

Abstract
The pool of abundant chiral terpene building blocks (i.e. “chiral pool terpenes”) has long served as a starting point for the chemical synthesis of complex natural products, including many terpenes themselves. As inexpensive and versatile starting materials, such compounds continue to influence modern synthetic chemistry. This review highlights 21st century terpene total syntheses which themselves use small, terpene-derived materials as building blocks. An outlook to the future of research in this area is highlighted as well.
Graphical abstract
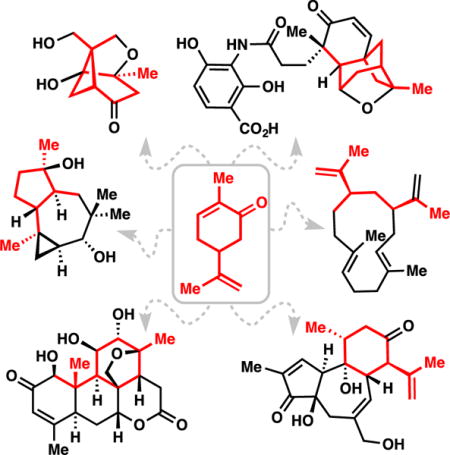
1. Introduction
Naturally occurring terpenes and their derivatives have profoundly impacted the human experience.1 As flavors, fragrances, poisons, and medicines, nearly every human on earth has experienced their effects. As potential fuels,2 monomers for polymer synthesis,3 biochemical signaling agents,1 sources of chirality for synthetic reagents and catalysts,4 and starting materials for organic synthesis, terpenes have also impacted virtually every area of modern chemistry. Along with carbohydrates and amino acids, small chiral terpenes collectively form what is commonly referred to as the “chiral pool,” that is, the collection of abundant chiral building blocks provided by nature. Owing to their low cost, high abundance, and general renewability, the chiral pool has been extensively utilized by synthetic chemists in the synthesis of both natural products as well as pharmaceutical agents and dozens of reviews, books, and highlights exist on this topic.5–11 In particular, the ability to convert one terpene into another was recognized long before the biogenetic “isoprene rule” was formally delineated.12–14 Coupled with advances in spectroscopy and separation techniques, the last 50 years have witnessed an explosion in synthetic terpene research resulting in the total synthesis of many complex terpene natural products, the rise of the semisynthetic steroid field, and the FDA-approval of a variety of terpene-based drugs.15 Even considering the enormous advances in asymmetric synthesis developed during the 20th century,16 the use of chiral terpenes as starting materials for terpene synthesis continues unabated today. Multiple recent reviews on the total synthesis of complex terpenes exist.17–20 This review focuses on complex terpene total syntheses utilizing the chiral pool of terpenes as starting materials and effort has been made to avoid overlap with an excellent 2012 review by Gaich and Mulzer on this topic.21 In addition, the material discussed herein is limited solely to total syntheses appearing in the 21st century and also largely omits meroterpenes, terpene/alkaloid hybrids, and other compounds of “mixed” biosynthetic origins. The semisynthesis of steroid derivatives, to which multiple books and reviews have been devoted, are also not highlighted herein.22,23
2. Starting Points and Historical Perspective
Chiral pool terpene syntheses are influenced by three main factors: (i) the current availability of the starting terpene building blocks, (ii) the current state of the art in synthetic methodology, and (iii) the creativity of the practitioner. With regard to the first point, Figure 1 presents a general depiction of the most frequently utilized chiral pool terpenes in total synthesis. In addition, their current lowest available prices from Sigma-Aldrich are also shown.24 It should be noted that the enantiomeric purity of many terpene-building blocks are variable depending on the source and this information is not always stated.10 As many terpenes are liquids or oils, they cannot be crystallized to enantiopurity directly. Moreover, even if a terpene starting material is of high enantiomeric excess, it may be only available as one enantiomer. Sometimes this is not a problem as a convenient asymmetric method exists to prepare the needed enantiomer, or a related terpene can be converted into the scarcer enantiomer. Many of these points will be further discussed below.
Figure 1.
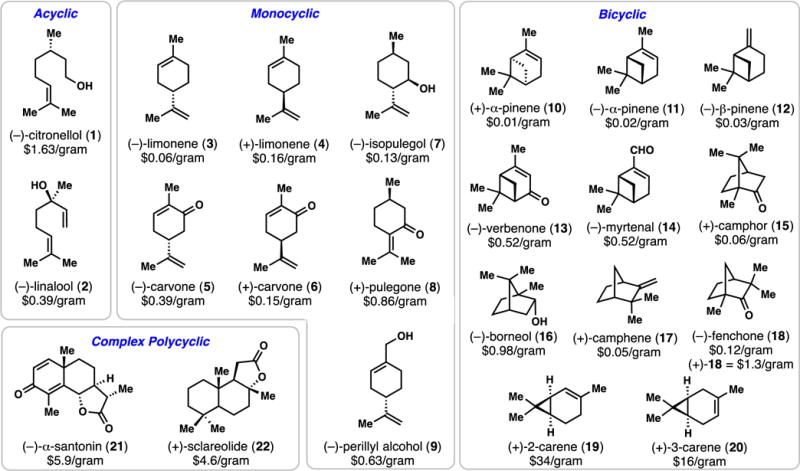
Chiral pool terpenes of both historical and modern use in natural product synthesis.
(−)-Citronellol (1) serves as a common acyclic, chiral pool terpene building block and is easily transformed into both citronellal and citronellic acid, two useful synthetic derivatives, via oxidation. A review on the use of citronellal in synthesis has been reported.25 While the (+)-enantiomer of 1 is approximately twenty times more expensive, either enantiomer is readily prepared from geraniol via enantioselective reduction.26 Similarly, linalool (2), which is most readily available as the (−) enantiomer, can be easily prepared in either enantiomeric form though asymmetric epoxidation of geraniol, mesylation, and reductive ring opening.27
The monocyclic monoterpenes represent widely utilized building blocks in polycyclic terpene synthesis and many chemical transformations.10,11 The chiral hydrocarbon limonene (see 3 and 4) is a commodity chemical, available as both (+) and (−) enantiomers, and is exceedingly inexpensive in either mirror image form. Its allylic oxidation product carvone (see 5, 6), however, represent the most useful and versatile building blocks in this series and the most frequently utilized chiral pool terpene employed in this review. A review on the use of carvone in natural product synthesis has also recently appeared.28 (−)-Isopulegol (7), a monoterpene of the menthane subtype, also finds use in total synthesis owing to its altered oxygenation pattern, as does (−)-perillyl alcohol (9). Pulegone (8), whose reactive enone system is readily functionalized, has found extensive use in terpene synthesis; it is of note that the (−) enantiomer of 8 is prohibitively expensive. Although somewhat less frequently employed in total synthesis, the bicyclic family of monoterpenes (see 10–20) offer unique synthetic possibilities in synthesis owing to the ring strain present in many members.10,11 α-Pinene (see 10 and 11) is perhaps the flagship member, and it is also one of the most inexpensive terpenes in general. Its β-isomer (12), however, is inexpensive only as the (−) enantiomer. While more costly, verbenone (13) and myrtenal (14) offer more possibilities in synthesis owing to the presence of increased functionality. (+)-Camphor (15), (−)-borneol (16), (+)-camphene (17), and (−)-fenchone (18) represent inexpensive building blocks containing the bicyclo [2.2.1] heptane nucleus. The chemistry of camphor is especially extensive.29 Notably, oxidation of 16 serves as a way of accessing (−)-camphor. Finally, the carenes (see 19 and 20), which have proven especially useful in the synthesis of cyclopropane-containing terpenes (vide infra), round out this series. Notably, 2-carene can be prepared in either enantiomer from carvone.30 Bulk 3-carene of unreported optical purity is exceedingly inexpensive ($0.04 USD/gram). Besides steroid systems, which lie outside the scope of this review, several complex, higher-order terpenes have found general use in the synthesis of natural products. Two examples are (−)-α-santonin (21) and sclareolide (22), the former of which has been utilized extensively in the synthesis of guaianolide natural products.21,31
With an abundance of terpene building blocks available for use, where does one start in designing a chiral pool-based terpene synthesis? While there are no general flowcharts for such activities, chiral pool syntheses can be roughly grouped based on the similarity of the terpene building block to the target molecule (Figure 2). In the most common scenario (denoted here as “level 1”), the entire uninterrupted carbon skeleton of the starting terpene can be directly identified within the skeleton of the target. Notably, structural database searching tools (i.e. Reaxys, Scifinder, etc.) can be easily employed for identifying such relationships, in addition to the capable human mind which is adept at pattern recognition.32 Corey’s landmark 1979 synthesis of picrotoxinin (23) from carvone,33 and the Hoffman La Roche synthesis of artemisinin (24) from isopulegol,34 exemplify level 1 syntheses. It should be noted however, that this classification has no bearing on the actual tools, tactics, and exact starting terpene employed.35 For instance, the skeleton of a monocyclic monoterpene can be easily identified within the carbon framework of the marine-derived anticancer agent eleutherobin (25), yet the Nicolaou and Danishefsky groups identified different starting terpenes, namely (+)-carvone and (−)-α-phellandrene respectively, and completely different synthetic strategies en route to this target.36,37
Figure 2.
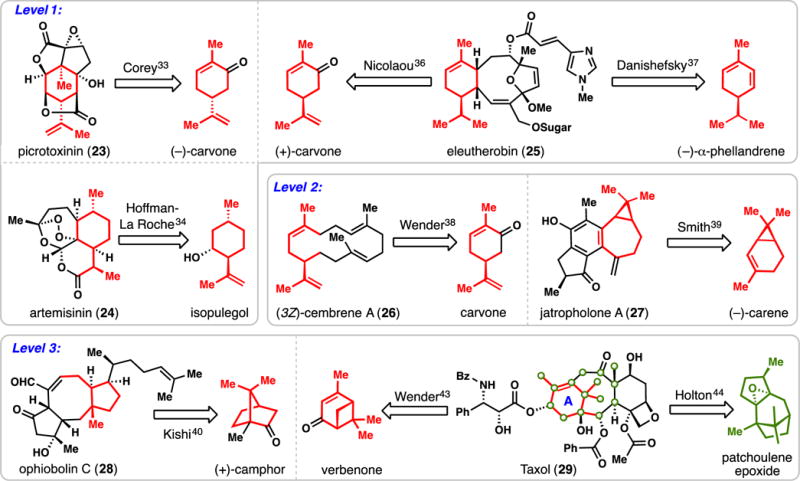
Selected terpene syntheses of the 20th century. Terpene syntheses can be roughly grouped according to the structural similarity of the starting terpene with that of the final product.
On level 2, one can find a partial, but substantial, structural match between the starting terpene and the target. For instance, while (3Z)-cembrene A (26) does not directly contain an uninterrupted monocyclic monoterpene unit, it is only one bond removed from doing so. Wender and co-workers exploited this similarity in their pioneering synthesis of 26 from carvone wherein a C–C bond of carvone was ultimately broken.38 Similarly, jatropholone A (27) does not contain the carbon skeleton of (−)-carene, but its dimethylcyclopropane unit is suggestive of this unique monoterpene and this recognition was leveraged by Smith in a concise total synthesis of this compound.39
Finally, on level 3, there is a significant disconnect between the structure of the starting terpene and the placement of the carbon atoms in the final target. Moreover, not all of the carbons of the starting terpene may be found in the final structure. Level 3 syntheses are often only possible by having in-depth knowledge of the unique chemistry of a particular terpene family. For instance, the chemistry of camphor and its many fascinating rearrangements have been studied in detail,29 and such knowledge was utilized by Kishi in a historic synthesis of ophiobolin C (28).40 Taxol (29), perhaps the most important synthetic terpene target of the 20th century, is another interesting case study.41,42 By understanding and exploiting the photochemistry of verbenone and the acid-mediated rearrangement chemistry of patchoulene epoxide respectively, the Wender and Holton groups were able to accomplish innovative total syntheses of this venerable anticancer agent.43,44 In both the case of ophiobolin C and Taxol, it is not easy to “map” the structures of the starting terpenes onto the final target owing to deep-seated molecular rearrangements.
