

Abstract
Purpose of review
Long-acting cabotegravir may provide a novel therapeutic option for both the treatment and prevention of HIV-1 infection that does not necessitate adherence to a daily regimen. The present review will highlight the unique formulation properties and pharmacologic attributes of long-acting cabotegravir nanosuspension.
Recent findings
Cabotegravir is a potent integrase strand transfer inhibitor that has been formulated as an oral tablet for daily administration and as a long-acting injectable nanosuspension. Long-acting cabotegravir is readily absorbed following intramuscular and subcutaneous administration and has an elimination half-life of approximately 40 days, allowing for administration on a monthly or less frequent schedule. Repeat-dose pharmacokinetic studies and population pharmacokinetic modeling indicate monthly and bi-monthly dosing achieves clinically relevant plasma concentrations considered effective for HIV maintenance therapy and that quarterly injections are appropriate for investigation as preexposure prophylaxis. Cabotegravir is primarily metabolized by uridine diphosphate glucuronosyltransferase 1A1 and is unlikely to be impacted by the cytochrome P450 metabolic pathway. In vitro and in vivo data suggest cabotegravir has a low propensity to cause, or be subject to, significant drug interactions.
Summary
The pharmacologic profile of long-acting cabotegravir supports its continued development for both treatment and prevention of HIV-1 infection.
Keywords: cabotegravir, formulation, long-acting, nanosuspension, pharmacokinetics
INTRODUCTION
The advent of highly active antiretroviral therapy has drastically improved both the length and quality-of-life for HIV-infected patients. There are now over 20 antiretrovirals from six drug classes and multiple effective first-line regimens for HIV-1 treatment [1]. Despite these remarkable advances, strict adherence to daily antiretroviral therapy remains critical to maintaining viral suppression, preventing the emergence of drug resistance, and reducing the risk of HIV transmission [2,3]. Long-acting antiretrovirals capable of achieving prolonged exposures at therapeutic concentrations may enable simplified treatment regimens that do not necessitate daily administration.
Cabotegravir (GSK1265744) is a potent integrase strand transfer inhibitor and a structural analogue of dolutegravir. Cabotegravir's unique physiochemical and pharmacokinetic properties have permitted its formulation and delivery both as an oral tablet for daily administration and as a long-acting nanosuspension for monthly to quarterly intramuscular (i.m.) injection. These attributes have supported the ongoing clinical development of cabotegravir for both the treatment and prevention of HIV-1 infection. For HIV treatment, the novel two-drug regimen of oral cabotegravir and rilpivirine, which has also been formulated as a long-acting injection, has demonstrated potent antiviral activity for the maintenance of viral suppression in antiretroviral-naive patients [4]. The safety and efficacy of this dual–drug regimen administered intramuscularly once monthly or once every 2 months is currently under investigation in a phase 2 clinical trial [5]. In addition, data from nonhuman primate rectal and vaginal simian/human immunodeficiency virus (SHIV) challenge models indicate that cabotegravir long acting is a promising candidate for HIV prevention [6▪–8▪]. Cabotegravir long acting, dosed once quarterly, circumvents the need for daily or coitally dependent dosing and may provide a useful alternative for HIV preexposure prophylaxis (PrEP). This review will focus on the formulation properties and manufacturing considerations that enable nanosuspension delivery, and highlight the pharmacologic profile of cabotegravir long acting.
Box 1.

no caption available
FORMULATION AND MANUFACTURING PROCESS
Cabotegravir long acting is a sterile aqueous nanosuspension containing 200 mg/ml cabotegravir free acid as crystalline nano-sized particles with an average particle size of approximately 200 nm. It is manufactured by a wet-bead milling process and terminally sterilized by gamma irradiation [9].
Formulation
Cabotegravir long acting is made with the crystalline-free acid form of cabotegravir, which exhibits low aqueous solubility, a long systemic half-life (approximately 40 h after oral administration), and high antiviral potency. These properties are optimal for a nanosuspension delivery system and permit high drug loading, which in turn minimizes injection volume for a specified dose. Inactive ingredients include polysorbate 20 and polyethylene glycol 3350 as components of a nonionic stabilizing system and mannitol to maintain isotonicity.
