

Supplemental digital content is available in the text.
Key words/Abbreviations: disability, disease activity, genetics, mental health, pain, rheumatoid arthritis, CARDERA = Combination Anti-Rheumatic Drugs in Early RA, CBT = cognitive-behavioral therapy, DAS28 = disease activity score on a 28-joint count, ESR = erythrocyte sedimentation rate, GWAS = genome-wide association study, HAQ = health assessment questionnaire, HRQoL = health-related quality of life, HWE = Hardy-Weinberg equilibrium, LOCF = last observation carried forward, MAF = minor allele frequency, MCS = mental component summary score, MDD = major depressive disorder, MH = mental health, PGA = patient global assessment, pT = p value threshold, RA = rheumatoid arthritis, RCT = randomized controlled trial, REC = Research Ethics Committee, RF = rheumatoid factor, SE = standard error, SF-36 = short form-36, SJC = swollen joint count, SNP = single nucleotide polymorphism, TJC = tender joint count, VAS = visual analog scale, wGRS = weighted genetic risk score
ABSTRACT
Objective
Reduced mental health (MH) is prevalent in rheumatoid arthritis (RA). Although longitudinal studies are limited, there is evidence that depression is associated with worse disease outcomes. We evaluated reciprocal relationships between MH, RA severity, and genetic risks for depression for 2 years in a well-characterized cohort of RA patients.
Methods
We evaluated 520 early RA patients previously enrolled to two clinical trials. MH was measured using the short form-36 MH domain and mental component summary scores (MCS). MCS/MH associations over 2 years with disease activity (disease activity score on a 28-joint count), disability (health assessment questionnaire), pain visual analog scale scores, and a weighted genetic risk score for depression were tested using linear mixed-effects and regression models.
Results
Poorer MH was associated with worse RA outcomes. Lower MCS scores (indicating worse MH) were seen in patients with a greater genetic risk for depression (weighted genetic risk score: coefficient = −1.21, p = .013). Lower baseline MCS was associated with lower 2-year improvements in disease activity score on a 28-joint count (coefficient = −0.02, p < .001), pain (coefficient = −0.33, p < .001), and health assessment questionnaire (coefficient = −0.01, p = .006). Baseline MCS was associated with changes in the swollen joint count (coefficient = −0.09, p < .001) and patient global assessment (coefficient = −0.28, p < .001) but not the tender joint count (p = .983) and erythrocyte sedimentation rate (p = .973). Only baseline pain visual analog scale (coefficient = −0.07, p = .002) was associated with 2-year changes in MCS.
Conclusions
Reduced baseline MH was associated with lower improvements in disease activity, disability, and pain for 2 years, supporting current national guidelines recommending screening for depression in RA. Pain had a bidirectional relationship with MH. Depression genetic risk had a significant association with MH.
INTRODUCTION
Reduced mental health (MH) is prevalent in rheumatoid arthritis (RA), with major depression present in 16.8% of patients (1). The cause of this excess burden of MH impairment is uncertain. Comorbid depression also seems to have a detrimental impact on the disease course of RA, being associated with increased healthcare use and costs (2) and representing an independent risk factor for nonsuicide-related mortality (3). Determining the cause and effect of depression in RA is, therefore, a key research goal.
Research in this area has mainly involved cross-sectional studies in patients with long-standing RA. These identified associations between depression and pain (4), disability (5), and arthritis disease activity (6). Their cross-sectional nature, however, made it impossible to infer causality. Although longitudinal studies are limited, there is some evidence for a bidirectional effect with pain in patients with musculoskeletal disorders, whereby depression influences pain and vice versa (7,8). There is also some evidence that depression predicts the subsequent disease activity of RA, with an analysis of established RA patients finding a slower rate of decline in disease activity over time in patients with a history of depression (9).
Depression also has a substantial genetic component (10), with several variants associated with depression identified in a genome-wide association study (GWAS) (11). This is consistent with previous twin studies of the heritability of major depressive disorder (MDD), which found that the disorder has a heritability of 48% to 75%, depending on assumptions made on the prevalence of MDD in the general population (12). The role of these in determining MH in RA has not previously been evaluated.
The aim of our study was to evaluate the relationship between MH and disease activity, disability, pain, and genetic risk for depression for 2 years in a well-characterized clinical trial cohort of patients with early RA. The direction of any associations was tested by examining the impact of baseline MH on changes in disease activity, disability and pain, and vice versa.
METHODS
Participants
We studied patients in the Combination Anti-Rheumatic Drugs in Early RA (CARDERA) genetics cohort. It has been described in detail previously (13). In brief, it comprises European ancestry RA patients enrolled to two multicenter randomized controlled trials (RCTs): CARDERA-1 and CARDERA-2 (14,15). Both recruited patients with early RA (<2-year duration) and active disease defined as three of three or more swollen joints, six or more tender joints, 45-minute or more morning stiffness, or erythrocyte sedimentation rate (ESR) of 28 mm/h or greater. CARDERA-1 recruited patients between 2000 and 2002; CARDERA-2 recruited patients between 2003 and 2010. CARDERA-1 randomized patients to receive either (1) methotrexate; (2) methotrexate and ciclosporin; (3) methotrexate and prednisolone; or (4) methotrexate, ciclosporin, and prednisolone. CARDERA-2 randomized patients to receive either (1) methotrexate or (2) methotrexate and anakinra. Because the original aim of the CARDERA studies was to investigate the performance of combination therapy with reference to monotherapy, a placebo group was not assigned. Rheumatoid factor (RF), a biomarker providing clinical information on the antibody composition of patient serum, was assayed as described previously (16). Follow-up was 2 years. The current analysis is restricted to the 520 patients with baseline MH data available.
