS<0.2 | Stdev_period_LS<0.2 |
|---|---|---|---|
| CLN-pulse CLB rep1 | 615 | 95.4 | 26.4 |
| CLN-pulse CLB rep2 | 617 | 96.4 | 26.8 |
| CLN-pulse clbΔ rep1 | 971 | 99.4 | 25.8 |
| CLN-pulse clbΔ rep2 | 909 | 100.1 | 25.1 |
These results indicated that the dominant periods in the “most periodic” gene set are 100 ± 30 minutes. This new range eliminates the original lower bound of 50 minutes used by Rahi et al (2016). However, the period length of cycling wild-type cells in rich media is longer than 50 minutes. Although Rahi et al (2016) did not report bud emergence timing for these experiments, the cells were cultured in synthetic media but not rich media. Therefore, an average period length of 100 minutes for CLN-pulse clbΔ and CLN-pulse CLB cells seemed to be a reasonable estimate. Thus, we ran DL at the average period length of 100 minutes, and re-ran LS at a range of 70–130 minute periods. We used the second set of DL and LS results to search for and rank periodic genes in each experiment from Rahi et al.33 (Table 1 and Table S2).
Identification of periodic genes in the cdc14–3 mutant
As described above, the time-series microarray data corresponding to the cdc14–3 mutant were examined using the Lomb-Scargle (LS) and de Lichtenberg (DL) algorithms.37,42,43 By visual inspection (Figure S7B), we chose a range of period length from 60–90 minutes for LS and the period length of 75 minutes for DL. At the p-value cutoffs chosen in a previous study28 (pLS≤ 0.5 and pDL_Per≤ 0.2), 673 genes were reported as periodic in the cdc14–3 mutant. We further intersect these genes with the high-confidence periodic genes in wild type.28 The remaining 249 genes were used for heat maps shown in Fig. 6C.
Yeast strains and cell culture synchronization
The cdc14–3 and cdc15–2 strains are derivatives of S. cerevisiae BF264–15D (ade1 his2 leu2–3,112 trp1–1a). Strain A1268 (W303 cdc14–3 PDS1-HA-LEU2::pds1) was provided by Angelika Amon47 and outcrossed with BF264–15D 5 times. Yeast cultures were grown in standard YEP medium (1% yeast extract, 2% peptone, 0.012% adenine, 0.006% uracil supplemented with 2% sugar). For synchronization by α-factor, cultures were grown in YEP-galactose medium at 25°C and incubated with 50 ng/ml α-factor for 180 minutes. Synchronized cultures were then resuspended in YEP-dextrose medium at 37°C. Aliquots were taken at each time point for subsequent analysis.
RNA extraction and microarray assay
Total RNA was isolated by standard acid phenol protocol and cleaned up by RNA Clean and Concentrator (Zymo Research) if necessary. Samples were submitted to Duke Microarray Facility for labeling, hybridization, and image collection. mRNA was amplified and labeled by Ambion MessageAmp Premier kit (Ambion Biosystems) and hybridized to Yeast Genome 2.0 Array (Affymetrix).
Normalization of microarray data
Previously published data sets used in this study are GEO: GSE8799, GEO: GSE32974, and GEO: GSE49650. All CEL files analyzed in this study were normalized together using the dChip method from the Affy package in Bioconductor as described previously.28
Microscopy
Cells were fixed in 2% paraformaldehyde for 5 minutes at room temperature, washed with PBS, and then resuspended in 30% glycerol for mounting on glass slides. All imaging was performed on Zeiss Axio Observer.
Flow cytometry
Cells were prepared for flow cytometric analysis using SYTOX Green staining as described.65 Graphs were generated using the FlowViz package in Bioconductor in R.
Materials and methods
Literature evidence for cell-cycle regulations relevant to this study. Evidence for the regulations shown in the network diagrams was compiled from previous literature as discussed in the text and additional references 66-76.
Quantitative modeling of SIC1 activation
Justifications and full methodology of the mathematical modeling can be found in Document S1.
Data availability
Newly generated array data and the normalized data matrix have been submitted to GEO: GSE96997.
Supplementary Material
Disclosure of potential conflicts of interest
There are no conflicting interests over the research findings shared in this manuscript.
Acknowledgments
We thank Daniel J. Lew and Adam R. Leman for helpful discussions and critical reading of the manuscript. We are grateful to Angelika Amon for generously providing the yeast strain W303 cdc14–3.