Cabotegravir long acting is designed as a dissolution-controlled depot formulation [10]. The rate-limiting step of drug absorption is dissolution of drug particles in interstitial fluid, surrounding the drug depot. In addition to its solubility, the particle size of cabotegravir has an effect on drug dissolution and absorption. Reduction in particle size increases the surface area of drug particles and therefore the dissolution rate. The nanoparticles not only enhance cabotegravir dissolution and absorption, but also improve the syringeability of the formulation.
Manufacturing process
Cabotegravir drug substance is presterilized by gamma irradiation and is manufactured by conventional wet-bead milling. Sterile cabotegravir drug substance undergoes a clean wet-bead milling process with mannitol, polysorbate 20, polyethylene glycol 3350, and water for injection to a target average cabotegravir particle size of 200 nm. The nanosuspension is filled into presterilized depyrogenated glass vials. The vials are stoppered with pre-sterilized stoppers and then sealed. Finally, the sealed vials are sterilized using gamma irradiation at a lethal dose of 25 kGy.
PHARMACOKINETICS
Cabotegravir long-acting pharmacokinetics following intramuscular and subcutaneous administration were first described in a single-dose, dose-ascending study in 72 healthy volunteers [11▪]. In this study, intramuscular gluteal injections were administered at doses of 100, 200, 400, and 800 mg (2 equally split injections of 400 mg), and subcutaneous abdominal injections at doses of 100, 200, and 400 mg (two equally split injections of 200 mg) to cohorts of eight patients (six active/two placebo). Sixteen volunteers received 400 mg i.m. either as a single injection or two equally split injections of 200 mg to assess the differences in cabotegravir disposition following a single versus split dose administration.
The plasma concentration–time profiles were similar between intramuscular and subcutaneous administration, with comparable maximal concentrations (C max) and total plasma exposures achieved with each route of administration (Fig. 1) [11▪,12]. Rapid absorption from the intramuscular and subcutaneous depot was observed, with detectable plasma cabotegravir concentrations observed 4 h after dosing (first sampling time) in all patients. The median time to C max following a single 400-mg dose was approximately 69 days, although this parameter was highly variable among patients, and ranged from 2 to 213 days. Geometric mean concentrations observed 4 weeks after dosing remained over four times the protein-adjusted 90% inhibitory concentration, 0.66 μg/ml, following intramuscular and subcutaneous split injections of 400 mg (2 × 200 mg) and split intramuscular injections of 800 mg (2 × 400 mg). This concentration is comparable with the mean trough concentration (C τ) observed in a 10-day monotherapy study of oral cabotegravir 5 mg in HIV-infected antiretroviral-naive adults [13]. In this study, a C τ of 0.57 μg/ml was associated with a mean –2.2 log10 reduction in HIV RNA, providing a clinically relevant pharmacodynamic target for cabotegravir long acting.
FIGURE 1.
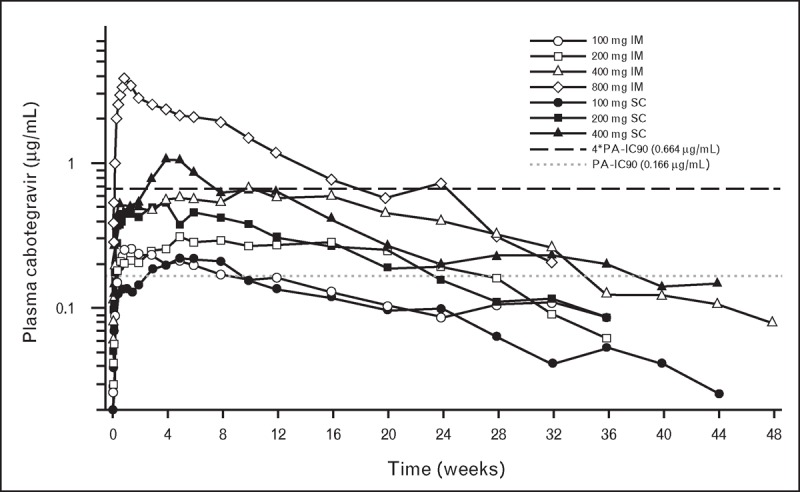
Long-acting cabotegravir pharmacokinetics following single-dose administration in healthy adult individuals. Mean cabotegravir plasma concentration–time profiles following a single long-acting intramuscular (gluteal) or subcutaneous (abdominal) injection in healthy volunteers. The dotted line represents the PA-IC90. The black dashed line represents 4∗PA-IC90. 4∗PA-IC90, four times the PA-IC90; PA-IC90, protein-adjusted concentration required for 90% viral inhibition. Adapted with permission from [11▪,12].