Disease Outcomes
The following disease outcomes were captured. First, disease activity (how active a patient's arthritis is) was recorded using the disease activity score on a 28-joint count (DAS28). This composite score combines information on the number of swollen and tender joints (assessed by a clinician from 28 joint counts), the patient global assessment (PGA) of disease activity (which involves a patient rating his or her overall disease activity on a 100-mm visual analog scale [VAS]), and the ESR in a mathematical formula to give an assessment of RA activity ranging from 0 to 10. Lower scores indicate less active disease, with scores of higher than 5.1, lower than 3.2, and lower than 2.6 indicating high disease activity, low disease activity, and remission, respectively. Second, disability was recorded using the health assessment questionnaire (HAQ), a patient-completed questionnaire giving a score of function ranging from 0 to 3. HAQ scores of lower than 1, 1 to 2, and higher than 2 indicate mild, moderate, and severe disability, respectively. Third, pain was recorded using a 100-mm patient completed pain VAS, a method for quantifying the severity of self-reported pain (17). Fourth, health-related quality of life (HRQoL) was recorded using the short form-36 (SF-36), which is described in detail hereinafter. In CARDERA-1, the aforementioned outcomes were captured every 6 months. In CARDERA-2, they were captured at 0, 6, 12, and 24 months.
Mental Health
The SF-36 is a generic measure of health status, capturing HRQoL across eight domains (4 physical and 4 mental) (18). These domains are scored from 0 to 100, with higher scores indicating better HRQoL. They can be normalized, z-transformed, and combined into mental component summary (MCS) and physical component summary scores providing summary measures of overall mental and physical health, relative to a population mean score of 50 (SD = 10) (19).
We used the MH domain score and MCS as measures of MH in our analysis. Both have been used to screen for depression, with an MCS cutoff of 42 having a sensitivity and specificity of 74% and 81%, respectively, for detecting depressive disorder (20). They also associate with depression severity, both cross-sectionally and over time (21).
Genotyping
CARDERA patients were genotyped on the Illumina ImmunoChip array (described in detail previously (13)). Single nucleotide polymorphism (SNP) markers were removed that had more than 5% missingness, were duplicates, were not in Hardy-Weinberg equilibrium (HWE, p < .00001), and had a minor allele frequency (MAF) of less than 0.01. From 196,524 pre–quality control markers, 138,873 were available in the final data set. Imputation was subsequently performed using IMPUTE2 (22) and the 1000 Genomes Phase 1 integrated variant version 3 (March 2012) reference panel (variants filtered with a European MAF <0.01). Postimputation SNPs were removed with low INFO scores (<0.7), MAF (<0.05), HWE (p < .000001), and genotyping rate (<0.1), resulting in 429,193 available markers.
Genetic Risk for Depression
The Psychiatric Genomics Consortium MDD GWAS mega-analysis failed to find a locus of genome-wide significance, likely reflecting limited power caused by the genetic architecture of MDD (small effect sizes of individual genetic variants) and the high prevalence of MDD, which increases the difficulty in recruiting large samples of screened, low-risk controls (23). We therefore tested a weighted genetic risk score (wGRS), combining loci of nominal association with MDD for an association with MH in CARDERA. This approach is commonly used in studies of polygenic disorders, whose genetic architecture comprises thousands of very small effect common alleles (24,25). We used a p value threshold (pT) for SNPs to include in the wGRS of 0.05 (representing nominal association with MDD). A continuous wGRS based on MDD GWAS results has been shown to predict depression in independent cohorts, with a pT value of .05 demonstrated to generate a wGRS that most strongly predicts MDD risk (26). After linkage disequilibrium pruning, 3010 SNPs were included in the wGRS. The wGRS was generated for each individual in CARDERA by calculating the number of nominally associated risk alleles they carried, weighted by the log odds ratio from the MDD mega-analysis, summed across SNPs.
Statistical Analysis
Associations with MH
Two different modeling approaches were used to evaluate the relationship between MH, RA severity measures, and genetic risk for depression. The first approach established whether MH was associated with either RA severity measures or the wGRS for depressive disorder over time. This used a linear mixed-effects model, which incorporated either MCS or MH measured at each time point as the response variable regressed on the corresponding predictors (wGRS, DAS28 and its components, HAQ, or pain VAS) from each time point. Effect size estimates (coefficients) for predictor variables provided information on the average differences in the MCS or MH score for the 2-year period relative to the average predictor variable score. We have included a level 1 random effect of individual, fitting random intercepts for each individual. These models have a level 2 random effect for time point, modeling deviation from the overall effect of time point on outcome within each individual as random slopes. We specify this correlation structure using the lme4 package in R (27). The following variables were tested for their associations with MCS: age, sex, disease duration, and RF status. Of these, only sex improved the model fit—as determined using a stepwise selection approach, with the optimal model determined using the Akaike information criterion—and thus, only this variable was included as a covariate (Supplementary Tables A.1 and A.2, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380). The wGRS was standardized to a z score to provide interpretable β values. Examination of residuals from a model containing time, sex, and treatment only confirmed a good model fit (Supplementary Figure A.1, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380), and variance inflation factors calculated for each predictor-ensured multicollinearity between RA outcomes and DAS28 components were not an issue (Supplementary Table A.3, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380).
The second approach evaluated the direction of associations between MH and RA severity by testing whether MH at study baseline was associated with 2-year changes in RA outcomes over time or vice versa. This used linear regression models to look at the association between (a) baseline MCS or MH and 2-year changes in RA severity measures and (b) baseline RA severity measures and 2-year changes in MH scores. These models included the baseline response variable score, treatment, and sex as covariates. Examination of model residuals confirmed good model fits (Supplementary Figure A.1, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380).
Missing Data Imputation
In the original CARDERA-1 trial, missing data at each time point had previously been imputed using last observation carried forward (LOCF) analysis for study end points (DAS28, HAQ, and SF-36). LOCF is a commonly used procedure to address missing data in clinical trials with repeated measures over multiple time points. For each individual, missing values at a time point are replaced by the last observed value of that variable (14,28). In the original CARDERA-2 trial, missing data were not imputed (15). For consistency in the current analysis, we imputed missing, previously nonimputed CARDERA-1 data (swollen joint count [SJC], tender joint count [TJC], ESR, PGA, pain VAS) and missing CARDERA-2 data using LOCF. The largest amount of missing data was seen for pain VAS (11% of observations missing across all time points). We repeated our analysis with nonimputed data only; this excluded a significant impact of the LOCF assumption (Supplementary Table A.4, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380).