Throughout this review, which will highlight only selected syntheses from the 21st century, we will see a variety of approaches to complex terpenes on all three previously discussed levels. The efficiency of the syntheses covered depends less on the correct choice of starting terpene, but more on the combination of this material with the synthetic strategy and methods employed. If the correct terpene and strategy are chosen, redox operations can often be minimized leading to short step-counts and minimal use of protecting groups.45–49
3. Syntheses from the 21st Century
3.1. Monoterpene Targets
While the ten carbon-containing family of monoterpenes represent important sources of flavors and fragrances,1 as well as the majority of commercially available terpenes utilized for synthesis, they themselves are the least important group of terpenoids from a human health and medicinal perspective. Accordingly, such targets have received much less synthetic attention than their larger sesquiterpene (C-15) and diterpene (C-20) counterparts. Nevertheless their densely packed structures, which are often highly hydroxylated, make the synthetic construction of such compounds by no means trivial. Two representative works are discussed below. MacMillan’s elegant 2004 synthesis of brasoside and littoralisone,50 while fitting for this section, was highlighted in Gaich and Mulzer’s 2012 review.21
3.1.1. Bermejo’s Synthesis of (+)-Paeonisuffrone (2008) (Scheme 1)
Scheme 1.
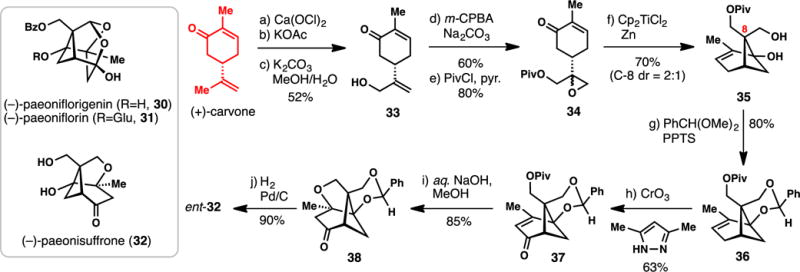
Bermejo’s Synthesis of (+)-Paeonisuffrone from (+)-Carvone (2008)
The plant family Paeoniaceae produces a variety of highly-oxygenated pinene-derived monoterpenes which have been extensively used in traditional Chinese medicine.51,52 Isolated from the roots of the chinese peony, paeoniflorigenin (30), its β-glucoside paeoniflorin (31), and paeonisuffrone (32) are representative of this monoterpene class and have proven popular and challenging synthetic targets (Scheme 1). To date, two total syntheses of 31 have been reported by the groups of Corey and Takano,53,54 and two of 32 by Hatakeyama and Bermejo.55,56 Bermejo’s 10-step, chiral pool-based synthesis of paeonisuffrone will be discussed below.
The synthesis of 32 begins with carvone and in 3 steps arrives at 33 via allylic chlorination of the isopropenyl group with calcium hypochlorite, chloride displacement with potassium acetate, and ester hydrolysis. The allylic alcohol (33) was then epoxidized (m-CPBA) and protected (PivCl) arriving at epoxide 34. In the key step of the synthesis, the strained cyclobutane-containing ring system was constructed by a reductive, titanocene-mediated cyclization initiated by homolytic epoxide-opening.57,58 This transformation afforded 35 in a remarkable 70% isolated yield with 2:1 diastereoselectivity at the newly forged quaternary center (C-8). From a historic perspective, it is of note that the strained cyclobutane unit found in pinene-type monoterpenes is often strategically broken during a total synthesis while in this case, it is constructed.10,21 With the pinene ring system in hand, only 4 additional transformations were required to complete the target. The two free hydroxyl groups were protected (see 36), allowing for subsequent chromium-mediated allylic C–H oxidation leading to enone 37. Upon deprotection of the pivaloyl group with sodium hydroxide, the primary hydroxyl group was found to spontaneously engage the neighboring enone system in a conjugate addition reaction leading to ketone 38. Finally, hydrogenolysis of 38 (H2, Pd/C) completed a synthesis of (+)-paeonisuffrone (ent-32) in only 10 operations and further solidified the power of Ti(III)-mediated radical transformations in natural product synthesis.59
3.1.2. Maimone’s Synthesis of (+)-Cardamom Peroxide (2014) (Scheme 2)
Scheme 2.
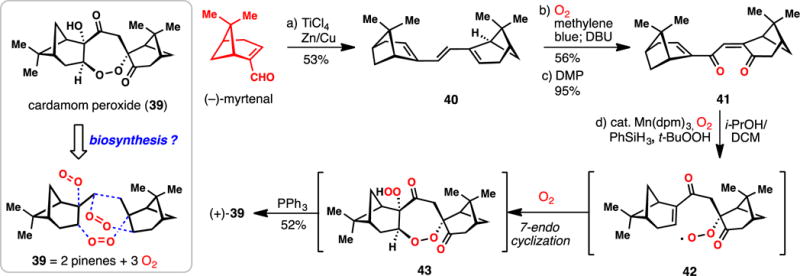
Maimone’s Synthesis of (+)-Cardamom Peroxide from (−)-Myrtenal (2014)
In 1995 Clardy and co-workers isolated an unusual endoperoxide natural product (see 39) from Amomum krervanh Pierre (Siam Cardamom) (Scheme 2).60 As with most O–O bond-containing molecules,61–63 the cardamom peroxide (39) was found to possess significant inhibitory activity against P. falciparum, the major causative agent of malaria. Given the symmetry of 39 and the observation that it was isolated alongside a variety of monoterpenes, Maimone and co-workers suggested this terpene might arise in nature from the coupling of two pinene fragments and three equivalents of molecular oxygen (Scheme 2). This hypothesis guided a 2014 synthesis of 39 in four steps.64
The monoterpene (−)-myrtenal was first dimerized using the venerable McMurray coupling leading to triene 40 in 53% isolated yield. This C2-symmetric compound was then subjected to singlet oxygen (1O2), inducing a [4+2] cycloaddition reaction,65 and after exposure to DBU, a Kornblum-DeLaMare fragmentation ensued. Following Dess-Martin periodinane (DMP) oxidation, enone 41 was obtained. Taking inspiration from the hydroperoxidation reaction of Mukaiyama and Isayama,66 and the enone conjugate reduction of Magnus,67 41 was treated with catalytic quantities of Mn(dpm)3 in the presence of oxygen and phenylsilane, presumably leading to peroxyradical intermediate 42. This species underwent an unusual and diastereoselective 7-endo peroxyradical cyclization,68,69 followed by trapping with an additional molecule of oxygen and reduction, ultimately affording hydroperoxide 43. Addition of triphenylphosphine then led to chemoselective hydroperoxide reduction and formation of the cardmom peroxide (39) in 52% isolated yield from 41. It is notable that the chirality of the pinene nucleus subtlety orchestrates all aspects of selectivity in this tandem process, which also serves to further showcase the power of metal-catalyzed, radical-based hydrofunctionalization chemistry in the rapid assembly of molecular complexity.70 Moreover, this work highlights the power of biosynthetic planning in the efficient chemical synthesis of terpenes.71
3.2 Sesquiterpenes
15-Carbon sesquiterpenes represent a historically popular class of targets for total synthesis and many chiral pool strategies have been documented.10 A handful of excellent 21st century chiral pool-based sesquiterpene syntheses were disclosed in Gaich and Mulzer’s 2012 review and will not be duplicated herein. These include Danishefsky’s synthesis of peribysin E,72 Ward’s synthesis of lairdinol,73 Nicolaou’s synthesis of zingiberene and biyouyanagin A,74 Fürstner’s synthesis of α-cubebene,75 Ley’s synthesis of thapsivillosin F,76 Xu’s synthesis of 8-epi-grosheimin,77 Altmann’s synthesis of valerenic acid,78 and Zhai’s synthesis of absinthin.79
3.2.1. Bachi’s Synthesis of (+)-Yingzhaosu A (2005) (Scheme 3)
Scheme 3.
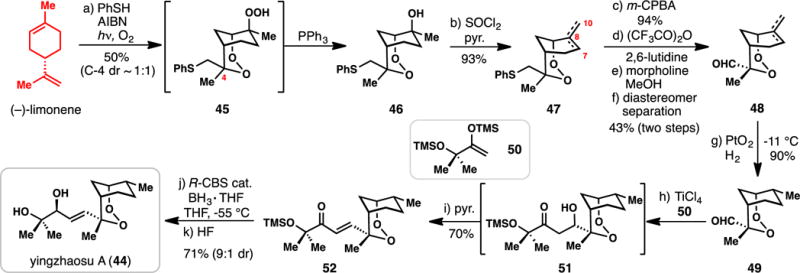
Bachi’s Synthesis of (+)-Yingzhaosu A from (−)-Limonene (2005)
The sesquiterpene endoperoxide yingzhaosu A (44) was isolated in 1979 from the plant Artabotrys uncinatus, extracts of which have been used to treat malaria in traditional Chinese medicine (Scheme 3).80 Two total syntheses of this compact natural product have been reported to date, both of which utilize chiral pool terpenes as starting materials.81,82 Herein, we discuss Bachi’s 2005 synthesis of yingzhaosu A starting from limonene.82
To construct the bridging endoperoxide ring system, Bachi and co-workers turned to the classic thiol-oxygen cooxidation (TOCO) reaction, which has found extensive use in the synthesis of peroxides.68,69 In this reaction, a thiyl radical is generated which adds to an olefin, producing a carbon-centered radical that rapidly reacts with O2. Thus treatment of (−)-limonene with thiophenol and O2 led to a cascade peroxidation forming bicyclic hydroperoxide 45 as an approximate 1:1 mixture of inseperable C-4 diastereomers. As in the synthesis of 39, the hydroperoxide group could be chemoselectively reduced in situ with triphenylphosphine leading to endoperoxide 46. The extraneous tertiary alcohol could then be eliminated (SOCl2/pyridine) leading to 47 as a mixture of Δ7,8 and Δ8,10 alkene isomers. The thiol group was then oxidized to a sulfoxide with m-CPBA, which upon treatment with trifluoroacetic anhydride and 2,6-lutidine, underwent Pummerer rearrangement. The thiohemiacetal ester thus formed was then cleaved (morpholine/MeOH), resulting in aldehyde 48. Notably at this stage in the synthesis, the C-4 diastereomers could be separated. Remarkably, under very careful temperature control, the double bond of 48 could be hydrogenated in the presence of the sensitive peroxide and aldehyde groups. With aldehyde 49 in hand, the authors then installed the final five carbons of the target through a TiCl4-mediated Mukaiyama aldol reaction with silyl enol ether 50.83 With added pyridine, the initial aldol product 51 could be funneled into enone 52. The final reduction of 52 into protected yingzhaosu A (44), however, proved challenging as achiral reducing agents showed little preference for producing a single secondary alcohol diastereomer. Ultimately, the Corey-Bakshi-Shibata reduction was found to impart good stereoselectivity (~9:1 dr) to this process,84 affording 44 after desilylation with HF. Again, the ability to perform a reduction of this type in the presence of an endoperoxide is notable; moreover, the fact that an endoperoxide was carried through an entire total synthesis speaks to the synthetic acumen of the practitioners.85 Finally, the conciseness of this route allowed for the procurement of sufficient material to further quantify the antimalarial activity of 44.
3.2.2. Vosburg’s Synthesis of (+)-Artemone (2015) (Scheme 4)
Scheme 4.