Funding
This work is supported by the Defense Advanced Research Projects Agency (DARPA) Grant #D12AP00025.
References
- [1].Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive Identification of Cell Cycle–regulated Genes of the Yeast Saccharomyces cerevisiae by Microarray Hybridization. Mol Biol Cell. 1998;9:3273-97. doi: 10.1091/mbc.9.12.3273. PMID:9843569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rustici G, Mata J, Kivinen K, Lió P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bähler J. Periodic gene expression program of the fission yeast cell cycle. Nat Genet. 2004;36:809-17. doi: 10.1038/ng1377. PMID:15195092 [DOI] [PubMed] [Google Scholar]
- [3].Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L, Hampton G, Elledge SJ, Davis RW, Lockhart DJ. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27:48-54. doi: 10.1038/83751. PMID:11137997 [DOI] [PubMed] [Google Scholar]
- [4].Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al.. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977-2000. doi: 10.1091/mbc.02-02-0030. PMID:12058064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kelliher CM, Leman AR, Sierra CS, Haase SB. Investigating Conservation of the Cell-Cycle-Regulated Transcriptional Program in the Fungal Pathogen, Cryptococcus neoformans. PLoS Genet. 2016;12:e1006453. doi: 10.1371/journal.pgen.1006453. PMID:27918582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Haase SB, Wittenberg C. Topology and control of the cell-cycle-regulated transcriptional circuitry. Genetics. 2014;196:65-90. doi: 10.1534/genetics.113.152595. PMID:24395825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koch C, Schleiffer A, Ammerer G, Nasmyth K. Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes Dev. 1996;10:129-41. doi: 10.1101/gad.10.2.129 [DOI] [PubMed] [Google Scholar]
- [8].Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993-1007. [DOI] [PubMed] [Google Scholar]
- [9].Cross FR. Two redundant oscillatory mechanisms in the yeast cell cycle. Dev Cell. 2003;4:741-52. doi: 10.1016/S1534-5807(03)00119-9. PMID:12737808 [DOI] [PubMed] [Google Scholar]
- [10].Chen KC, Calzone L, Csikász-Nagy A, Cross FR, Novák B, Tyson JJ. Integrative Analysis of Cell Cycle Control in Budding Yeast. Mol Biol Cell. 2004;15:3841-62. doi: 10.1091/mbc.E03-11-0794. PMID:15169868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].de Bruin RAM McDonald WH, Kalashnikova TI, Yates J III, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117:887-98. doi: 10.1016/j.cell.2004.05.025. PMID:15210110 [DOI] [PubMed] [Google Scholar]
- [12].Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117:899-913. doi: 10.1016/j.cell.2004.05.024. PMID:15210111 [DOI] [PubMed] [Google Scholar]
- [13].Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454:291-6. doi: 10.1038/nature07118. PMID:18633409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pic-Taylor A, Darieva Z, Morgan BA, Sharrocks AD. Regulation of Cell Cycle-Specific Gene Expression through Cyclin-Dependent Kinase-Mediated Phosphorylation of the Forkhead Transcription Factor Fkh2p. Mol Cell Biol. 2004;24:10036-46. doi: 10.1128/MCB.24.22.10036-10046.2004. PMID:15509804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reynolds D, Shi BJ, McLean C, Katsis F, Kemp B, Dalton S. Recruitment of Thr 319-phosphorylated Ndd1p to the FHA domain of Fkh2p requires Clbkinase activity: a mechanism for CLB cluster gene activation. Genes Dev. 2003;17:1789-802. doi: 10.1101/gad.1074103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743-58. [DOI] [PubMed] [Google Scholar]
- [17].Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682-6. doi: 10.1126/science.1172867. PMID:19779198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859-64. doi: 10.1038/nature02062. PMID:14574415 [DOI] [PubMed] [Google Scholar]
- [19].Landry BD, Mapa CE, Arsenault HE, Poti KE, Benanti JA. Regulation of a transcription factor network by Cdk1 coordinates late cell cycle gene expression. The EMBO J. 2014;33:1044-60. doi: 10.1002/embj.201386877. PMID:24714560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hara K, Tydeman P, Kirschner M. A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc Natl Acad Sci USA. 1980;77:462-6. doi: 10.1073/pnas.77.1.462. PMID:6928638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Murray AW, Kirschner MW. Dominoes and clocks: the union of two views of the cell cycle. Science. 1989;246:614-21. doi: 10.1126/science.2683077. PMID:2683077 [DOI] [PubMed] [Google Scholar]