Statistical Software
Analyses were performed in the statistical environment R (R Foundation for Statistical Computing, Vienna, Austria), PRSice (Version 1.2) (29), IMPUTE2 (22), and PLINK (Version 1.9) (30).
Ethics, Consent, and Permissions
CARDERA-1 (South Thames Multicentre Research Ethics Committee (REC) reference: MREC (1) 99/04) and CARDERA-2 (South East REC reference: MREC 02/1/089) were ethically approved. Approval was obtained to genotype archived DNA (National Research Ethics Service Committee East of England-Essex REC reference: 11/EE/0544). All patients provided consent.
RESULTS
Patient Baseline Characteristics
Most patients were female (69%, Table 1) and possessed RF in their serum (67%). Baseline disease activity was high (mean DAS28 = 5.88), moderate disability (mean HAQ = 1.56), and short disease duration (mean duration = 3.3 months). Baseline MH was reduced relative to the general population (mean MCS score = 40.6, which is 9.4 U lower than the general population mean).
TABLE 1.
CARDERA Genetics Cohort Baseline Characteristics

Disease Severity Associations With MH
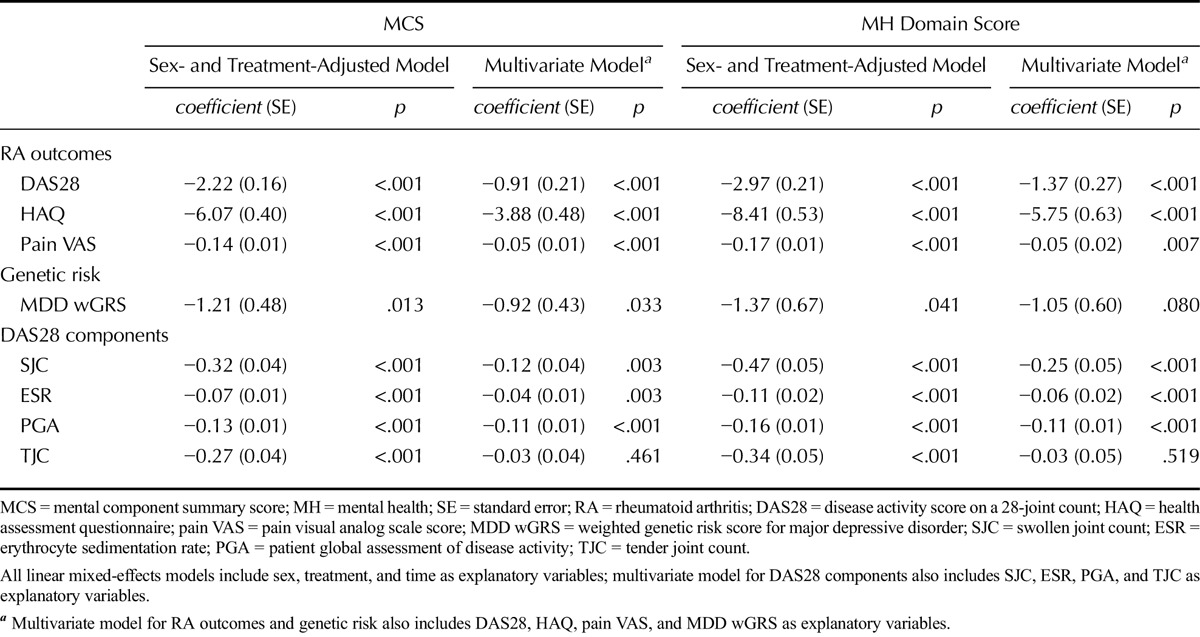
In a sex- and treatment-adjusted linear mixed-effects model, DAS28 (p < .001), HAQ (p < .001), and pain VAS (p < .001) were significantly associated with MCS (Table 2). On average, for 2 years, MCS scores were 2.22, 6.07, and 0.14 units lower per unit increase in DAS28, HAQ, and pain VAS scores, respectively. This indicates that the higher a patient's disease activity, disability, and pain levels, the worse their MH. In multivariate models, the following three disease severity measures retained a highly significant association with MCS (Table 2): HAQ (coefficient = −3.88, p < .001), DAS28 (coefficient = −0.91, p < .001), and pain VAS (coefficient = −0.05, p < .001). Similar associations were seen with the MH domain.
TABLE 2.
Longitudinal Associations Between MH, RA Outcomes, and Depression Genetic Risk Score
Major Depressive Disorder Genetic Risk Score Associations With MH
A significant association was seen between the wGRS for depression and MCS (p = .013) and MH (p = .041) (Table 2). The association with MCS (p = .033) but not MH (p = .080) was retained in multivariate models including DAS28, HAQ, and pain VAS as covariates. Higher wGRS scores, which indicate a greater genetic risk for depression, were associated with worse MH (lower MCS and MH scores) (MCS: coefficient = −1.21; MH: coefficient = −1.37). Repeating the analysis with a linear mixed-effects model that incorporated a wGRS-time interaction term provided some evidence that genetic risk for depression also predicted the rate at which MH improved across time points, with a significant association seen between the wGRS-time term and MH (p = .039, coefficient = −0.83) but not MCS (p = .330, coefficient = −0.30).
Disease Activity Score on a 28-Joint Count Component Associations With MH
In a sex- and treatment-adjusted linear mixed-effects model, all four DAS28 components—SJC, TJC, ESR, and PGA—were associated with MCS and MH scores when tested individually (Table 2). Higher scores in each DAS28 component were associated with lower MCS and MH scores; this indicates that more active disease is linked with poorer MH. On average, for 2 years, MCS scores were 0.32, 0.07, 0.13, and 0.27 units lower per unit increase in SJC, ESR, PGA, and TJC scores, respectively. In multivariate models including all four DAS28 components, the TJC failed to retain a significant association with MCS (p = .461) and MH (p = .519).