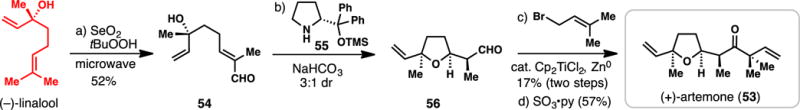
Vosburg’s 4-step Synthesis of (+)-Artemone from (−)-Linalool (2015)
The oil extract of the Indian sage Artemisia pallens (Davana oil) contains a multitude of sesquiterpene natural products characterized by a tetrahydrofuran ring system and various members have proven popular synthetic targets.86 Artemone (53) is one such natural product and despite its small size, early syntheses of 53 required up to 20 synthetic steps.87–89 Vosburg and co-workers have devised two syntheses of this molecule,86,87 one of which employs the chiral pool monoterpene linalool as starting material (Scheme 4).87
Allylic oxidation of (−)-linalool (cat. SeO2/tBuOOH) under microwave heating afforded enal 54 in 52% yield. In the bioinspired key step of the synthesis, 54 was stirred for one week in presence of the catalytic quantities of the Hiyashi-Jørgensen organocatalyst (55) and sodium bicarbonate.90 These conditions promoted oxy-Michael addition of the hindered tertiary alcohol to the enal system as well as controlled formation of the α-methyl stereocenter after enolate protonation (3:1 ratio of 56: the sum of other isomers). In the final step, reverse prenylation of the chiral aldehyde using Ashfield’s conditions91 followed by oxidation led to (+)-artemone (53). Incredibly, only 4 steps were required to access this target highlighting the power of chiral pool synthesis in concert with the judicious employment of reagent-controlled methodology.
3.2.3. Romo’s Synthesis of (+)-Omphadiol (2011) (Scheme 5)
Scheme 5.
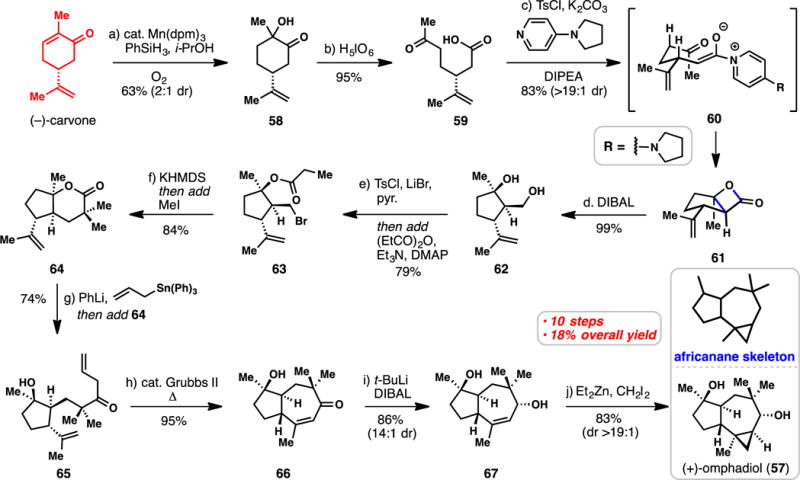
Romo’s 10-step Synthesis of (+)-Omphadiol from (−)-Carvone (2011)
The sesquiterpene omphadiol (57) was isolated from the fungus Clavicorona pyxidata and the basidiomycete Omphalotus illudens.92,93 As a member of the biologically active africanane sesquiterpenes, 57 possesses a complex and synthetically challenging 5,7,3-fused tricyclic ring system (Scheme 5).94 In 2011, the Romo research group reported the inaugural total synthesis of this natural product starting from (−)-carvone.95
Utilizing Magnus’s formal enone hydration conditions,67 carvone could be converted into hydroxyl ketone 58 which served as a substrate for a periodic acid-mediated oxidative cleavage reaction affording ketoacid 59. In a key step of the synthesis, 59 was activated with tosyl chloride and upon addition of the nucleophilic promoter 4-pyrrolidinopyridine and base (DIPEA), pyridinium enolate 60 was presumably generated. Through the chair transition state depicted, this compound underwent a tandem aldol/lactonization cascade, generating β-lactone 61 in high yield and with excellent diastereoselectivity (83%, >19:1 dr).96 Reduction of this strained compound with DIBAL afforded diol 62. The primary hydroxyl group in 62 was converted to the corresponding alkylbromide (TsCl, LiBr) and the tertiary alcohol acylated leading to ester 63. Treating this compound with strong base (KHMDS) induced intramolecular enolate alkylation, which was then followed by an intermolecular alkylation with added methyl iodide. The lactone product formed (see 64) was then opened with allyllithium (generated in situ from allyltriphenyltin and phenyllithium) leading to ketone 65. The critical 7-membered ring was then forged in near quantitative yield via ring closing metathesis of 65 catalyzed by Grubbs’ second-generation ruthenium catalyst;97 notably, one of the olefins first isomerizes into conjugation prior to the metathesis event. Stereoselective reduction of 66 with the DIBAL/t-BuLi “ate” complex (see 67) followed by non-directed Simmons-Smith cyclopropanation afforded (+)-57. Remarkably only 10 steps were required to reach this complex target, no protecting groups were necessary,47,48 and all relevant transformations proceeded with high levels of stereocontrol and efficiency, resulting in an impressive 18% overall yield. Moreover, the conversion of a cyclic monoterpene’s six-membered ring to that of a cyclopentane is a recurring theme in chiral pool terpene syntheses and will be utilized in several additional syntheses (vide infra).10,11,28
3.2.4. Liu’s Synthesis of (+)-Onoseriolide and (−)-Bolivianine (2013) (Scheme 6)
Scheme 6.
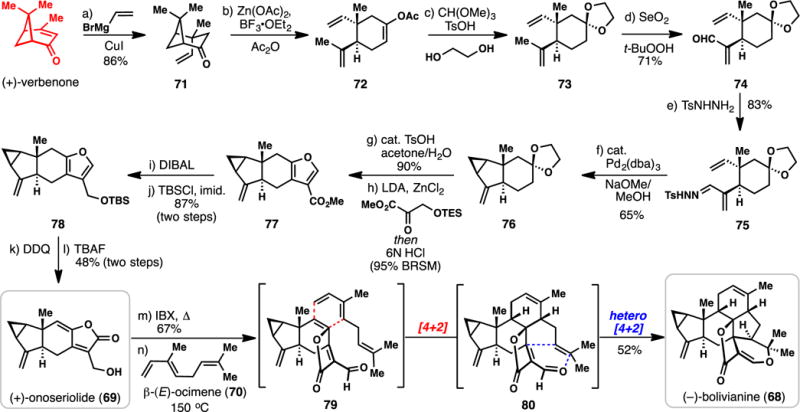
Liu’s Synthesis of (+)-Onoseriolide and (−)-Bolivianine from (+)-Verbenone (2013)
The flowering plant family Chloranthaceae has been widely used in traditional Chinese folk medicine and produces an array of complex lindenane-type sesquiterpenes.94,98,99 In 2007, the architecturally interesting 25-carbon metabolite bolivianine (68) was isolated from the Chloranthaceae species Hedyosmum angustifolium (Scheme 6).98 It was initially hypothesized that 68 resulted from the coupling of an oxidized form of the sesquiterpene onoseriolide (69) with geranylpyrophosphate followed by an ene-type cyclization and hetero Diels-Alder reaction.98 Owing to the observation that β-(E)-ocimene (70) is also detected in Hedyosmum angustifolium, Liu et al. proposed that this diene might be capable of engaging the unsaturated butenolide unit directly in a Diels-Alder cycloaddition reaction. Herein we highlight Liu’s successful execution of this idea resulting in a highly concise route to 68 and 69 from verbenone.99,100
Stereoselective copper-mediated conjugate addition of a vinyl group to verbenone (see 71) following by Lewis acid-mediated cyclobutane cleavage afforded enol acetate 72.101 This material could be directly converted to ketal 73 (ethylene glycol, acid) allowing for a subsequent allylic oxidation leading to enal 74. Conversion of 74 to its tosylhydrazone proceeded cleanly, setting the stage for one of several key steps in the synthesis. Decomposition of 75 with base in the presence of Pd2(dba)3, presumably generating an unusual allylic palladium carbenoid, led to a highly diastereoselective cyclopropanation reaction and the formation of 76 in good yield (65%).102 More commonly utilized metals in diazo-based cyclopropanation chemistry,103 such as rhodium and copper, were less effective for this transformation.99 Following deketalization (cat. TsOH, Me2CO), the ketone formed engaged the TES-protected pyruvate derivative shown in an aldol condensation, and following treatment with strong acid, furan 77 was formed. DIBAL reduction of the ester and silylation afforded 78. At this stage the furan was oxidized directly to the unsaturated butenolide system (an alkylidene-5H-furan-2-one) with DDQ, and after fluoride-mediated desilylation (+)-onoseriolide (69) was obtained. It was discovered that this dienophile was thermally unreactive toward β-(E)-ocimene (70) at temperatures up to 150 °C; however, once oxidized to the corresponding aldehyde (IBX, Δ), a smooth cycloaddition took place, presumably through transition state 79 wherein the diene approaches the butenolide from its less hindered α-face. After this initial [4+2] cycloaddition occurs, a facile intramolecular hetero Diels-Alder reaction ensues, affording (−)-bolivianine (68) in 52% yield for this pericyclic cascade. In parallel studies, it was found that 80 cyclizes to 68 at ambient temperatures.99 Overall, only 12 and 14 steps were needed to access 68 and 69 respectively, and the choice of verbenone, along with knowledge of its fragmentation chemistry, were crucial in this regard.101 Aside from giving credence to a pericyclic-based biogenesis of 68,104–106 this work once again shows the unquestionable power of the Diels-Alder reaction in the rapid assembly of complex polycyclic molecules.107
3.2.5. Total Syntheses of Jiadifenolide
Since their isolation beginning in the late 1960’s, sesquiterpenes from the Illicium family of plants have proven popular synthetic targets.108 Among this large family, jiadifenolide (81, Scheme 7) has recently attracted significant synthetic attention owing to its compact and highly oxidized molecular framework coupled with its ability to promote neurite outgrowth at very low concentrations.109 To date, total syntheses of 81 have been disclosed by the groups of Theodorakis,110,111 Paterson,112 Sorensen,113 Shenvi,114 and Zhang,115 in addition to a recent formal synthesis by Gademann.116 Herein we discuss the three chiral pool-based total syntheses of 81 by Sorensen (2014), Zhang (2015), and Shenvi (2015).
Scheme 7.