The mean apparent terminal-phase half-life ranged from 25 to 54 days across long-acting dosing cohorts. This is approximately 25-fold longer than the 40-h elimination half-life observed following oral cabotegravir administration [13] and suggests the prolonged concentration–time profiles observed with parenteral administration reflect rate-limiting absorption from the depot rather than elimination from the central compartment. The prolonged apparent plasma half-life noted in this study resulted in detectable cabotegravir concentrations for approximately 52 weeks in some patients.
Splitting the 400-mg dose equally into 2 × 200-mg injections resulted in more than twofold increases in the partial area under the concentration–time curve (AUC) from predose to week 4 (AUC[0–4 wk]), concentration observed 4 weeks after dose (C wk4), and C max relative to unsplit dosing. The total cabotegravir exposures extrapolated to infinity, however, were similar between split and unsplit administration, indicating that dose splitting increases the rate, but not the extent, of cabotegravir absorption. This increase in absorption rate may be because of the increased exposure of nanoparticles to solubilizing biological fluids and subsequent permeation area that results with dose splitting [14].
Pharmacokinetics following repeat doses of cabotegravir long acting have been assessed in a Phase 1, randomized, open-label study [15▪▪]. Healthy volunteers were randomized to receive one of four cabotegravir long-acting dosing regimens: 200 mg subcutaneously monthly × 3 doses, 200 mg i.m. monthly × 3 doses, 400 mg i.m. monthly × 3 doses, or 800 mg i.m. quarterly (every 12 weeks) × 2 doses. All monthly regimens were preceded by an 800-mg i.m. loading dose and the cohorts receiving intramuscular cabotegravir (200 and 400 mg) also received long-acting rilpivirine concomitantly with the third and fourth dose. Forty-seven healthy adults were enrolled, with 40 adults receiving at least one cabotegravir injection (37 adults completed all planned injections).
The mean plasma concentration–time profiles of cabotegravir administered subcutaneously or intramuscularly on monthly or quarterly dosing schedules are provided in Fig. 2. By Day 3, all regimens achieved mean plasma concentrations in excess of 0.66 μg/ml (four times the protein-adjusted 90% inhibitory concentration). Cabotegravir concentrations remained above this threshold throughout the monthly or quarterly dosing interval for each of the tested regimens. These data suggest that a two-drug regimen of cabotegravir long acting and rilpivirine long acting, dosed once monthly, is potentially clinically feasible, was generally well tolerated, and achieved plasma concentrations associated with therapeutic efficacy.
FIGURE 2.