Direction of Association Between RA Outcomes and MH
Association Between Baseline Disease Severity and Changes in MH
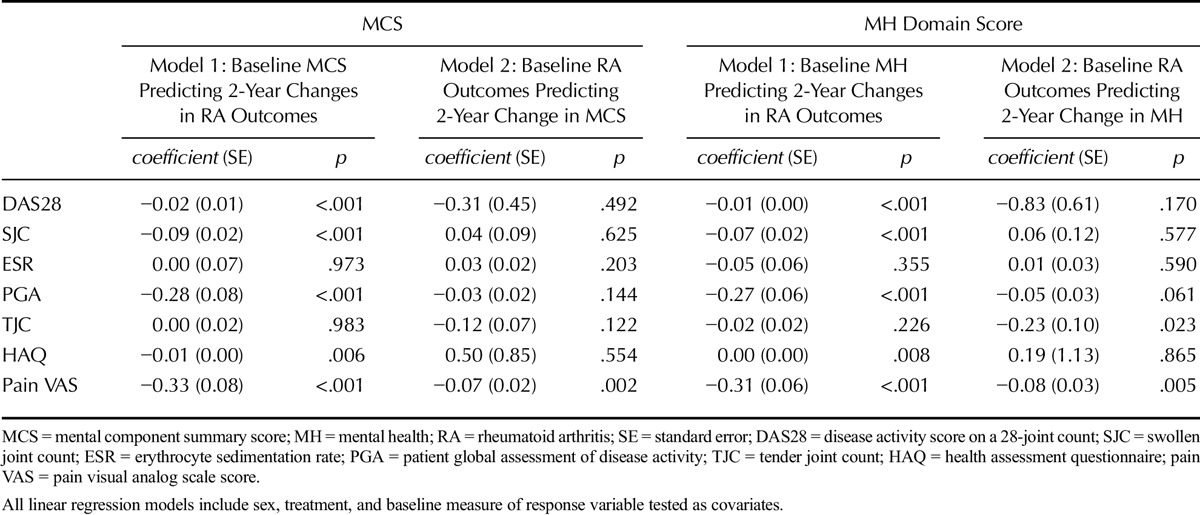
The only baseline RA severity measure that had a significant association with 2-year changes in both MCS and MH scores was pain VAS (Table 3). Higher baseline pain VAS scores (indicating greater levels of pain) were associated with lesser increases in MCS and MH scores (indicating lower improvements in MH). The increase in MCS was 0.07 units less per 1-mm increase in baseline pain VAS. A significant association between the baseline TJC and 2-year changes in MH domain scores was also seen (p = .023), although this variable was not significantly associated with 2-year changes in MCS scores (p = .122).
TABLE 3.
Direction of Associations Between MH and RA Outcomes
Association Between Baseline MH and Changes in RA Outcomes
Baseline MCS and MH domain scores had significant inverse associations with 2-year changes in DAS28 (MCS and MH, p < .001), pain VAS (MCS and MH, p < .001), and HAQ (MCS, p = .006; MH, p = .008) (Table 3). Lower baseline MCS and MH scores (indicating poorer MH) were associated with lesser improvements in DAS28, pain VAS, and HAQ scores. The effect sizes were, however, modest: per 10 unit increase in baseline MCS, the 2-year reductions in DAS28, HAQ, and pain VAS were 0.20, 0.10, and 3.30 units greater, respectively (Table 3).
Dividing patients into octiles based on their baseline MCS and plotting the mean disease severity measure in each octile demonstrated the effect of baseline MCS on RA outcomes (Figure 1). Trends toward (a) worse disease outcomes at each time point and (b) lower improvements in disease outcomes for 2 years across increasing baseline MCS octiles were seen (Figure 1). For 2 years, the mean DAS28, HAQ, and pain VAS scores changed by −1.14, −0.23, and −8.11 U, respectively, in the lowest MCS octile (group 1) and −1.94, −0.49, and −18.49 units, respectively, in the highest MCS octile (group 8).
FIGURE 1.
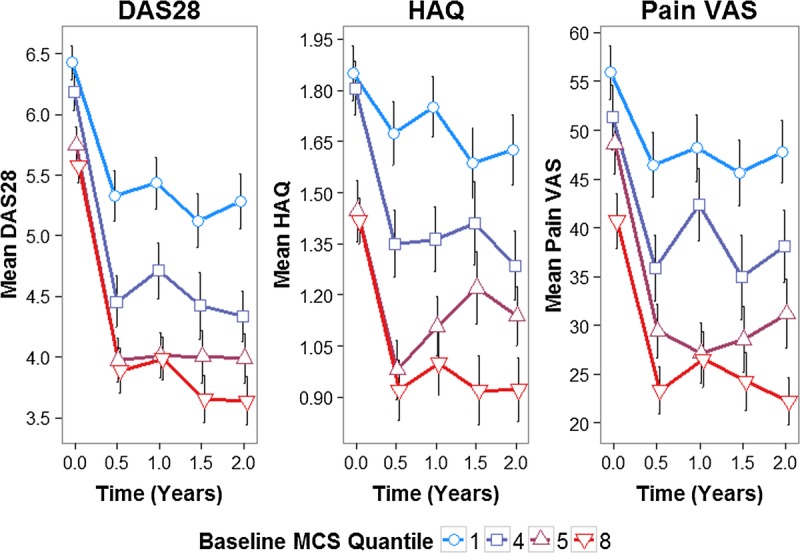
Mean DAS28, HAQ, and pain VAS stratified by baseline MCS octile. MCS divided into octiles (8 quantiles); mean scores with SE bars for octiles 1, 4, 5, and 8 plotted at each time point; to facilitate visual interpretation, octiles 2, 3, 6, and 7 are not plotted, although the same trends are observed (Supplementary Figure A.2, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380). Color image is available only in online version (www.psychosomaticmedicine.org).