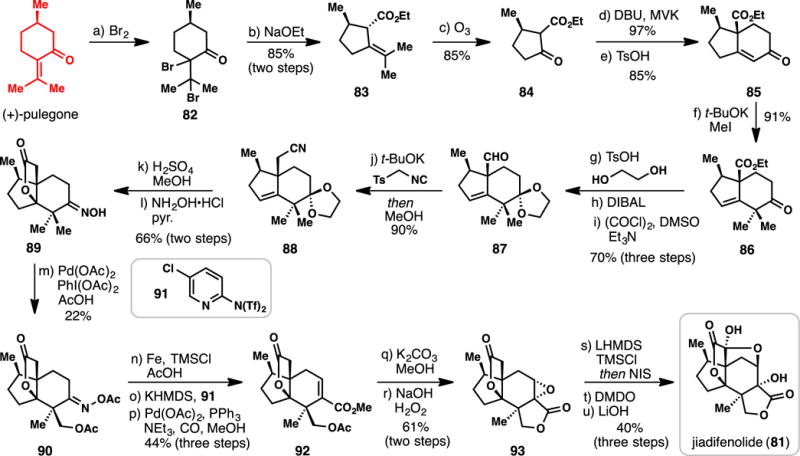
Sorensen’s Synthesis of (−)-Jiadifenolide Employing (+)-Pulegone (2014)
3.2.5.1. Sorensen’s Synthesis of (−)-Jiadifenolide (2014) (Scheme 7)
The Sorensen synthesis commenced with dibromination of pulegone (producing 82), ethoxide-induced Favorskii-type ring contraction leading to ethyl pulegenate (83),117 and finally ozonolysis of the resulting tetrasubstitued alkene.118 This decades-old sequence gives rise to optically active keto-ester 84 which has seen use in multiple terpene syntheses.10,118 Subjecting 84 to the venerable Robinson annulation produced enone 85,119 a building block employed in the classic 1990 synthesis of the Illicium sesquiterpene anisatin by Niwa and co-workers.120 Thermodynamic enolate formation and double α-alkylation yielded ketone 86. Protection of the ketone (ethylene glycol, H+), ester reduction, and reoxidation afforded aldehyde 87. Utilizing toluenesulfonylmethyl isocyanide (TosMIC), the authors were able to effect an unusual one-carbon Van Leusen-type homologation of an aldehyde,121 arriving directly at nitrile 88. Treating this material with acid brought about three transformations: deprotection of the masked ketone, nitrile hydrolysis, and cyclization to the jiadifenolide γ-lactone system. Subsequent oxime formation lead to the production of 89, setting up a key step in the synthesis. Taking inspiration from the work of Sanford,122–124 treatment of 89 with catalytic quantities of Pd(OAc)2 and stoichiometric PhI(OAc)2 promoted C–H bond acetoxylation resulting in the formation of acetyl oxime 90 in 22% yield. A lack of differentiation between the two oxidizable methyl groups, combined with the formation of bis-acetoxylated material, accounted for the relatively low isolated yield of product. Nevertheless, gram quantities of 90 could be procured through this sequence demonstrating the robustness of this chemistry. The oxime was then reductively cleaved (Fe, TMSCl) and the resulting ketone converted to its corresponding vinyl triflate with Comins’ reagent (91). A Pd-mediated methoxycarbonylation reaction then afforded ester 92. Treating 92 with basic methanol assembled the second lactone ring, and a nucleophilic epoxidation (H2O2/NaOH) then arrived at 93. Iodination of the silyl ketene acetal of γ-lactone 93, following by oxidation with dimethyldioxirane, afforded an intermediate α-keto lactone (not shown). Treatment of this material with lithium hydroxide completed a total synthesis of jiadifenolide (81) by an epoxide-opening/ketalization sequence. This synthesis is a beautiful demonstration of the successful merger of classic, scalable carbonyl-based chemistry combined with cutting-edge C-H activation synthetic methodology.125–130
3.2.5.2 Zhang’s Synthesis of (−)-Jiadifenolide (2015) (Scheme 8)
Scheme 8.
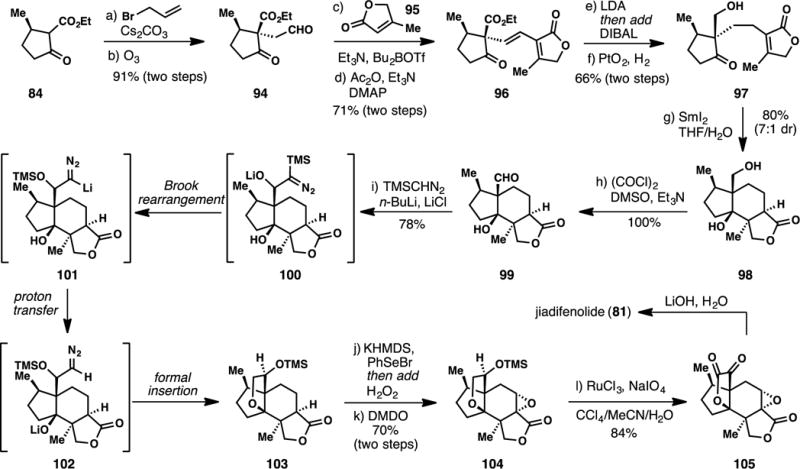
Zhang’s Synthesis of (−)-Jiadifenolide from Pulegone-derived Building Block 84 (2015)
In 2015, Zhang and co-workers reported a synthesis of jiadifenolide (81) (Scheme 8) which also employed the pulegone-derived building block 84.115 Diastereoselective alkylation of ketone 84 with allyl bromide, followed by ozonolytic alkene cleavage, afforded aldehyde 94. The extended boron enolate of butenolide 95 was then coupled with this material via an aldol reaction, and following treatment with acetic anhydride to induce dehydration, compound 96 was produced (a similar disconnection was utilized by Paterson in an earlier 2014 synthesis of 81).112 Treating 96 with excess LDA masked both the butenolide and cyclopentenone carbonyl groups as transient enolates, thereby allowing for reduction of the ester group with DIBAL. Following hydrogenation (PtO2, H2), alcohol 97 was forged, setting up a key step in the synthesis. Taking inspiration from Paterson and coworkers, the authors closed the central 6-membered ring of the target through a reductive radical cyclization.112 Thus treating 97 with the powerful reductant SmI2/H2O accomplished this transformation,131–133 producing tricycle 98 in excellent yield (80%) and with good diastereoselectivity (7:1). Swern oxidation of 98 led to aldehyde 99, thus setting the stage for a second pivotal annulation reaction wherein the authors envisioned formally “inserting” one-carbon to construct the final γ-lactone ring in the target. Thus addition of the anion derived from trimethylsilyldiazomethane to aldehyde 99 led to lithium alkoxide 100, which underwent Brook rearrangement to form anion 101. A proton transfer event then led to intermediate 102 which was converted into the product (103), possibly via a carbene intermediate. Advanced tetracycle 103 was then subjected to one-pot phenylselenation and oxidative elimination sequence furnishing an intermediate α, β-unsaturated ester, which could be epoxidized with DMDO. The epoxide intermediate thus formed (see 104) could be converted into jiadifenolide (81) in only two additional steps. First 104 was directly oxidized to α-keto lactone 105 with RuCl3/NaIO4, and finally the bridging lactol motif was constructed via base-mediated epoxide opening as previously demonstrated in Sorensen’s synthesis. Overall only 15-steps were needed to access 81, and the synthesis pathway was devoid of protecting group manipulations.47,48
3.2.5.3. Shenvi’s Synthesis of (−)-Jiadifenolide (2015) (Scheme 9)
Scheme 9.
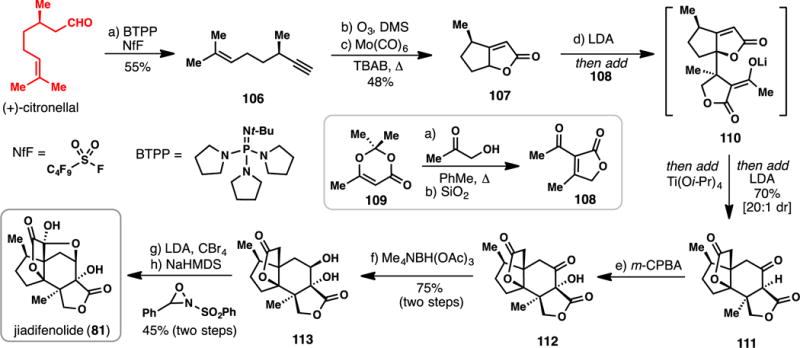
Shenvi’s 8-step Synthesis of (−)-Jiadifenolide from (+)-Citronellal (2015)
In 2015, Shenvi and co-workers reported an exceedingly concise route to 81 utilizing the chiral pool terpene (+)-citronellal (Scheme 9).114 Dehydration of citronellal was achieved in one step using the activating agent nonafluorobutanesulfonyl fluoride (NfF) and the bulky phosphazine base tert-butylimino-tri(pyrrolidino)phosphorane (BTPP).134 The resulting alkyne substrate (106) was then subjected to ozone, resulting in cleavage of the double bond and formation of an aldehyde capable of undergoing a subsequent molybdenum-mediated hetero Pauson-Khand reaction. In a separate sequence, diketene acetone adduct 109 was converted into known butenolide 108 in two steps.135 In the key step of the synthesis, butenolide 107 was deprotonated with LDA and the resulting enolate reacted with butenolide 108. This butenolide coupling presumably first formed intermediate 110, the product of a direct Michael-type addition. When Ti(Oi-Pr)4 was added to this intermediate followed by additional LDA, a second Michael-type process ensued leading to tetracyclic lactone 111 in 70% isolated yield (20:1 dr). Thus in a single step sequence, the entire carbocyclic core of the natural product was constructed and only redox manipulations were required to access the target. α-Oxidation of the 1,3-dicarbonyl motif with m-CPBA afforded lactone 112 and a subsequent directed reduction of the ketone group gave 113.112 To complete the synthesis of 81 the authors first brominated the α-position of the lactone (LDA, CBr4), which upon further enolate oxidation with Davis’ racemic oxaziridine afforded jiadifenolide (81). This total synthesis required only 8 linear operations, was devoid of protecting group use,47,48 and enabled the production of 1 gram of jiadifenolide in a single synthetic pass.136 Moreover, the Shenvi route to 81 is a model for convergency in complex terpene synthesis.20
3.2.6 Total Syntheses of Englerin A
In 2009, Beutler and coworkers isolated the complex guaianane sesquiterpenoid englerin A (114) from the East African plant Phyllanthus engleri.137 This natural product immediately attracted the attention of both chemists and biologists due its high potency and selectivity toward renal cancer cell lines (GI50 values = 1–87 nM). Not surprisingly, myriad synthetic groups have pursued syntheses of this target,138 and in the eight years since its isolation, total and formal syntheses have already been reported by the groups of Christmann,139 Nicolaou,140 Theodorakis,141 Ma,142 Echavarran,143 Chain,144 Hatakeyama,145 Parker,146 Cook,147 Metz,148 Sun and Lin,149 Shen,150 Hashimoto and Anada,151 Iwasawa,152 and Mascareñas.153 Among these works, five have utilized chiral pool terpenes: Christmann’s synthesis using cis,trans-nepetalactone,139 Ma’s synthesis employing citronellal,142 Chain’s synthesis from citronellal,144 Metz’s synthesis from isopulegol,148 and Shen’s carvone-based route.150 The Christmann and Ma syntheses were recently highlighted in Gaich and Mulzer’s 2012 review;21 herein we will discuss the Chain and Metz routes to englerin A (114).
3.2.6.1. Chain’s Synthesis of (−)-Englerin A (2011) (Scheme 10)
Scheme 10.
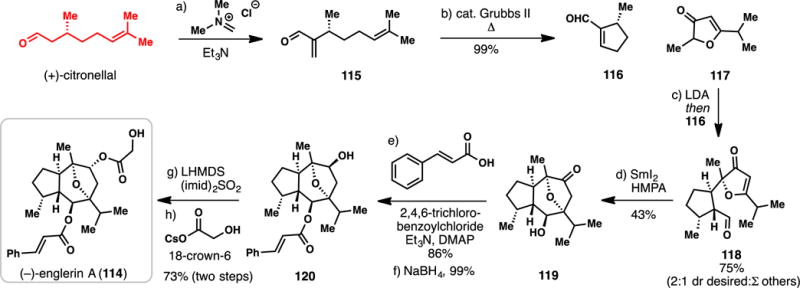
Chain’s 8-step Total Synthesis of (−)-Englerin A (2011)
In 2011, Chain and co-workers reported an exceedingly concise route to englerin A (114) (Scheme 10).144 Chiral pool monoterpene (+)-citronellal was converted into cyclopentenal 116 via a previously developed, two-step procedure involving α-methylenation (see 115) following by ring closing metathesis with Grubbs’ second generation catalyst.154,155 This chiral aldehyde was then ingeniously merged with the lithium enolate of butenolide 117 via a diastereoselective Michael addition which afforded coupling product 118 in 75% yield and with 2:1 selectivity (118: sum of other isomers = 2:1). In a second powerful bond-forming step, the authors constructed the central 7-membered ring of the target via a SmI2-mediated reductive cyclization.131–133 This transformation was conducted using the diastereomeric mixture of aldehydes containing 118, and while the isolated yield is moderate (43%), the theoretical maximum yield is only ~66%. Moreover, polycycle 119, which bears the entire guaianane core, is remarkably assembled in only 4 linear steps. To complete the synthesis of 114, the cinnamyl ester sidechain was attached using Yamaguchi’s protocol,156 and the ketone group was stereoselectvely reduced with sodium borohydride leading to 120. Finally, the secondary alcohol was converted into its corresponding sulfonate imidazole (LHMDS, (imid)2SO2) and this activated species displaced with cesium hydroxyacetate completing the synthesis of englerin A (114). Overall, the Chain synthesis required only 8 steps and was devoid of protecting group use. This work showcases highly creative synthetic planning in the convergent assembly of complex terpenes,20 and like Shenvi’s route to 81, highlights the timeless power of fundamental carbonyl chemistry in the rapid, assembly of polycyclic ring systems.