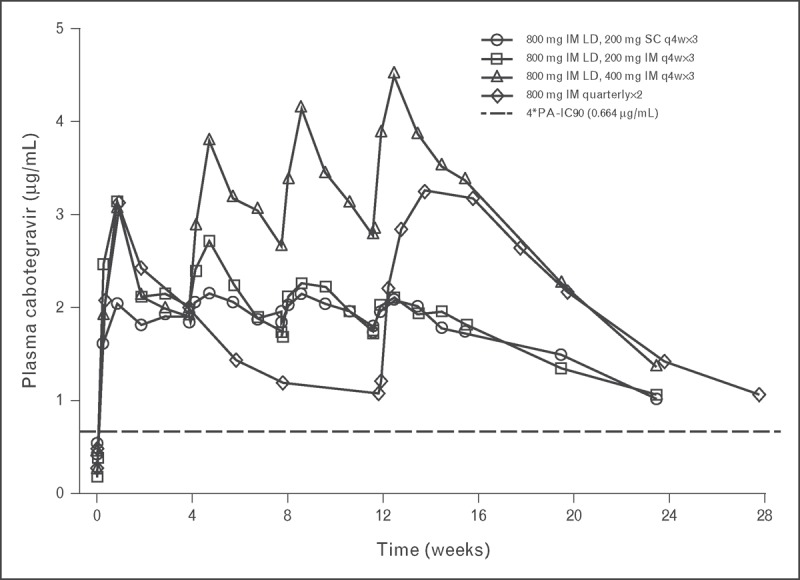
Repeat-dose administration of monthly or quarterly injections of cabotegravir long acting. Mean cabotegravir concentration–time curves following monthly (q4w) or quarterly (every 12 week) repeat-dose administration in healthy individuals. Monthly dosing regimens were preceded by an 800 mg (split 2 × 400 mg) intramuscular LD. 4∗PA-IC90, four times the protein-adjusted concentration required for 90% viral inhibition; LD, loading dose; q4w, every 4 weeks. Adapted with permission from [15▪▪].
Factors affecting long-acting absorption
Wide variability in the apparent absorption rate has been observed following cabotegravir long-acting administration, as evidenced by coefficient of variation of 26.0–96.6% associated with early pharmacokinetic parameter estimates (i.e., partial AUC[0–4wks], C wk4, and C max). The mechanistic explanation and ultimate clinical significance of the variability is currently unknown.
Current phase 1 data suggest concentration–time profiles differ between male and female individuals. Some female individuals appear to have flat concentration–time curves, with blunted C max and a prolonged time to C max relative to male individuals. Despite the difference in the early shape of the concentration–time profile observed in some females, both males and females achieved similar total exposure (AUC[0–∞]), following a single cabotegravir long-acting dose, suggesting the rate, and not the extent of absorption, may be different between males and females. This finding is consistent with a population pharmacokinetic analysis performed using pharmacokinetic data from 346 participants from eight phase 1/2 cabotegravir clinical trials [16]. This analysis showed that both body mass index (BMI) and sex were clinical covariates with significant impact on the rate of absorption following long-acting administration. Slower absorption rates following cabotegravir long-acting administration were associated with increasing BMI [−28% at 90th percentile (30.8 kg/m2) relative to median BMI (25.8 kg/m2)] and female sex (−35.2% relative to males). The converse was observed with lower BMI, with individuals in the 10th percentile BMI (21.6 kg/m2) predicted to have 39% faster absorption rate than those in the 50th percentile BMI. A similar effect of sex and BMI was observed on rilpivirine C max, a measure of absorption rate, following long-acting administration [17]. Current hypotheses surrounding the source of variability in cabotegravir long-acting apparent absorption rate include differences in body fat distribution, muscle mass, injection technique, and physical activity. Additional data obtained following cabotegravir long acting in phase 2 studies will be incorporated into the model to better understand pharmacokinetic variability, particularly given the small sample size of patients receiving single or repeat doses of cabotegravir long acting (n = 121) in phase 1 studies.
Distribution to tissue
As a potential agent for HIV prevention, there is interest in describing the distribution of cabotegravir into the anatomical tissues associated with sexual HIV transmission. The penetration of cabotegravir long acting to vaginal, cervical, and rectal tissue was assessed following a single dose in healthy adults [11▪]. Males (n = 8) and females (n = 8) were administered cabotegravir long acting 400 mg i.m. as either a single injection or as 2 × 200-mg split injections and underwent serial and intermittent pharmacokinetic sampling in blood plasma, vaginal tissue, cervical tissue, and rectal tissue. Distribution into these compartments was limited throughout the 12 weeks of sampling, with median tissue-to-plasma ratios ranging from 16 to 28% in the female genital tract and ≤ 8% in rectal tissue. The relatively low penetration into mucosal tissue is likely attributed to extensive plasma protein binding (>99%) and the low volume of distribution associated with oral cabotegravir dosing. Although partitioning into the sampled matrices was low relative to plasma, the tissue-to-plasma ratios noted in humans were comparable with those observed in the nonhuman primate models for PrEP. Despite limited penetration into these tissues in macaques, protection has been demonstrated in rectal and vaginal SHIV challenge experiments, suggesting adequate drug concentrations are achieved and providing proof-of-concept for long-acting cabotegravir as a PrEP development candidate [18].