- [22].Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al.. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799-804. doi: 10.1126/science.1075090. PMID:12399584 [DOI] [PubMed] [Google Scholar]
- [23].Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, Wyrick JJ, Zeitlinger J, Gifford DK, Jaakkola TS. Serial Regulation of Transcriptional Regulators in the Yeast Cell Cycle. Cell. 2001;106:697-708. doi: 10.1016/S0092-8674(01)00494-9. PMID:11572776 [DOI] [PubMed] [Google Scholar]
- [24].Pramila T, Wu W, Miles S, Noble WS, Breeden LL. The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev. 2006;20:2266-78. doi: 10.1101/gad.1450606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hillenbrand P, Maier KC, Cramer P, Gerland U. Inference of gene regulation functions from dynamic transcriptome data. Elife. 2016;5:e12188. doi: 10.7554/eLife.12188. PMID:27652904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sevim V, Gong X, Socolar JES. Reliability of Transcriptional Cycles and the Yeast Cell-Cycle Oscillator. PLoS Comput Biol. 2010;6:e1000842. doi: 10.1371/journal.pcbi.1000842. PMID:20628620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Orlando DA, Lin CY, Bernard A, Wang JY, Socolar JES, Iversen ES, Hartemink AJ, Haase SB. Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature. 2008;453:944-7. doi: 10.1038/nature06955. PMID:18463633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bristow SL, Leman AR, Simmons Kovacs LA, Deckard A, Harer J, Haase SB. Checkpoints couple transcription network oscillator dynamics to cell-cycle progression. Genome Biol. 2014;15:446. doi: 10.1186/s13059-014-0446-7. PMID:25200947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Simmons Kovacs LA, Mayhew MB, Orlando DA, Jin Y, Li Q, Huang C, Reed SI, Mukherjee S, Haase SB. Cyclin-dependent kinases are regulators and effectors of oscillations driven by a transcription factor network. Molecular Cell. 2012;45:669-79. doi: 10.1016/j.molcel.2011.12.033. PMID:22306294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Simmons Kovacs LA, Orlando DA, Haase SB. Transcription networks and cyclin/CDKs: the yin and yang of cell cycle oscillators. Cell Cycle. 2008;7:2626-9. doi: 10.4161/cc.7.17.6515. PMID:18758238 [DOI] [PubMed] [Google Scholar]
- [31].Lu Y, Cross FR. Periodic Cyclin-Cdk Activity Entrains an Autonomous Cdc14 Release Oscillator. Cell. 2010;141:268-79. doi: 10.1016/j.cell.2010.03.021. PMID:20403323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Oikonomou C, Cross FR. Frequency control of cell cycle oscillators. Curr Opin Genet Dev. 2010;20:605-12. doi: 10.1016/j.gde.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rahi SJ, Pecani K, Ondracka A, Oikonomou C, Cross FR. The CDK-APC/C Oscillator Predominantly Entrains Periodic Cell-Cycle Transcription. Cell 2016;165:475-87. doi: 10.1016/j.cell.2016.02.060. PMID:27058667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Haase SB, Winey M, Reed SI. Multi-step control of spindle pole body duplication by cyclin-dependent kinase. Nat Cell Biol. 2001;3:38-42. doi: 10.1038/35050543. PMID:11146624 [DOI] [PubMed] [Google Scholar]
- [35].Haase SB, Reed SI. Evidence that a free-running oscillator drives G1 events in the budding yeast cell cycle. Nature [Internet] 1999;401:394-7. Available from: http://www.nature.com/doifinder/10.1038/43927. doi: 10.1038/43927 [DOI] [PubMed] [Google Scholar]
- [36].Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, et al.. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65-73. doi: 10.1016/S1097-2765(00)80114-8. PMID:9702192 [DOI] [PubMed] [Google Scholar]
- [37].de Lichtenberg U Jensen LJ, Fausboll A, Jensen TS, Bork P, Brunak S. Comparison of computational methods for the identification of cell cycle-regulated genes. Bioinformatics. 2005;21:1164-71. doi: 10.1093/bioinformatics/bti093. PMID:15513999 [DOI] [PubMed] [Google Scholar]