3.2.6.2. Metz’s Synthesis of (−)-Englerin A (2013) (Scheme 11)
Scheme 11.
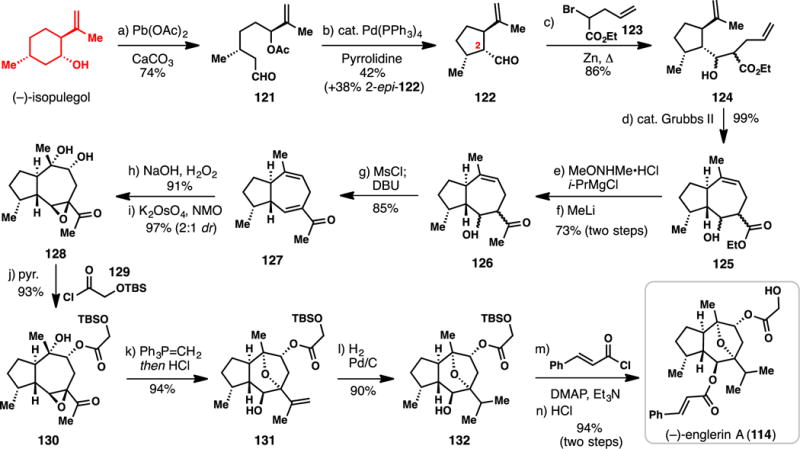
Metz’s Total Synthesis of (−)-Englerin A from (−)-isopulegol (2013)
In 2013, the group of Metz reported a chiral pool approach to 114 (Scheme 11).148 As in many guaianane and guaianolide syntheses of the past,10,21,31,138 the Metz approach relies on the ring contraction of a 6-membered cyclic monoterpene to a stereodefined cyclopentane ring system. Their requisite building block, known aldehyde 122, was constructed in two-steps from (−)-isopulegol via a novel pathway. Oxidative cleavage of isopulegol with Pb(OAc)4 produced aldehyde 121 which could be reclosed to 122 via palladium-catalyzed allylic alkylation of an in-situ formed enamine.157 A significant quantity of C-2 epi-122 was also produced in this reaction. A Reformatzky reaction between aldehyde 122 and α-bromoester 123 furnished 124 as an inconsequential mixture of diastereomers. This C–C bond-forming step was immediately followed by a high yielding ring-closing metathesis reaction, thus completing the hydroazulene core of the natural product (see 125) in only 4 steps. A two-step process transformed ethyl ester 125 into methyl ketone 126, which was then dehydrated to enone 127 via the intermediacy of a mesylate. At this point, several oxygen atoms were stereoselectively installed via nucleophilic epoxidation of the enone group and dihydroxylation of the remaining double bond (see 127 to 128). The diastereoselectivity of the second step was modest, and various attempts to increase the selectivity were unsuccessful. The first ester side-chain was attached to the free secondary alcohol group via coupling with acid chloride 129, and the methyl ketone moiety of 130 was converted to an isopropenyl group via Wittig olefination. Treating this material with hydrochloric acid forged the natural product’s hallmark bridging ether by nucleophilic opening of the reactive allylic epoxide. With intermediate 131 in hand, the natural product was procured in three additional steps: hydrogenation of the isopropenyl group, cinnamoylation of the secondary alcohol, and acidic deprotection of the primary alcohol. Overall, this synthetic pathway constructed (−)-englerin A (114) in only 14 steps from abundant (−)-isopulegol and featured many high-yielding transformations. The ablity to cleave isopulegol and rapidly reforge 122 in only two steps was particularly noteworthy. It should be noted that Shen and co-workers reported a conceptually similar metathesis-based total synthesis of 114, also employing building block 122, in 2014.150
3.3. Diterpene Targets
Owing to their vast numbers, significant biological activities, and enormous structural diversity, diterpenes have historically been the most heavily investigated group of terpenes from a total synthesis perspective.1,10,11,21,158 A variety of informative 21st century chiral pool-based diterpene syntheses were disclosed in Gaich and Mulzer’s 2012 review and will not be duplicated herein.21 These include Deslongchamps’ synthesis of chatancin,159 Overman’s syntheses of briarellin E and F,160 Sorensen’s synthesis of guanacastepene E,161 Ghosh’s synthesis of platensimycin,162 Harrowven’s synthesis of colombiasin A,163 Halcomb’s synthesis of phomactin A,164 Mulzer’s synthesis of platencin,165 Rutjes synthesis of platencin,166 Molander’s synthesis of deacetoxyalcyonine acetate,167 and Chen’s synthesis of nanolobatolide.168
3.3.1. Overman’s Synthesis of (−)-Aplyviolene (2012) (Scheme 12)
Scheme 12.

Overman’s Chiral Pool-based Synthesis of (−)-Aplyviolene from (+)-Fenchone (2012)
Marine nudibranchs and sponges produce a variety of rearranged spongiane-type diterpenes with interesting biological properties and unique structures, and many members have proven to be attractive synthetic targets.169 One such natural product is aplyviolene (133), isolated in 1986 from the purple encrusting sponge Chelonaplysilla violacea.170 Aplyviolene possesses two complex ring systems linked by a central C–C σ-bond (shown in blue)–such motifs pose unique challenges to the field of stereoselective synthesis (Scheme 12).171 In 2011, the group of Overman reported the first chemical solution to this highly challenging problem in terpene synthesis,172 and in 2012, disclosed a second-generation, chiral pool-based strategy which will be discussed below.173
The bicyclic monoterpene (+)-fenchone, who carbon atoms are not straightforwardly mapped onto 133, was converted to its corresponding oxime and then subjected to Beckmann fragmentation affording nitrile 134.174 DIBAL reduction of 134 produced an intermediate aldehyde which underwent Wittig olefination and a subsequent deprotection with hydrochloric acid. These three operations required only a single chromatographic event. Primary alcohol 135 was then converted to nitroalkane 136 via an Appel reaction (I2, PPh3) followed by iodide displacement with silver nitrite. Dehydration of 136 with phenylisocyanate and base generated a reactive nitrile oxide, which participated in a diastereoselective, intramolecular dipolar cycloaddition. The isoxazoline formed (see 137), was directly reduced to keto alcohol 138, which possesses the 5,7-fused ring system found in the western sector of aplyviolene. This material could then be dehydrated (TsOH, Δ), forming an enone which underwent copper-mediated 1,4-addition of a vinyl group producing ketone 139 in good yield. Addition of (trimethylsilyl)methyllithium to this ketone, followed by ozonolysis of the vinyl group and treatment with hydrofluoric acid produced an exomethylene aldehyde product which could be converted into activated ester 140 via Pinnick oxidation and DCC coupling with N-hydroxyphthalimide. With activated ester 140 in hand, this material was subjected to a decarboxylative radical coupling with chloroenone 141 under photoredox-mediated conditions.175 In this transformation, a tertiary radical is generated on fragment 140 which then undergoes diastereoselective radical conjugate addition to 141 forming an α-keto radical which abstracts a hydrogen atom from Hantzsch ester 142. Considering the steric congestion surrounding the newly formed C–C bond in this process, the 61% isolated yield is quite remarkable. Reductive dehalogenation of 143 with dilithium dimethyl(cyano)cuprate led to the formation of an enolate which could be trapped with tert-butyldimethylsilyl chloride to form enol silane 144. Takai-Lombardo olefination of 144 afforded an intermediate methyl enol ether, which underwent selective hydrolysis with oxalic acid to deliver methyl ketone 145. The silyl enol ether double bond was selectively cleaved via osmylation (cat. OsO4/NMO), followed by scission of the resulting crude α-hydroxycyclopentanone with Pb(OAc)4. With aldehyde 146 in hand, the TBS-protected alcohol was then removed with TBAF to provide a hemiacetal, which could be converted to fluoride 147 upon reaction with diethylaminosulfur trifluoride (DAST). Hydrolysis of the methyl ester (NaOH) produced a carboxylic acid product that underwent lactonization in the presence of SnCl2 thus unveiling the hallmark dioxabicyclo[3.2.1]octan-3-one motif (see 148). The anomeric fluoride was crucial in this process as it allowed for lactonization to proceed under mild conditions tolerant of the acid sensitive exo-methylene group. In a bold final maneuver, the sensitive α-acetoxy acetal functionality was introduced via Baeyer-Villger oxidation thus completing the total synthesis of (−)-aplyviolene (133). This work testifies to the power of radical-based coupling strategies in the convergent synthesis of complex terpenes featuring highly sterically congested chiral fragments.175
3.3.2. Vanderwal and Alexanian’s Synthesis of (+)-Chlorolissoclimide (2015) (Scheme 13)
Scheme 13.
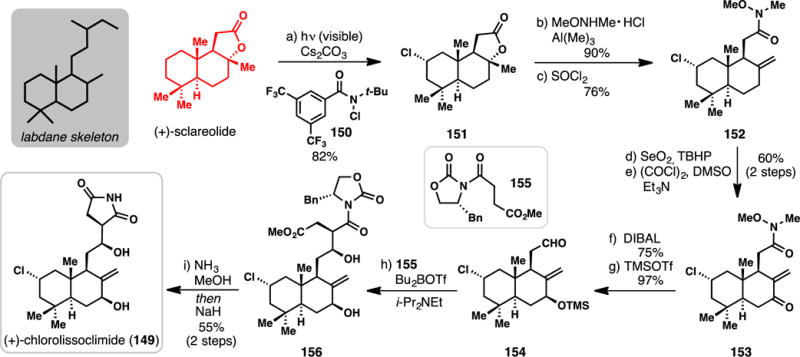
Vanderwal and Alexanian’s Synthesis of (+)-Chlorolissoclimide from (+)-Sclareolide (2015).
Owing to their interesting biological profiles, which often include antineoplastic effects,176 labdane diterpenes have proven popular synthetic targets.177 In the 1990’s, the groups of Malochet-Grivois and Roussakis described an interesting halogenated class of labdanes which included chlorolissoclimide (149) (Scheme 13).178,179 Moreover, 149 and congeners were shown to possess potent cytotoxicity toward a number of tumor cell lines.178, 179 In 2015, a total synthesis of 149 was reported by a collaborative effort between the groups of Vanderwal and Alexanian.180 Utilizing (+)-sclareolide as a chiral pool-derived building block, this work represents the first synthesis of a member of this class of labdanes.
The installation of the remote chlorine stereocenter poses an obvious challenge to the synthesis of 149 and this hurdle was cleared in the first step of the synthesis. Visible light irradiation of a solution of sclareolide and bulky N-chloroamide 150 promoted radical C-H chlorination leading to 2-chlorosclareolide (151). While alkane free radical halogenation is one of the oldest organic reactions, and many conditions are known to effect this process,181 the use of 150, which arose from prior work on C–H bromination,182 was superior to all other reagents examined in terms of yield, scalability, selectivity, and ease of use. The regio- and stereoselectivity in this process is in accordance with previous reports on the C–H oxidation,183–187 and in particular, C–H halogenation,188–195 of sclareolide. Weinreb aminolysis of lactone 151 and subsequent tertiary alcohol dehydration then afforded amide 152. The less hindered allylic position of 152 could be oxidized with selenium dioxide and a subsequent Swern oxidation converted this material into 153. Treating this material with DIBAL led to both reduction of the Weinreb amide (producing an aldehyde), and stereoselective formation of the C-7 hydroxyl group, which was consequently protected with trimethylsilyl trifluoromethanesulfonate. To install the succinimide portion of the target, previously employed imide 155 was merged with aldehyde 154 using Evans boron-aldol methodology.196 These conditions also fortuitously removed the trimethylsilyl protecting group. Coupled product 156 could be converted into chlorolissoclimide (149) by auxiliary removal with ammonia/MeOH and cyclization to the succinimide with sodium hydride. Overall this 9-step route to 149 proceeded in an impressive 14% overall yield, further demonstrating the power of C(sp3)–H bond oxidation in the synthesis of complex terpenoids.125–130,184
3.3.3. Lindel’s Synthesis of (+)-Cubitene (2012) (Scheme 14)
Scheme 14.