DRUG INTERACTION ASSESSMENT
The pathways for cabotegravir metabolism, transport, and excretion have been investigated in both in-vitro and in-vivo studies of the oral formulation [19,20]. It is predicted that the results of clinical mass balance and drug-interaction studies using the oral formulation can be extrapolated to the long-acting product, as the pathways of metabolism and route of elimination are believed to be the same among cabotegravir formulations.
Cabotegravir is primarily metabolized by uridine diphosphate glucuronosyltransferase (UGT) 1A1 with a minor contribution from UGT1A9 [20]. In a single-dose mass balance study utilizing a radiolabeled 30-mg oral dose to investigate the recovery, excretion, and pharmacokinetics of cabotegravir, it was found that cabotegravir is primarily eliminated in the feces (58.5% mean recovery) as unchanged drug [19]. Both unchanged cabotegravir and the glucuronide metabolite were excreted in bile, suggesting that enterohepatic recirculation may contribute to the prolonged pharmacokinetic profiles associated with cabotegravir. A small proportion of radioactivity (26.8%) was recovered in the urine, primarily as the glucuronide metabolite. There was no unchanged cabotegravir detected in the urine.
In-vitro studies demonstrate that cabotegravir has a very low potential to be a significant perpetrator or victim of clinically relevant drug interactions [20]. Cabotegravir is a P-glycoprotein and breast cancer resistance protein substrate; however, the impact of these transporters on cabotegravir disposition is predicted to be minimal provided the compound's high passive membrane permeability. Cabotegravir does not induce the activity of cytochrome P450 (CYP) 1A2, 2B6, or 3A4 in human hepatocytes, nor is there evidence that it inhibits CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 at clinically relevant concentrations. With the exception of organic anion transporter 1 and 3 (OAT1/3), cabotegravir is not a significant inhibitor of the hepatic, intestinal, or renal drug transporters summarized in Table 1. Although further characterization of the effect of cabotegravir on OAT1/3 substrates, specifically those with narrow therapeutic index (e.g., methotrexate), is needed, it is anticipated that cabotegravir has a low potential for causing, or being subject to, clinically significant drug interactions involving drug transporters.
Table 1.
In-vitro assessment of cabotegravir's ability to inhibit hepatic, intestinal, and renal drug transporters
| Hepatic and intestinal drug transporters | |
| Transporter | Half-maximal inhibitory concentration (μmol/l) |
| P-glycoprotein (Pgp) | >30 |
| Breast cancer resistance protein (BCRP) | >30 |
| Bile salt export pump (BSEP) | >30 |
| Multidrug resistance associated protein 2 (MRP2) | >30 |
| Organic cation transporter 1 (OCT1) | >30 |
| Organic anion transporting polypeptide 1B1 (OATP1B1) | >30 |
| Organic anion transporting polypeptide 1B3 (OATP1B3) | >30 |
| Renal drug transporters | |
| Multidrug and toxin extrusion transporter 1 (MATE1) | 18 |
| Multidrug and toxin extrusion transporter 2-K (MATE2-K) | 14 |
| Multidrug resistance associated protein 4 (MRP4) | >30 |
| Organic anion transporter 1 (OAT1) | 0.81 |
| Organic anion transporter 3 (OAT3) | 0.41 |
| Organic cation transporter 2 (OCT2) | >30 |
In-vitro transporter assays were conducted using cells transfected with Pgp, BCRP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1, or OCT2, and vesicles expressing BSEP, MRP2, or MRP4. Established radiolabeled probe substrates and liquid scintillation counting were utilized to determine transporter inhibition at cabotegravir concentrations up to 30 μmol/l [20].