Examining individual DAS28 components revealed that baseline MCS and MH scores had significant inverse associations with 2-year changes in the SJC (MCS and MH, p < .001) and PGA (MCS and MH, p < .001) but not the TJC (MCS, p = .983; MH, p = .226) and ESR (MCS, p = .973; MH, p = .355) (Table 3). This differential impact on DAS28 components is shown in Figure 2. For 2 years, the mean SJC, PGA, TJC, and ESR levels changed by −0.17, −13.91, −8.02, and −11.98 units, respectively, in the lowest MCS octile (group 1) and −4.69, −20.46, −5.97, and −11.03, respectively, in the highest baseline MCS octile (group 8).
FIGURE 2.
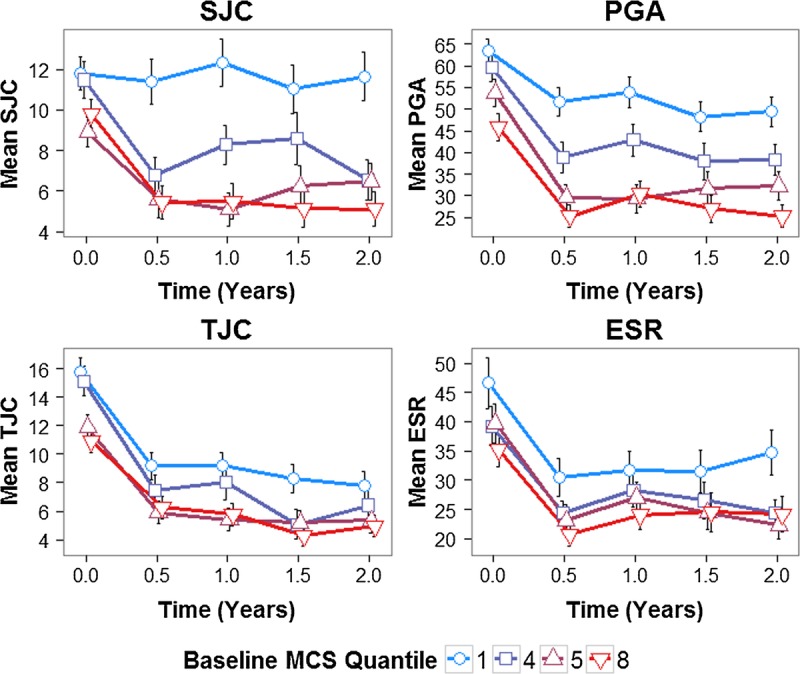
Mean DAS28 components stratified by baseline MCS octile. MCS divided into octiles (8 quantiles); mean scores with SE bars for octiles 1, 4, 5, and 8 plotted at each time point; to facilitate visual interpretation, octiles 2, 3, 6, and 7 are not plotted, although the same trends are observed (Supplementary Figure A.3, Supplemental Digital Content 1, http://links.lww.com/PSYMED/A380). Color image is available only in online version (www.psychosomaticmedicine.org).
DISCUSSION
Our study evaluated the relationship between MH and disease activity, disability, pain, and genetic risk for depression for 2 years in a well-characterized clinical trial cohort of patients with early RA. It has three key findings. The first, and most clinically important, is that low MH was associated with poorer disease outcomes. In a repeated measures analysis, lower MCS and MH scores had significant associations with more active disease, increased disability, and greater pain for 2 years; because MCS and MH scores increased over time, DAS28, HAQ, and pain levels fell. Lower baseline MCS and MH scores (indicating worse MH) were associated with a reduced improvement in disease activity and disability, suggesting that depression predicts the degree to which RA improves over time. The relationship between pain and MH seemed bidirectional, with baseline pain associating with lower improvement in MCS and MH domain scores and vice versa; this is in keeping with existing studies of musculoskeletal disorders (7,8).
The second finding was that swollen, but not TJCs, had a significant association with reduced MH. In a multivariate model incorporating all four DAS28 components, the tender joint component of the DAS28 (TJC) failed to retain a significant association with MCS and MH scores. In established RA patients attending routine clinics, the opposite relationship seems true, with an analysis of the CORRONA registry reporting that a lifetime depression history was associated with slower improvements in the TJC but not the SJC (9). One explanation for the lack of association between MCS/MH scores and the TJC in CARDERA is that the short disease duration of patients means the pain pathway sensitization characterizing fibromyalgic RA—which could be particularly influenced by MH—is yet to occur. An explanation for the association observed between MCS/MH scores and the SJC is that overlapping proinflammatory cytokines, which are present in high levels in early active RA, play important roles in mediating both reduced MH and RA activity. Although this hypothesis is supported by evidence that administering interleukin 1β and tumor necrosis factor α induces depressive behavior in mice (31) and that these cytokines are elevated in the serum of depressed patients (32,33), it fails to explain why baseline MH scores did not predict changes in ESR levels. There is an extensive body of literature investigating the pathophysiology of inflammation-related depression (34), with one proposed mechanism being the activation of the enzyme indoleamine 2,3-dioxygenase by inflammatory cytokines, which catabolizes tryptophan leading to a downstream depletion in serotonin (35)—indeed, inflammation-related depression seems to be dependent on the activation of indoleamine 2,3-dioxygenase (36). In light of this, anti-inflammatory medication has been proposed as a treatment for inflammation-related depression; however, its efficacy is still contested (37); therefore, further research is required in other early active RA cohorts to confirm the generalizability of our results.
Our third finding was that genetic risk for MDD was a significant predictor of MH. We tested a wGRS combining 3010 SNPs of nominal association with MDD in the publicly available Psychiatric Genomics Consortium GWAS for its association with MH in CARDERA. Although a significant association with lower MCS and MH scores was observed, the comparatively large standard error (SE) of the wGRS variable makes any conclusions on its relative importance challenging. The significant interaction term for wGRS-time predicting MH, indicating slower improvements in MH among individuals with high MDD genetic risk, is consistent with previous work indicating that depression genetic risk increases an individual's sensitivity to adverse environmental effects (38,39). Taken together, these findings support the notion that depression is a complex disorder with a modest, albeit important, genetic contribution comprising thousands of alleles of a small effect size.