- [38].Eser P, Demel C, Maier KC, Schwalb B, Pirkl N, Martin DE, Cramer P, Tresch A. Periodic mRNA synthesis and degradation co‐operate during cell cycle gene expression. Mol Syst Biol. 2014;10. doi: 10.1002/msb.20140001. PMID:24489117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Granovskaia MV, Jensen LJ, Ritchie ME, Toedling J, Ning Y, Bork P, Huber W, Steinmetz LM. High-resolution transcription atlas of the mitotic cell cycle in budding yeast. Genome Biol. 2010;11:R24. doi: 10.1186/gb-2010-11-3-r24. PMID:20193063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241-57. doi: 10.1091/mbc.11.12.4241. PMID:11102521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Slavov N, Botstein D. Coupling among growth rate response, metabolic cycle, and cell division cycle in yeast. Mol Biol Cell. 2011;22:1997-2009. doi: 10.1091/mbc.E11-02-0132. PMID:21525243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lomb NR. Least-squares frequency analysis of unequally spaced data. Astrophys Space Sci. 1976;39:447-62. doi: 10.1007/BF00648343 [DOI] [Google Scholar]
- [43].Scargle DJ. Studies in astronomical time series analysis. II – Statistical aspects of spectral analysis of unevenly spaced data. Astrophys J. 1982;263:835-853. doi: 10.1086/160554 [DOI] [Google Scholar]
- [44].Deckard A, Anafi RC, Hogenesch JB, Haase SB, Harer J. Design and analysis of large-scale biological rhythm studies: a comparison of algorithms for detecting periodic signals in biological data. Bioinformatics. 2013;29:3174-80. doi: 10.1093/bioinformatics/btt541. PMID:24058056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Acar M, Pando BF, Arnold FH, Elowitz MB, van Oudenaarden A. A general mechanism for network-dosage compensation in gene circuits. Science. 2010;329:1656-60. doi: 10.1126/science.1190544. PMID:20929850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, Hogenesch JB. Network features of the mammalian circadian clock. PLoS Biol. 2009;7:e52. doi: 10.1371/journal.pbio.1000052. PMID:19278294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709-18. doi: 10.1016/S1097-2765(00)80286-5. PMID:9885559 [DOI] [PubMed] [Google Scholar]
- [48].Kraikivski P, Chen KC, Laomettachit T, Murali TM, Tyson JJ. From START to FINISH: computational analysis of cell cycle control in budding yeast. NPJ Systems Biology and Applications. 2015;1:15016-. doi: 10.1038/npjsba.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bäumer M, Braus GH, Irniger S. Two different modes of cyclin clb2 proteolysis during mitosis in Saccharomyces cerevisiae. FEBS Lett. 2000;468:142-8. doi: 10.1016/S0014-5793(00)01208-4. PMID:10692575 [DOI] [PubMed] [Google Scholar]
- [50].Yeong FM, Lim HH, Padmashree CG, Surana U. Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28-Clb2 mitotic kinase and the role of Cdc20. Mol Cell. 2000;5:501-11. doi: 10.1016/S1097-2765(00)80444-X. PMID:10882135 [DOI] [PubMed] [Google Scholar]
- [51].Amon A. A decade of Cdc14 – a personal perspective. FEBS J. 2008;275:5774-84. doi: 10.1111/j.1742-4658.2008.06693.x. PMID:19021755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Weiss EL. Mitotic Exit and Separation of Mother and Daughter Cells. Genetics. 2012;192:1165-202. doi: 10.1534/genetics.112.145516. PMID:23212898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shirayama M, Tóth A, Gálová M, Nasmyth K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 1999;402:203-7. doi: 10.1038/46080. PMID:10647015 [DOI] [PubMed] [Google Scholar]
- [54].Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207-20. doi: 10.1016/S0092-8674(02)00618-9. PMID:11832211 [DOI] [PubMed] [Google Scholar]
- [55].Sullivan M, Uhlmann F. A non-proteolytic function of separase links the onset of anaphase to mitotic exit. Nat Cell Biol. 2003;5:249-54. doi: 10.1038/ncb940. PMID:12598903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Charbonneau H, Deshaies RJ. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233-44. doi: 10.1016/S0092-8674(00)80733-3. PMID:10219244 [DOI] [PubMed] [Google Scholar]
- [57].Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398:818-23. doi: 10.1038/19775. PMID:10235265 [DOI] [PubMed] [Google Scholar]
- [58].de Bruin RAM Kalashnikova TI, Chahwan C, McDonald WH, Wohlschlegel J, Yates J III, Russell P, Wittenberg C. Constraining G1-Specific Transcription to Late G1 Phase: The MBF-Associated Corepressor Nrm1 Acts via Negative Feedback. Mol Cell. 2006;23:483-96. doi: 10.1016/j.molcel.2006.06.025. PMID:16916637 [DOI] [PubMed] [Google Scholar]