A clinical study utilizing midazolam as a probe substrate confirmed in vivo that cabotegravir does not significantly inhibit or induce the CYP3A metabolic pathway [20]. In this study, 12 healthy individuals received single doses of midazolam 3 mg alone and following 14 days of oral cabotegravir 30 mg/day. The geometric least-squares mean ratios (midazolam and cabotegravir/midazolam alone) and the corresponding 90% confidence intervals for midazolam AUC(0–∞) and C max were 1.08 (0.962, 1.22) and 1.09 (0.944, 1.26), respectively, demonstrating that repeat doses of cabotegravir had no significant effect on the CYP3A pathway and midazolam pharmacokinetics.
A clinical drug-interaction study with repeat doses of etravirine was conducted to assess the impact of CYP450 induction on cabotegravir disposition [21]. CYP-mediated metabolism of cabotegravir is expected to be minimal, as evidenced by the lack of effect on cabotegravir pharmacokinetic parameters when co-administered with repeat doses of etravirine, a known CYP3A inducer in 12 healthy volunteers. The C τ, AUC0-τ, and C max geometric least-squares mean ratios (90% confidence intervals) for cabotegravr plus etravirine/cabotegravir were 0.999 (0.942, 1.06), 1.01 (0.956, 1.06), and 1.04 (0.987, 1.09), respectively.
Cabotegravir also has no significant impact on the pharmacokinetics of rilpivirine, a nonnucleoside reverse transcriptase inhibitor [22]. Co-administration of oral rilpivirine 25 mg and cabotegravir 30 mg daily for 12 days had no significant effect on the pharmacokinetic concentration-time profiles of either drug.
CONCLUSION
Cabotegravir long acting is a sterile aqueous nanosuspension containing 200 mg/ml cabotegravir-free acid as crystalline particles. It is manufactured by a wet-bead milling process and terminally sterilized by gamma irradiation. Cabotegravir possesses a number of favorable pharmacokinetic characteristics, including rapid achievement of clinically relevant concentrations, low risk of drug interactions, and potential for monthly or less frequent dosing. Injectable antiretroviral therapies that are less reliant on patient compliance to daily administration are promising, next-generation HIV treatment and prevention strategies.
Acknowledgements
The authors thank Elizabeth Gould (GSK Clinical Study Operations), Yu Lou (GSK Statistical Support), and Gary Bowers (GSK Drug Metabolism and Pharmacokinetics) for their contributions to the early-phase development of cabotegravir. The authors also thank all of the research volunteers who have participated in the studies reviewed within this manuscript.
Financial support and sponsorship
This work was supported by ViiV Healthcare.
Conflicts of interest
C.T. was a postdoctoral fellow on assignment at GlaxoSmithKline at the time this manuscript was written. S.F., W.S., R.P., and S.P. are employees of and own stock in GlaxoSmithKline.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services 2014 http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf [Accessed 8 December 2014] [Google Scholar]
- 2. Paterson DL, Swindells S, Mohr J, et al. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann Intern Med 2000; 133:21–30. [DOI] [PubMed] [Google Scholar]
- 3. Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011; 365:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Margolis DA, Brinson CC, Eron JJ, et al. 744 and rilpivirine as two drug oral maintenance therapy: LAI116482 (LATTE) week 48 results. Proceedings of the 21st Conference on Retroviruses and Opportunistic Infections; 3–6 March 2014; Boston, MA; abstract 91 LB. [Google Scholar]
- 5. ClinicalTrials.gov. A phase IIb study to evaluate a long-acting intramuscular regimen for maintenance of virologic suppression (following induction with an oral regimen of GSK1265744 and abacavir/lamivudine) in human immunodeficiency virus type 1 (HIV-1) infected, antiretroviral therapy-naive adult subjects. http://clinicaltrials.gov/ct2/show/NCT02120352?term=gsk1265744 &rank=16 [Accessed 8 December 2014]. [Google Scholar]
- 6▪. Radzio J, Spreen W, Yueh YL, et al. The long-acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge. Sci Transl Med 2015; 7:270ra5. [DOI] [PubMed] [Google Scholar]; The efficacy of cabotegravir long-acting as PrEP was demonstrated in an established pigtail macaque model. Monthly intramuscular injections of cabotegravir long-acting protected female macaques from repeated intravaginal SHIV challenges.