Our study replicates existing research that depression and pain have a bidirectional relationship. In CARDERA, baseline MCS and MH scores predicted 2-year changes in pain VAS and vice versa. This finding has been documented in psoriatic arthritis; for example, Husted et al. (7) identified a small bidirectional relationship between MCS and pain in 394 patients followed up for a mean of 7.5 years. It has also been reported in patients with persistent back, hip, or knee pain (8), back pain (40,41), and pain from a variety of disorders (42). The complex nature of pain makes it difficult to discern mechanisms by which this pain-depression bidirectional relationship could occur. Possible mechanisms include the following: (1) low mood could affect pain through promoting maladaptive coping strategies, especially catastrophizing (perceiving a situation to be worse than it is) (43); (2) pain could affect MH through reducing daily activities (44) and reducing social activities (45); and (3) imbalances in shared neurotransmitters (serotonin and norepinephrine) in affective and nociceptive pathways could contribute to both mood and pain (44). Further research is required to better characterize the mechanisms underlying this complex relationship.
Supporting our finding that MH predicts disease outcomes across time points, other studies have reported a detrimental impact of reduced MH on patients' responsiveness to anti-inflammatory medication—specifically anti–tumor necrosis factor—as defined by DAS28 change (46,47). This effect is highly relevant to stratified medicine in RA. Although in CARDERA, the impact of baseline MCS on improvements in disease outcomes for 2 years was modest, if considered alongside other poor prognostic markers, such as anti-citrullinated protein antibody status (48), human leukocyte antigen variants (49), smoking, and sex (50), it could provide clinically useful prognostic information, guiding decisions on treatment intensity and facilitating a stratified approach to managing early RA patients.
Our study has several strengths. These include its large size, recruitment from multiple centers spanning two clinical trials, the measurement of multiple disease outcomes in a highly standardized manner, and the short disease duration of RA (mean = 3.3 months), leaving it well placed to examine the effects of MH in very early disease. It also has several weaknesses. As a secondary post hoc analysis of existing RCTs, it did not test a prespecified hypothesis according to a predetermined analysis plan. It evaluated a clinical trial cohort of severe RA patients, limiting its generalizability to patients seen in routine clinical practice. In addition, we only evaluated European ancestry individuals; the relevance of our findings to other ethnic populations is uncertain.
Current National Institute for Health and Care Excellence guidelines for RA management recognize the importance of assessing for comorbid depression, recommending this as part of an annual review process (51). Our findings strongly support this recommendation in early RA. One unresolved issue is the impact of treating depression on the disease course of RA. Although we did not evaluate the impact of MH therapies on RA outcomes, there is some evidence that psychological interventions (such as cognitive behavioral therapy, disclosure therapy, and biofeedback) are useful adjunctive management tools in RA patients. Two systematic literature reviews have evaluated the evidence base for this. Astin et al. (52) reported significant pooled effect sizes for psychological interventions at reducing postinterventional pain, disability, and psychological status across 25 trials. Similarly, Dissanayake and Bertouch (53) found evidence for the efficacy of disclosure therapy and cognitive behavioral therapy with maintenance therapy across four and five studies, respectively. The evidence base is, however, limited with both reviews noting that available trials had methodological limitations. Further research is required to better define the impact of specific psychological interventions at improving disease outcomes in large, well-conducted clinical trials of RA patients.
CONCLUSIONS
In this cohort of 520 early active RA patients, reduced MH (captured using the SF-36) was associated with worse disease outcomes. Lower MCS and MH scores (indicating poorer MH) were significantly associated with more active disease, increased disability, and greater pain for 2 years. Worse baseline MH was associated with lesser improvements in RA outcome measures, suggesting that depression predicts the rate at which RA improves over time. A bidirectional relationship was observed between MH and pain, replicating existing work in musculoskeletal disorders. Depression genetic risk had a significant, albeit modest, impact on MH. Our findings support the current National Institute for Health and Care Excellence RA management guidelines recommending the annual screening of RA patients for comorbid depression. Further research is needed to establish the impact of specific MH management strategies on improving RA outcomes.
Supplementary Material
Acknowledgments
Source of Funding and Conflicts of Interest: This study was supported by Arthritis Research UK (19739, to I.C.S.), the National Institute for Health Research (NIHR) (clinical lectureship, to I.C.S.), the Medical Research Council (studentship, to J.E.), the NIHR Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London, and the NIHR Mental Health Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King's College London (salary support, to F.M. and C.M.L.). It also presents research funded by the NIHR Programme Grants for Applied Research (http://www.ccf.nihr.ac.uk/PGfAR/Pages/Home.aspx) on “Treatment Intensities and Targets in Rheumatoid Arthritis Therapy: Integrating Patients' And Clinicians' Views—The TITRATE Programme (RP-PG-0610-10066).” The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. The funders had no role in the study design, data collection and analysis, data interpretation, the writing of the article, or the decision to submit the article for publication. The authors report no conflicts of interest.