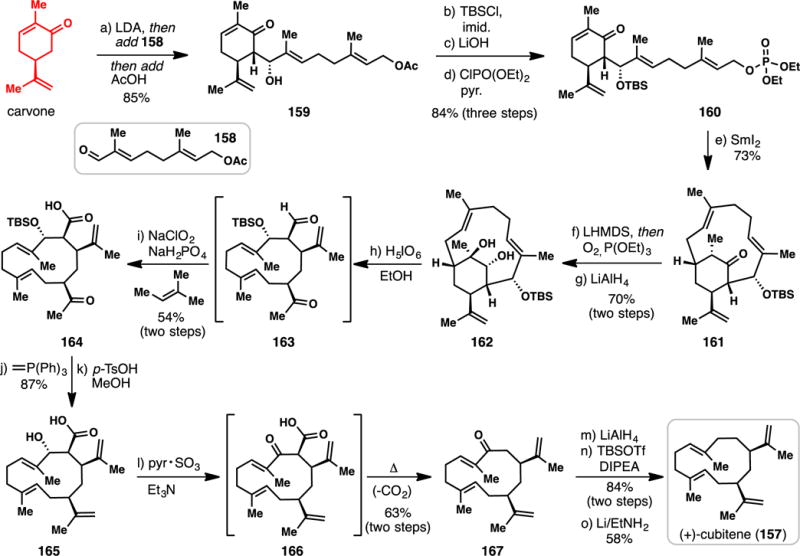
Lindel’s Total Synthesis of (+)-Cubitene from (+)-Carvone (2012)
Macrocyclic terpenes pose unique synthetic challenges in comparison to many of the rigid polycyclic structures discussed in this review; in many cases, the identification of a suitable chiral pool starting material is less obvious.197 (+)-Cubitene (157),198 a member of a small family of diterpenes containing a twelve-membered ring (i.e. cubatinoids),199 exemplifies these challenges (Scheme 14). In addition to its conformational flexibility, 157 is devoid of common functional groups, and a lack of such synthetic handles can complicate terpene syntheses.200 A non-stereoselective synthesis of 157 was first reported in 1980,201 followed by a stereoselective, racemic synthesis by Kodama in 1982.202 To date, two asymmetric routes to (+)-cubitene have been disclosed, both of which utilized chiral pool materials: Kodano’s 1996 synthesis from D-mannitol,203 and Lindel’s 2012 synthesis from carvone.204
The Lindel route begins with a stereoselective aldol reaction of the lithium enolate of carvone and geraniol-derived aldehyde 158, producing enone 159 in 85% yield. Protection of the resulting secondary alcohol (TBSCl, imidazole), ester hydrolysis, and phosphate ester formation afforded allylic phosphate 160, setting the stage for the key macrocyclization. When a solution of 160 was added slowly to a cold solution of SmI2, 12-membered macrocycle 161 was formed stereoselectively in 77% isolated yield. The presumed organosamarium intermediate showed high preference for 1,4-addition, possibly due to the intramolecular nature of this transformation.205 After having cleverly used the six-membered ring of carvone to template assembly of the bicyclo[8.2.2] tetradecane ring system, the authors then proceeded to dismantle it as cubitene possesses a single ring. Thus aerobic α-oxidation of ketone 161 (LHMDS, O2, P(OEt)3) followed by carbonyl reduction afforded diol 162 which could be oxidatively cleaved in the presence of H5IO6/EtOH. The crude keto aldehyde formed (see 163) was immediately subjected to Pinnick oxidation conditions resulting in a 54% isolated yield of 164. A Wittig olefination converted the methyl ketone into an isopropenyl group and the TBS protecting group was removed under acidic conditions (TsOH, MeOH). Oxidation of 165 under Parikh–Doering conditions (Pyr·SO3, DMSO/NEt3) produced keto acid 166, which was found to undergo smooth decarboxylation when heated, thus unveiling the full cubitene ring system. With 167 in hand, all that remained was the removal of a single oxygen atom. While many conditions can be envisioned to elicit this transformation, the authors obtained the best results via the following sequence: i) reduction of 167 with LiAlH4, ii) silylation of the resulting secondary alcohol (TBSOTf, DIPEA), and iii) careful deoxygenation via titration with Li/EtNH2. Under these conditions, overreduction and double bond migration could be minimized and (+)-cubitene (157) was isolated in 49% over three steps. Lindel’s synthesis was quite efficient (5.2% overall yield) and like Wender’s classic synthesis of (3Z)-cembrene A (26) (Figure 2), featured a very non-obvious use of carvone.38 Moreover, this work is an excellent example of Hoffman’s “overbred skeleton” concept wherein the skeletal complexity present in synthetic intermediates is greater than that of the final target.206
3.3.4. Hoppe’s Synthesis of (+)-Vigulariol (2008) (Scheme 15)
Scheme 15.
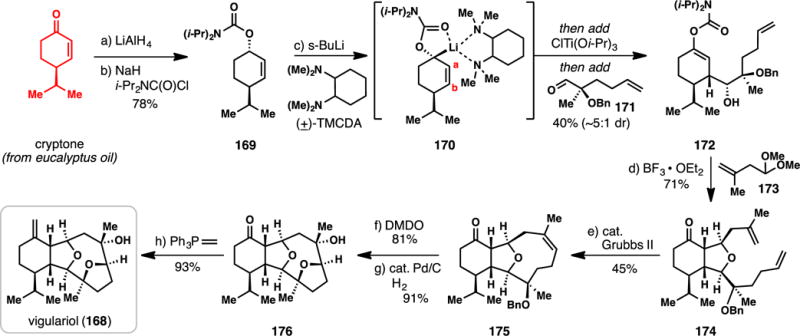
Hoppe’s Total Synthesis of (+)-Vigulariol from Cryptone (2008)
In 2005, the polycyclic diterpene (+)-vigulariol (168) was isolated from the sea pen Vigularia juncea.207 As a member of the cembrane-derived cladiellin diterpenes, 168 contains a hallmark 6,10-fused carbocyclic ring system.208,209 Along with the biogenetically related briarellin, asbestinin, and sarcodictyin diterpenes, members of this large family have proven popular targets for total synthesis and many creative strategies have been described.210 Of relevance to this review are 21st century chiral pool syntheses of deacetoxyalcyonine acetate (by Molander)167 and briarellins E and F (by Overman),160 both of which were highlighted in Gaich and Mulzer’s review.21 Paquette and co-workers first described a synthetic route to 168 during their studies toward the synthesis of related sclerophytin A.211 Notably this work was reported several years prior to 168 being discovered as a true natural product. Since then, “targeted” syntheses of vigulariol have been reported by the groups of Clarke (2007),212 Hoppe (2008),213 and Crimmins (2011).214
Hoppe’s chiral pool-based route to 168 (Scheme 15) begins with the conversion of cryptone, found in eucalyptus oil or easily prepared by asymmetric synthesis,215 to carbamate 169 via reduction and carbamoylation. When 169 was treated with sec-butyllithium and racemic, trans N,N,N′,N′-tetramethyl-1,2-diaminocyclohexane (TMCDA), stereoselective deprotonation occurred, presumably forming anion 170. Addition of ClTi(Oi-Pr)3 to this species resulted in lithium–titanium exchange, and this allylic nucleophile was then added to chiral aldehyde 171 resulting in the formation of 172 in 40% yield (5:1 dr). Enol carbamate 172 was then found to engage acetal 173 in a BF3-mediated condensation leading to tetrahydrofuran 174 in an excellent 71% yield.216 From a strategic perspective, this clever two-step protocol allows for both carbons a and b of 170 (shown in red) to function as nucleophilic sites. The oxacyclononene ring was then constructed using ring closing metathesis with Grubbs’ second generation catalyst to afford 175. The trisubstituted olefin was epoxidized with dimethyldioxirane (DMDO), and following benzyl group removal (H2, Pd/C), the final tetrahydrofuran ring instantly assembled via transannular epoxide opening. In the final step, 176 was converted into (+)-vigulariol via Wittig olefination. Overall, only 8 linear steps were required to access this complex diterpene, magnificently showcasing the synthetic utility of lithiated carbamates in organic synthesis.217,218
3.3.5. Reisman’s Synthesis of (+)-Ryanodol (2016) (Scheme 16)
Scheme 16.
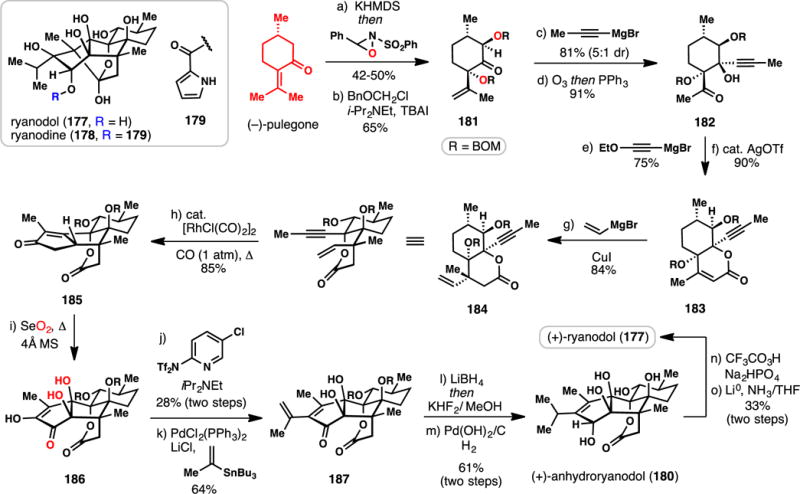
Reisman’s Synthesis of (+)-Ryanodol from (−)-Pulegone (2016)
Polyhydroxylated terpenes present unique challenges and opportunities to synthetic chemists. On the one hand, their highly oxidized structures often represent the ultimate testing ground for new chemoselective chemical transformations and methodologies. On the other, a judiciously chosen synthetic strategy can greatly increase the accessible structural variations of a natural product family, paving the way for future biologically relevant discoveries. The pyrrole ester-containing diterpene ryanodine (178),219,220 and its hydrolysis product ryanodol (177),221 have caught the eye of synthetic chemists for precisely these reasons (Scheme 16). As modulators of the ryanodine receptors (RyRs), these compounds markedly influence intracellular calcium-ion flux.222,223 As such, 178 and its derivatives represent both potent biochemical tools as well as potential medicinal and former agrochemical agents.224–226 Three syntheses of ryanodol (177) have been reported by the groups of Deslongchamps (1979),227–231 Inoue (2014),232 and very recently Reisman (2016).233 The Deslongchamps route to 177 was a landmark achievement in 20th century terpene synthesis. Of these works, only Inoue’s synthesis has proven capable of furnishing synthetic ryanodine (178).234,235 Both Deslongchamps’ and Reisman’s syntheses utilize chiral pool terpene starting materials (carvone and pulegone respectively), and both target a degradation product anhydroryanodol (180) as a key intermediate en route to ryanodol (177).