- 7▪. Andrews CD, Yueh YL, Spreen WR, et al. A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transl Med 2015; 7:270ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cabotegravir long acting was protective against repeat high-dose intravaginal SHIV challenges in a rhesus macaque animal model, providing support for continued development of cabotegravir as PrEP in women.
- 8▪. Andrews C, Spreen W, Mohri H, et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science 2014; 343:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors establish proof-of-concept for the protective efficacy of cabotegravir long acting against intrarectal SHIV challenge in a nonhuman primate model.
- 9. Mundhra D, Pan R. US patent application 20130171214. VIIV Healthcare Company, assignee. Issued 4 July 2013. [Google Scholar]
- 10. Bari H. A prolonged release parenteral drug delivery system: an overview. Int J Pharm Sci Rev Res 2010; 3:1–11. [Google Scholar]
- 11▪. Spreen W, Ford SL, Chen S, et al. GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr 2014; 67:481–486. [DOI] [PubMed] [Google Scholar]; This study is the first report of cabotegravir long-acting single-dose pharmacokinetics in healthy volunteers.
- 12. Spreen WR, Margolis DA, Pottage JC., Jr Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS 2013; 8:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spreen W, Min S, Ford SL, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials 2013; 14:192–203. [DOI] [PubMed] [Google Scholar]
- 14. Zuidema J, Kadir F, Titulaer HAC, Oussoren C. Release and absorption rates of intramuscularly and subcutaneously injected pharmaceuticals (II). Int J Pharm 1994; 105:189–207. [Google Scholar]
- 15▪▪. Spreen W, Williams P, Margolis D, et al. Pharmacokinetics, safety, and tolerability with repeat doses of GSK1265744 and rilpivirine (TMC278) long-acting nanosuspensions in healthy adults. J Acquir Immune Defic Syndr 2014; 67:487–492. [DOI] [PubMed] [Google Scholar]; This study describes the pharmacokinetics of cabotegravir long acting and rilpivirine long acting following repeat-dose administration in healthy volunteers. The prolonged plasma exposures achieved in this phase 1 study support treatment and prevention regimens that are administered on a monthly or less frequent basis.
- 16. Ford SL, Chiu J, Lovern M, et al. Population PK approach to predict cabotegravir (CAB, GSK1265744) long-acting injectable doses for phase 2b. Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 5–9 September 2014; Washington, DC; abstract H-645. [Google Scholar]
- 17. Jackson AG, Else LJ, Mesquita PM, et al. A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for preexposure prophylaxis. Clin Pharmacol Ther 2014; 96:314–323. [DOI] [PubMed] [Google Scholar]
- 18. Spreen W, Rinehart A, Smith K, et al. HIV PrEP dose rationale for cabotegravir (GSK1265744) long-acting injectable nanosuspension. Proceedings of the HIV Research for Prevention; 28–31 October 2014; Cape Town, South Africa; abstract A-671-0009-01071. [Google Scholar]
- 19. Culp A, Bowers G, Gould E, et al. Metabolism, excretion, and mass balance of the HIV integrase inhibitor, cabotegravir (GSK1265744) in humans. Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 5–9 September 2014; Washington, DC; abstract H-1010. [Google Scholar]
- 20. Reese M, Ford S, Bowers G, et al. In vitro drug interaction profile of the HIV integrase inhibitor, GSK1265744, and demonstrated lack of clinical interaction with midazolam. Proceedings of the 15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy; 19–21 May 2014; Washington, DC; abstract PP_20. 2014. [Google Scholar]
- 21. Ford SL, Gould E, Chen S, et al. Effects of etravirine on the pharmacokinetics of the integrase inhibitor S/GSK1265744. Antimicrob Agents Chemother 2013; 57:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ford SL, Gould E, Chen S, et al. Lack of pharmacokinetic interaction between rilpivirine and integrase inhibitors dolutegravir and GSK1265744. Antimicrob Agents Chemother 2013; 57:5472–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]