Footnotes
Supplemental Content
REFERENCES
- 1.Matcham F, Rayner L, Steer S, Hotopf M. The prevalence of depression in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology (Oxford) 2013;52:2136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce AT, Smith P, Khandker R, Melin JM, Singh A. Hidden cost of rheumatoid arthritis (RA): estimating cost of comorbid cardiovascular disease and depression among patients with RA. J Rheumatol 2009;36:743–52. [DOI] [PubMed] [Google Scholar]
- 3.Ang DC, Choi H, Kroenke K, Wolfe F. Comorbid depression is an independent risk factor for mortality in patients with rheumatoid arthritis. J Rheumatol 2005;32:1013–9. [PubMed] [Google Scholar]
- 4.Kojima M, Kojima T, Suzuki S, Oguchi T, Oba M, Tsuchiya H, Sugiura F, Kanayama Y, Furukawa TA, Tokudome S, Ishiguro N. Depression, inflammation, and pain in patients with rheumatoid arthritis. Arthritis Rheum 2009;61:1018–24. [DOI] [PubMed] [Google Scholar]
- 5.Peck JR, Smith TW, Ward JR, Milano R. Disability and depression in rheumatoid arthritis. A multi-trait, multi-method investigation. Arthritis Rheum 1989;32:1100–6. [DOI] [PubMed] [Google Scholar]
- 6.Cordingley L, Prajapati R, Plant D, Maskell D, Morgan C, Ali FR, Morgan AW, Wilson AG, Isaacs JD, Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) Barton A. Impact of psychological factors on subjective disease activity assessments in patients with severe rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Husted JA, Tom BD, Farewell VT, Gladman DD. Longitudinal study of the bidirectional association between pain and depressive symptoms in patients with psoriatic arthritis. Arthritis Care Res (Hoboken) 2012;64:758–65. [DOI] [PubMed] [Google Scholar]
- 8.Kroenke K, Wu J, Bair MJ, Krebs EE, Damush TM, Tu W. Reciprocal relationship between pain and depression: a 12-month longitudinal analysis in primary care. J Pain 2011;12:964–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathbun AM, Harrold LR, Reed GW. Temporal effect of depressive symptoms on the longitudinal evolution of rheumatoid arthritis disease activity. Arthritis Care Res (Hoboken) 2015;67:765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000;157:1552–62. [DOI] [PubMed] [Google Scholar]
- 11.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, Tung JY, Hinds DA, Perlis RH, Winslow AR. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016;48:1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGuffin P, Katz R, Watkins S, Rutherford J. A hospital-based twin register of the heritability of DSM-IV unipolar depression. Arch Gen Psychiatry 1996;53:129–36. [DOI] [PubMed] [Google Scholar]
- 13.Scott IC, Rijsdijk F, Walker J, Quist J, Spain SL, Tan R, Steer S, Okada Y, Raychaudhuri S, Cope AP, Lewis CM. Do genetic susceptibility variants associate with disease severity in early active rheumatoid arthritis? J Rheumatol 2015;42:1131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choy EH, Smith CM, Farewell V, Walker D, Hassell A, Chau L, Scott DL, CARDERA (Combination Anti-Rheumatic Drugs in Early Rheumatoid Arthritis) Trial Group Factorial randomised controlled trial of glucocorticoids and combination disease modifying drugs in early rheumatoid arthritis. Ann Rheum Dis 2008;67:656–63. [DOI] [PubMed] [Google Scholar]
- 15.Scott IC, Ibrahim F, Simpson G, Kowalczyk A, White-Alao B, Hassell A, Plant M, Richards S, Walker D, Scott DL. A randomised trial evaluating anakinra in early active rheumatoid arthritis. Clin Exp Rheumatol 2016;34:88–93. [PubMed] [Google Scholar]
- 16.Ma MH, Scott IC, Dahanayake C, Cope AP, Scott DL. Clinical and serological predictors of remission in rheumatoid arthritis are dependent on treatment regimen. J Rheumatol 2014;41:1298–303. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson AM. Assessment of chronic pain. I. Aspects of the reliability and validity of the visual analogue scale. Pain 1983;16:87–101. [DOI] [PubMed] [Google Scholar]
- 18.Ware JE, Jr., Gandek B. Overview of the SF-36 health survey and the international quality of life assessment (IQOLA) project. J Clin Epidemiol 1998;51:903–12. [DOI] [PubMed] [Google Scholar]
- 19.Ware JE, Jr., Kosinski M, Dewey J. How to Score Version 2 of the SF-36 Health Survey (Standard and Acute Forms). Lincoln, RI: Quality Metric, Inc; 2005. [Google Scholar]
- 20.Ware JE, Kosinski M, Keller SF. SF-36 Physical and Mental Health Summary Scales: A User's Manual. Boston, MA: The Health Institute; 1994. [Google Scholar]
- 21.Sturgeon JA, Zautra AJ. State and trait pain catastrophizing and emotional health in rheumatoid arthritis. Ann Behav Med 2013;45:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S, Heath AC, Holsboer F, Lucae S, Madden PA, Martin NG, McGuffin P, Muglia P, Noethen MM, Penninx BP, Pergadia ML, Potash JB, Rietschel M, Lin D, Müller-Myhsok B, Shi J, Steinberg S, Grabe HJ, Lichtenstein P, Magnusson P, Perlis RH, Preisig M, Smoller JW, Stefansson K, Uher R, Kutalik Z, Tansey KE, Teumer A, Viktorin A, Barnes MR, Bettecken T, Binder EB, Breuer R, Castro VM, Churchill SE, Coryell WH, Craddock N, Craig IW, Czamara D, De Geus EJ, Degenhardt F, Farmer AE, Fava M, Frank J, Gainer VS, Gallagher PJ, Gordon SD, Goryachev S, Gross M, Guipponi M, Henders AK, Herms S, Hickie IB, Hoefels S, Hoogendijk W, Hottenga JJ, Iosifescu DV, Ising M, Jones I, Jones L, Jung-Ying T, Knowles JA, Kohane IS, Kohli MA, Korszun A, Landen M, Lawson WB, Lewis G, Macintyre D, Maier W, Mattheisen M, McGrath PJ, McIntosh A, McLean A, Middeldorp CM, Middleton L, Montgomery GM, Murphy SN, Nauck M, Nolen WA, Nyholt DR, O'Donovan M, Oskarsson H, Pedersen N, Scheftner WA, Schulz A, Schulze TG, Shyn SI, Sigurdsson E, Slager SL, Smit JH, Stefansson H, Steffens M, Thorgeirsson T, Tozzi F, Treutlein J, Uhr M, van den Oord EJ, Van Grootheest G, Völzke H, Weilburg JB, Willemsen G, Zitman FG, Neale B, Daly M, Levinson DF, Sullivan PF. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013;18:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013;381:1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peyrot WJ, Milaneschi Y, Abdellaoui A, Sullivan PF, Hottenga JJ, Boomsma DI, Penninx BW. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry 2014;205:113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bates D, Machler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Statistic Soft 2015;67:48. [Google Scholar]