Reisman’s synthesis of ryanodol begins with a noteworthy opening sequence, a double hydroxylation of the monocyclic monoterpene (−)-pulegone in which two oxygen atoms (shown in red) are installed stereoselectively. First, γ-deprotonation of pulegone (KHMDS) forms an extended enolate, which reacts with Davis’ oxaziridine electrophile at the α-position. Then, a second enolization/oxidation sequence takes place at the α′-position, furnishing an intermediate diol as a single diastereomer. Straightforward protection of this compound as a bis BOM ether (BOMCl, i-Pr2NEt) afforded ketone 181. Addition of propynylmagnesium bromide to 181, followed by ozonolysis of the pendant isopropenyl group led to keto alcohol 182 in high yield. Ethoxyethynylmagnesium bromide addition to this ketone produced a tertiary alcohol that underwent a facile Ag-catalyzed cyclization/elimination cascade to produce lactone 183.236 Stereoselective, vinyl cuprate conjugate addition smoothly constructed enyne 184, which was poised to undergo an intramolecular Pauson-Khand reaction. After optimization, conditions were developed (1 mol% [RhCl(CO)2]2, CO) to produce cyclopentenone 185 in an impressive 85% yield.237 With the full ring system of anydroryanodol (180) now in hand, subsequent steps focused on tailoring this core to the precise structure of the natural product. A remarkable selenium dioxide-mediated oxidation of 185 installed the remaining hydroxyl groups of anhydroryanodol and generated diosphenol 186 in a single operation. 186 was then triflated (Comins’ reagent, i-Pr2NEt) and cross-coupled with tributyl(isopropenyl)stannane under standard Stille conditions to give anhydroryanodol precursor 187. Two reductions (LiBH4 then H2, Pd(OH)2/C) – the latter of which also removed the BOM protecting groups – completed the synthesis of anydroryanodol (180) in 13 steps. Conversion of this compound to ryanodol (177) itself was brought about using a slight modification of Deslongchamps’ two-step route featuring epoxidation (CF3CO3H) and reductive epoxide opening (Li0), thereby producing 177 in only 15 steps from pulegone. This work highlights a formidable combination of the strategic use of chiral pool material along with powerful metal-catalyzed C–C bond forming reactions. Moreover, both the strategic and serendipitous finding that five of the eight oxygen atoms of the target could be installed in only two steps was crucial in minimizing protecting group use, step count, and non-strategic redox manipulations.44–49
3.3.6. Williams’ Synthesis of (+)-Fusicoauritone (2007) (Scheme 17)
Scheme 17.
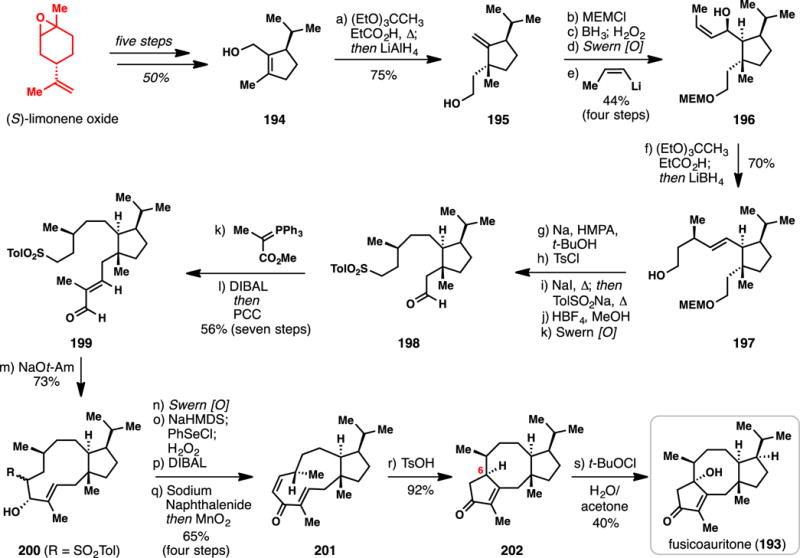
Williams’ Chiral Pool-based Synthesis of (+)-Fusicoauritone (2007)
The fusicoccanes constitute members of a large family of diterpenes containing 5,8,5-fused tricyclic ring systems, constituents of which includes the cotylenins (see cotylenol, 188), fusicoccin A (189), fusicoplagin A (191), and epoxydictymene (192) (Figure 3).238 Fusicoccanes and cotylenins have been shown to promote various biological effects, including activation of plasma membrane H+-ATPase and interaction with fusicoccin-binding proteins that play key roles in intracellular signal transduction pathways.239–241 Molecules of this class have proven to be powerful chemical tools for studying plant physiology.241 Several pioneering chiral pool-based syntheses of 5,8,5-fused diterpenes were accomplished in the 20th century, including Kato and Takeshita’s synthesis of 188,242,243 and total syntheses of 192 by the groups of Paquette and Schreiber.244,245 In 2007, Williams and coworkers disclosed a chiral pool-based synthesis of fusicoauritone (193),246 a natural product isolated in 1994 from the liverwort Anastrophyllum auritum.247 Utilizing biosynthetic logic, the Williams team first targeted a 5,11-fused macrocycle, which is believed to be a biogenetic precursor to 193 by way of a transannular ring closing step.248
Figure 3.

Various diterpenes containing 5,8,5-fused ring systems.
Starting with limonene oxide, a previously developed five-step sequence was used to construct cyclopentane building block 194,249 which has also found use in the synthesis of this class of molecules.242 A Johnson-Claisen rearrangement ((EtO)3CCH3, cat. EtCO2H, Δ) was used to set one of the key all-carbon quaternary centers in the target and following carbonyl reduction (LiAlH4), alcohol 195 was prepared in 75% yield. The bulky neighboring isopropyl group dictated the stereochemical course of this pericyclic reaction. A protection/hydroboration/oxidation sequence then produced an aldehyde to which (Z)-propenyllithium was added furnishing 196. A second Johnson-Claisen reaction was then cleverly employed, setting a remote methyl stereocenter, and after reduction (LiBH4) alcohol 197 was formed. Dissolving metal conditions (Na0, HMPA, tBuOH) were employed to reduce the lone olefin, which was prone to isomerize under more typical hydrogenation conditions. Tosylation of the free alcohol followed by Finkelstein reaction (NaI, Δ) afforded a primary iodide which could be displaced (NaSO2Tol, Δ) to yield a sulfone. Deprotection of the MEM protecting group and Swern oxidation fashioned intermediate 198. Wittig olefination, ester reduction (DIBAL), and Swern oxidation produced aldehyde 199, setting the stage for a critical macrocyclic Julia condensation. Treatment of 199 with sodium tert-amylate led to a very efficient (73–82%) ring closure, forming β-hydroxysulfone 200 as a 5:1 mixture of isomers. Swern oxidation then constructed an α-sulfonyl ketone which could be desaturated using an α-selenation/elimination sequence. Carbonyl reduction (DIBAL) led to an allylic alcohol, which could be desulfonylated with sodium naphthalenide. Mild allylic oxidation (MnO2) then produced (Z)-configured enone 201 setting the stage for the critical transannular cyclization event. In line with the authors biomimetic retrosynthesis, treating 201 with p-TsOH facilitated a Nazarov-type cyclization constructing 5,8,5-fused tricyclic enone 202 in high yield (92%).250 Serendipitously, the authors discovered that this material underwent slow air oxidation at C-6 to produce fusicoauritone (193) directly. A hypochlorite-mediated oxidation, however, was found to be superior for material throughput purposes, thus resulting in a 40% yield of 193. Overall, 25 steps were required to construct this complex diterpene from limonene, an exercise which not only further highlights the strength of biomimetic planning in complex molecule synthesis,251–255 but also showcased the timeless power of the classic Claisen rearrangement in stereocontrolled synthesis.256–258
3.3.7 Total Synthesis of Diterpenes from Euphorbiaceae
The “spurge” family of flowering shrubs (Euphorbiaceae) is found throughout the world and contains a wide range of highly complex, bioactive oxygenated terpenes.259–262 A particularly important class, from both a medicinal and synthetic perspective, are the biosynthetically related lathyrane, daphnane, tigliane, and ingenane diterpenes (Figure 4). Lathyranes possess a 5,11,3-fused tricyclic structure, and are believed to be the biosynthetic precursors to the 5,7,6,3-fused tiglianes by way of transannular C-8–C-9 bond formation.261 Cleavage of the tigliane cyclopropane (C-14–C-15 bond cleavage) presumably leads to the daphnanes (see 205),262 while 1,2-migration of the C-9–C-11 bond forges the ingenane ring system. Diterpenes from Euphorbiaceae possess great medicinal potential, with members exhibiting tumor promoting, anti-cancer, neurotrophic, anti-inflammatory, and anti-HIV activity among others.259–262 Most of these effects have been attributed to modulation of protein kinase C (PKC), a family of enzymes involved in myriad cell signaling processes.263 Esters of the tiglianes phorbol (203) and ingenol (206) are believed to chemically mimic diacylglycerols (DAG), the natural PKC secondary messengers. While many daphnanes are also believed to modulate PKC,262 the flagship member resiniferatoxin (205) activates the TRPV-1 receptors.264 These features, combined with their unique and highly complex molecular architectures, made diterpenes from Euphorbiaceae some of the most highly investigated classes of terpenes in the 20th century. Accordingly, completed total syntheses during this period remain landmark accomplishments in the field (vide infra).
Figure 4.
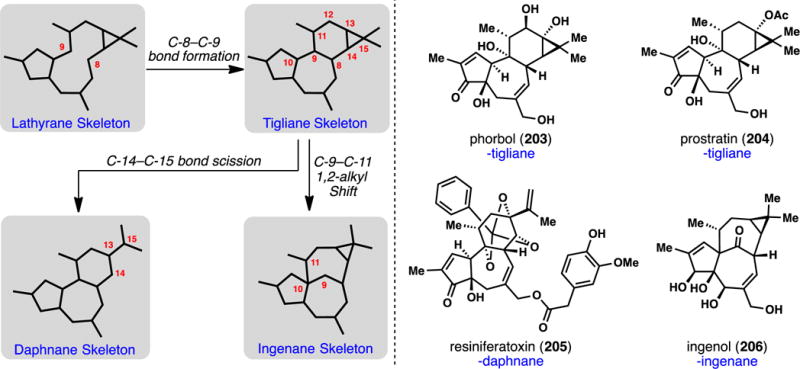
Complex diterpenes from Euphorbiaceae.
3.3.7.1 Wood’s Synthesis of Ingenol (2004) (Scheme 19)
Scheme 19.
Wood’s Total Synthesis of Ingenol (2004)
Ingenol (206), first isolated in 1968 by Hecker from Euphorbia ingens,265 and its esters have long been studied for their potent biological activity, including anti-cancer and anti-HIV potential.266–268 Furthermore, Picato® (ingenol mebutate) has recently been approved as a first-in-class treatment for the precancerous skin condition actinic keratosis.269 Ingenol has long served as a holy grail for total synthesis due to its complex oxygenation pattern and the unique “in,out” stereochemistry observed at the bridgehead positions of the bicyclo[4.4.1]undecane motif.270 While many groups have studied its synthesis,271 only Winkler (2002),272 Kuwajima (2003),273 Kigoshi (2004),274 Wood (2004),275 and Baran (2013)276 have published total or formal synthetic routes to ingenol, the latter two of which utilized the chiral pool of terpenes and will be discussed herein.
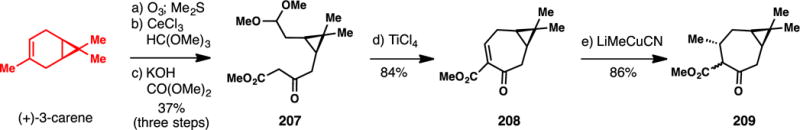
The in,out stereochemistry of ingenol has been one of the most difficult structural challenges to tackle en route to its synthesis, and was first solved by Funk and co-workers using a ring-contracting Claisen rearrangement strategy.277 While ultimately not leading to a final synthesis of 206, this work developed a five step sequence to convert chiral pool-derived (+)-3-carene, which contains the hallmark tigliane dimethylcyclopropane unit, into a suitable cycloheptanone precursor (Scheme 18). Ozonolysis, selective acetalization, and Claisen condensation afforded ester 207.30 A Lewis-acid mediated (TiCl4) intramolecular aldol condensation led to 208, which was transformed into keto ester 209 via diastereoselective methyl cuprate addition.