- [59].Schneider BL, Yang QH, Futcher AB. Linkage of replication to start by the Cdk inhibitor Sic1. Science. 1996;272:560-2. doi: 10.1126/science.272.5261.560. PMID:8614808 [DOI] [PubMed] [Google Scholar]
- [60].Tyers M. The cyclin-dependent kinase inhibitor p40SIC1 imposes the requirement for Cln G1 cyclin function at Start. Proc Natl Acad Sci USA. 1996;93:7772-6. doi: 10.1073/pnas.93.15.7772. PMID:8755551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Krylov DM, Nasmyth K, Koonin EV. Evolution of eukaryotic cell cycle regulation: stepwise addition of regulatory kinases and late advent of the CDKs. Current Biology. 2003;13:173-7. doi: 10.1016/S0960-9822(03)00008-3. PMID:12546794 [DOI] [PubMed] [Google Scholar]
- [62].Banyai G, di FBI, Coudreuse D, Szilagyi Z. Cdk1 activity acts as a quantitative platform for coordinating cell cycle progression with periodic transcription. Nat Commun. 2016;7:1-11. doi: 10.1038/ncomms11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21. doi: 10.1093/bioinformatics/bts635. PMID:23104886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46-53. doi: 10.1038/nbt.2450. PMID:23222703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Haase SB, Reed SI. Improved Flow Cytometric Analysis of the Budding Yeast Cell Cycle. Cell Cycle. 2001;1:117-21. doi: 10.4161/cc.1.2.114 [DOI] [PubMed] [Google Scholar]
- [66].Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne J-B, Reynolds DB, Yoo J, et al.. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99-104. doi: 10.1038/nature02800. PMID:15343339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Workman CT, Mak HC, McCuine S, Tagne J-B, Agarwal M, Ozier O, Begley TJ, Samson LD, Ideker T. A systems approach to mapping DNA damage response pathways. Science. 2006;312:1054-9. doi: 10.1126/science.1122088. PMID:16709784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].McGoff KA, Guo X, Deckard A, Kelliher CM, Leman AR, Francey LJ, Hogenesch JB, Haase SB, Harer JL. The Local Edge Machine: inference of dynamic models of gene regulation. Genome Biol. 2016;17:214. doi: 10.1186/s13059-016-1076-z. PMID:27760556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Di Talia S, Wang H, Skotheim JM, Rosebrock AP, Futcher B, Cross FR. Daughter-Specific Transcription Factors Regulate Cell Size Control in Budding Yeast. PLoS Biol. 2009;7:e1000221. doi: 10.1371/journal.pbio.1000221. PMID:19841732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhu G, Spellman PT, Volpe T, Brown PO, Botstein D, Davis TN, Futcher B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature. 2000;406:90-4. doi: 10.1038/35021046. PMID:10894548 [DOI] [PubMed] [Google Scholar]
- [71].Reimand J, Vaquerizas JM, Todd AE, Vilo J, Luscombe NM. Comprehensive reanalysis of transcription factor knockout expression data in Saccharomyces cerevisiae reveals many new targets. Nucl Acids Res. 2010;38:4768-77. doi: 10.1093/nar/gkq232. PMID:20385592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533-8. doi: 10.1038/35054095. PMID:11206552 [DOI] [PubMed] [Google Scholar]
- [73].Eser U, Falleur-Fettig M, Johnson A, Skotheim JM. Commitment to a Cellular Transition Precedes Genome-wide Transcriptional Change. Mol Cell. 2011;43:515-27. doi: 10.1016/j.molcel.2011.06.024. PMID:21855792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Horak CE, Luscombe NM, Qian J, Bertone P, Piccirrillo S, Gerstein M, Snyder M. Complex transcriptional circuitry at the G1/S transition in Saccharomyces cerevisiae. Genes Dev. 2002;16:3017-33. doi: 10.1101/gad.1039602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Knapp D, Bhoite L, Stillman DJ, Nasmyth K. The transcription factor Swi5 regulates expression of the cyclin kinase inhibitor p40SIC1. Mol Cell Biol. 1996;16:5701-7. doi: 10.1128/MCB.16.10.5701. PMID:8816483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pramila T, Miles S, GuhaThakurta D, Jemiolo D, Breeden LL. Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev. 2002;16:3034-45. doi: 10.1101/gad.1034302 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Newly generated array data and the normalized data matrix have been submitted to GEO: GSE96997.