- 28.Scott IC, Ibrahim F, Lewis CM, Scott DL, Strand V. Impact of intensive treatment and remission on health-related quality of life in early and established rheumatoid arthritis. RMD Open 2016;2:e000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Euesden J, Lewis CM, O'Reilly PF. PRSice: polygenic risk score software. Bioinformatics 2015;31:1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bluthé RM, Pawlowski M, Suarez S, Parnet P, Pittman Q, Kelley KW, Dantzer R. Synergy between tumor necrosis factor alpha and interleukin-1 in the induction of sickness behavior in mice. Psychoneuroendocrinology 1994;19:197–207. [DOI] [PubMed] [Google Scholar]
- 32.Owen BM, Eccleston D, Ferrier IN, Young AH. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand 2001;103:226–8. [DOI] [PubMed] [Google Scholar]
- 33.Tuglu C, Kara SH, Caliyurt O, Vardar E, Abay E. Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder. Psychopharmacology (Berl) 2003;170:429–33. [DOI] [PubMed] [Google Scholar]
- 34.Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 2016;16:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry 2002;7:468–73. [DOI] [PubMed] [Google Scholar]
- 36.O'Connor JC, André C, Wang Y, Lawson MA, Szegedi SS, Lestage J, Castanon N, Kelley KW, Dantzer R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci 2009;29:4200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller AH, Raison CL. Are anti-inflammatory therapies viable treatments for psychiatric disorders?: Where the rubber meets the road. JAMA Psychiatry 2015;72:527–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullins N, Power RA, Fisher HL, Hanscombe KB, Euesden J, Iniesta R, Levinson DF, Weissman MM, Potash JB, Shi J, Uher R, Cohen-Woods S, Rivera M, Jones L, Jones I, Craddock N, Owen MJ, Korszun A, Craig IW, Farmer AE, McGuffin P, Breen G, Lewis CM. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol Med 2016;46:759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keers R, Coleman JR, Lester KJ, Roberts S, Breen G, Thastum M, Bögels S, Schneider S, Heiervang E, Meiser-Stedman R, Nauta M, Creswell C, Thirlwall K, Rapee RM, Hudson JL, Lewis C, Plomin R, Eley TC. A genome-wide test of the differential susceptibility hypothesis reveals a genetic predictor of differential response to psychological treatments for child anxiety disorders. Psychother Psychosom 2016;85:146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurwitz EL, Morgenstern H, Yu F. Cross-sectional and longitudinal associations of low-back pain and related disability with psychological distress among patients enrolled in the UCLA Low-Back Pain Study. J Clin Epidemiol 2003;56:463–71. [DOI] [PubMed] [Google Scholar]
- 41.Meyer T, Cooper J, Raspe H. Disabling low back pain and depressive symptoms in the community-dwelling elderly: a prospective study. Spine 2007;32:2380–6. [DOI] [PubMed] [Google Scholar]
- 42.Geerlings SW, Twisk JW, Beekman AT, Deeg DJ, van Tilburg W. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol 2002;37:23–30. [DOI] [PubMed] [Google Scholar]
- 43.Quartana PJ, Campbell CM, Edwards RR. Pain catastrophizing: a critical review. Expert Rev Neurother 2009;9:745–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bair MJ, Robinson RL, Katon W, Kroenke K. Depression and pain comorbidity: a literature review. Arch Intern Med 2003;163:2433–45. [DOI] [PubMed] [Google Scholar]
- 45.Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford) 2001;40:1327–30. [DOI] [PubMed] [Google Scholar]
- 46.Hider SL, Tanveer W, Brownfield A, Mattey DL, Packham JC. Depression in RA patients treated with anti-TNF is common and under-recognized in the rheumatology clinic. Rheumatology (Oxford) 2009;48:1152–4. [DOI] [PubMed] [Google Scholar]
- 47.Kekow J, Moots R, Khandker R, Melin J, Freundlich B, Singh A. Improvements in patient-reported outcomes, symptoms of depression and anxiety, and their association with clinical remission among patients with moderate-to-severe active early rheumatoid arthritis. Rheumatology (Oxford) 2011;50:401–9. [DOI] [PubMed] [Google Scholar]
- 48.Seegobin SD, Ma MH, Dahanayake C, Cope AP, Scott DL, Lewis CM, Scott IC. ACPA-positive and ACPA-negative rheumatoid arthritis differ in their requirements for combination DMARDs and corticosteroids: secondary analysis of a randomized controlled trial. Arthritis Res Ther 2014;16:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Viatte S, Plant D, Han B, Fu B, Yarwood A, Thomson W, Symmons DP, Worthington J, Young A, Hyrich KL, Morgan AW, Wilson AG, Isaacs JD, Raychaudhuri S, Barton A. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality, and treatment response. JAMA 2015;313:1645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scott IC, Lewis CM, Cope AP, Steer S. Rheumatoid arthritis severity: its underlying prognostic factors and how they can be combined to inform treatment decisions. Int J Clin Rheumatol 2013;8:247–63. [Google Scholar]
- 51.National Institute for Health and Care Excellence. Rheumatoid arthritis: the management of rheumatoid arthritis in adults. NICE guidelines [CG79]. NICE Web site. Available at: http://www.nice.org.uk/TA130. Accessed February 11, 2015.
- 52.Astin JA, Beckner W, Soeken K, Hochberg MC, Berman B. Psychological interventions for rheumatoid arthritis: a meta-analysis of randomized controlled trials. Arthritis Rheum 2002;47:291–302. [DOI] [PubMed] [Google Scholar]
- 53.Dissanayake RK, Bertouch JV. Psychosocial interventions as adjunct therapy for patients with rheumatoid arthritis: a systematic review. Int J Rheum Dis 2010;13:324–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.