Scheme 18.
Conversion of (+)-3-Carene into Funk’s Keto Ester (209).
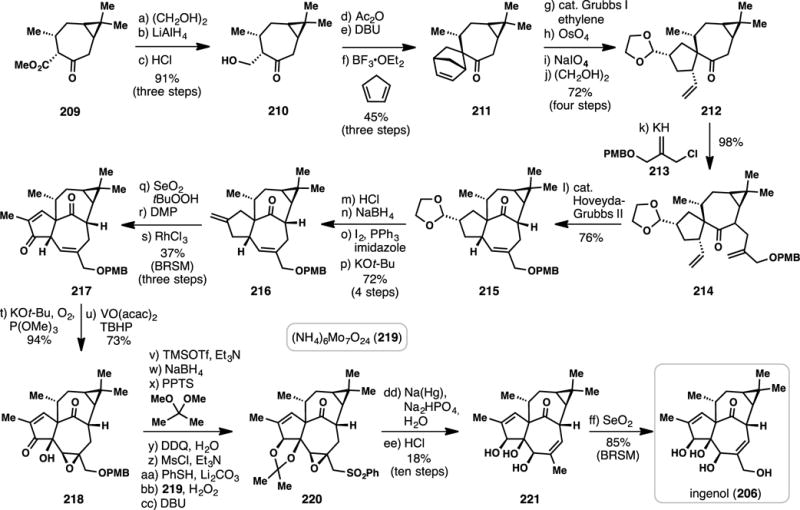
Wood’s synthesis began with conversion of 209 into alcohol 210 via ketalization, ester reduction, and deprotection (Scheme 19). Acetylation of 210 followed by elimination with DBU led to an enone that underwent facile Diels-Alder cycloaddition with cyclopentadiene assembling 211. Ring-opening metathesis of the [2.2.1] bicycle in 211 in the presence of ethylene, followed by selective olefin cleavage (OsO4/ NaIO4) and subsequent aldehyde protection generated spirocycle 212. This material underwent a high-yielding (98%) alkylation with allyl chloride 213, producing a substrate (see 214) poised to under a second metathesis event.278 Thus, treatment of 214 with Hoveyda-Grubbs’ second generation catalyst yielded [4.4.1]-bicycle 215, which possesses the challenging in,out stereochemistry, in 76% yield.97 A four-step sequence consisting of aldehyde deprotection and subsequent reduction, Appel reaction, and elimination converted 215 to 216. Allylic oxidation (SeO2/tBuOOH) followed by hydroxyl oxidation formed an enone, which could then be isomerized to 217 with RhCl3. Two of ingenols key hydroxyl groups were then installed in rapid, diastereoselective fashion via an enolate oxygenation with O2, followed by hydroxyl-directed epoxidation of the resultant allylic alcohol. The epoxide formed (see 218) was then taken through a seven-step sequence involving tertiary alcohol protection, ketone reduction, TMS hydrolysis with concomitant acetonide formation, PMB removal, a three-step conversion of the primary hydroxyl group to a sulfone, and double bond isomerization with DBU. Reduction of 220 with sodium amalgam followed by acidic removal of the acetonide group furnished deoxyingenol (221), which could be converted into the natural product via allylic oxidation with selenium dioxide. As in Smith’s classic synthesis of jatropholone A (27) (Figure 2), the identification and exploitation of a dimethylcyclopropane-containing chiral pool terpene was highly simplifying.39 This work also highlights how judicious retrosynthetic planning, in conjunction with two highly powerful metathesis-based events,279 can be leveraged in the construction of topologically and thermodynamically challenging polycyclic ring systems.
3.3.7.2 Baran’s Synthesis of Ingenol (2013) (Scheme 20)
Scheme 20.
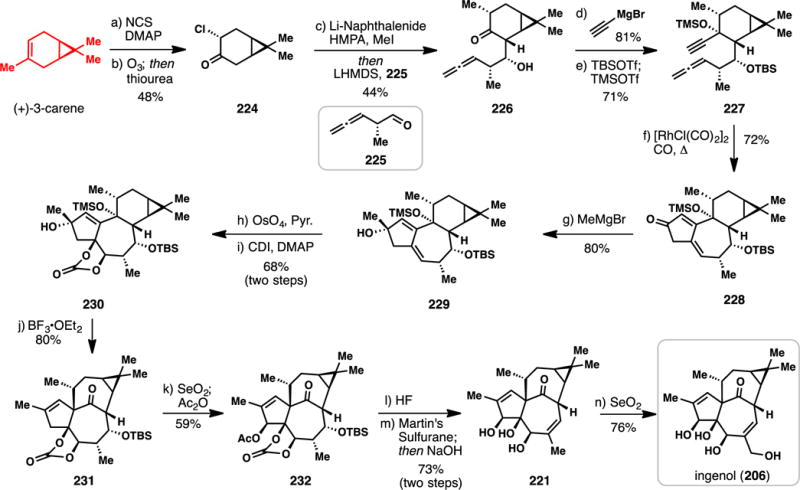
Baran’s Synthesis of Ingenol from (+)-3-Carene (2013)
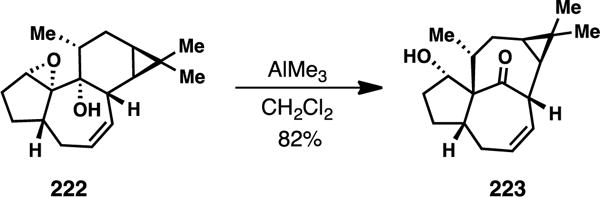
In 2013, Baran and coworkers developed an elegant 14-step route to ingenol (206), also utilizing (+)-3-carene as a starting material but with a distinctly different strategy to access the unusual in,out bicyclo[4.4.1]undecane ring system.276,280 The team was inspired by the work of Cha and coworkers who, in 2005,281 demonstrated that the 5,7,6-fused tigliane core (see 222) could be converted into the ingenane skeleton (see 223) via a Lewis acid-mediated rearrangement along plausible biosynthetic lines (Figure 5).
Figure 5.
Cha’s pinacol-type rearrangement to access the ingenane ring system.
The Baran synthesis begins with allylic chlorination and ozonolysis of (+)-3-carene to furnish α-chloro ketone 224 (Scheme 20). Reductive dechlorination of 224 with lithium naphthalenide produced an enolate that could be stereoselectively alkylated with methyl iodide. In the same pot, the resulting methylated ketone was deprotonated and subjected to an aldol coupling with chiral aldehyde 225 thus forming allene 226 in short order. Addition of ethynylmagnesium bromide to 226 and double protection afforded allene 227 which was primed for an allenic Pauson-Khand reaction.282 This transformation was realized using catalytic quantities of [RhCl(CO)2]2 under a carbon monoxide atmosphere, wherein enone 228 was formed in 72% yield. Methyl Grignard addition to this material furnished compound 229 – thus constructing the entire carbon skeleton of the tiglianes in only seven linear steps. After dihydroxylation of the trisubstituted alkene and subsequent carbonate formation, attention turned toward eliciting the key alkyl 1,2-shift reaction in analogy to Figure 5. Ultimately it was discovered that treating 230 with boron trifluoride diethyl etherate induced ionization of the tertiary alcohol and a subsequent high yielding (80%) ring shift.280 Ingenane core-containing ketone 231 was then oxidized at an allylic position with selenium dioxide and subsequently acetylated. Treatment of 232 with hydrofluoric acid removed the silyl protecting group, unveiling a secondary alcohol which could be dehydrated with Martin’s sulfurane. Ester and carbonate cleavage with sodium hydroxide furnished deoxyingenol (221). As in the Wood synthesis, the final step consisted of a selenium-mediated allylic oxidation, albeit under slightly modified conditions. At 14 steps, this work represents the shortest route to ingenol to date by a substantial margin. By using several powerful skeletal bond-forming steps and judicious incorporation of the oxygen atoms late stage, the authors were able to minimize functional group manipulations, thus resulting in an unusally concise synthesis of this complex terpene.45–49
3.3.7.3 Baran’s Synthesis of (+)-Phorbol (2016) (Scheme 21)
Scheme 21.
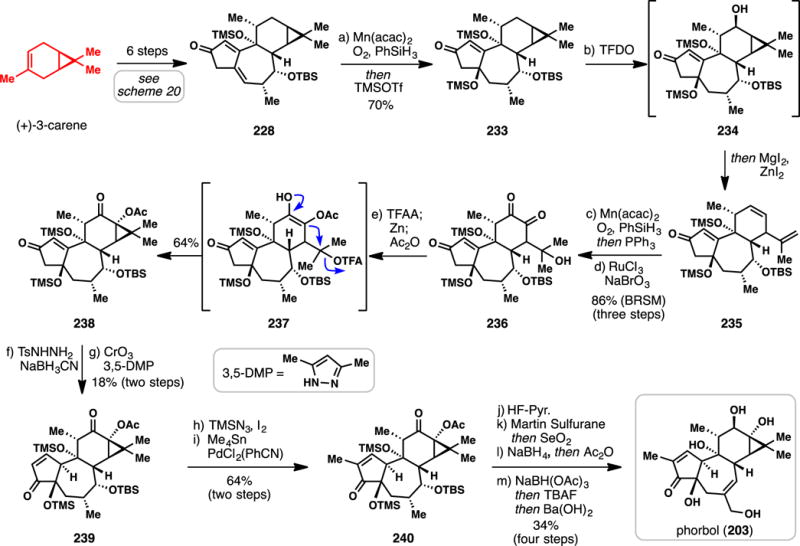
Baran’s Chiral Pool-based Synthesis of (+)-Phorbol (2016)
Phorbol (203), which was first isolated in 1934 by Bohm and coworkers as one of the principle constituents of croton oil,283 has been a target of intense synthetic interest for decades, particularly because of the unique biochemical and medicinal properties of the phorbol esters which have remained powerful tools for the study of PKC.284–287 Despite numerous synthetic studies directed towards phorbol,260,284 only syntheses by Wender,288–291 a formal synthesis by Cha,292 and a recent total synthesis by Baran have reached the final goal.293 Only the Baran synthesis utilizes the terpene chiral pool, and this work takes inspiration from their ingenol strategy (vide supra), which generates the tigliane framework en route to rearrangement. However, the presence of significant additional oxidation on the carene-derived C-ring fragment of the tiglianes (which is not found in ingenol) remained a key challenge to address in this synthetic campaign.
With large scale access to intermediate 228 in hand, the authors began with a Mukaiyama hydration of the trisubstituted alkene and subsequent protection affording enone 233.70 Impressively, and guided by NMR calculations, treating 233 with methyl(trifluoromethyl)dioxirane (TFDO) introduced a single hydroxyl group onto this complex scaffold in a regio- and stereoselective manner and on gram scale.294,295 Treating intermediate 234 with ZnI2/MgI2 led to ring-opened triene 235 and a second Mukaiyama hydration of the resulting isopropenyl group, followed by ruthenium-catalyzed alkene oxidation, afforded diketone 236. At this point, the cyclopropane was reassembled through a cascade process. Conversion of the tertiary alcohol to a trifluoroacetate group, followed by zinc-mediated reduction of the dione and acetylation, led to activated intermediate 237, which underwent a cyclopropane-forming displacement reaction to give 238. An enone reduction with concomitant alkene transposition, followed by chromium-mediated allylic oxidation, afforded enone 239. Iodination of this enone followed by methyl Stille coupling gave 240, which possesses the full tigliane carbon ring system. To complete the synthesis of phorbol, the following sequence was employed. First, selective deprotection of the TBS-protected secondary alcohol was accomplished with HF-pyridine, allowing for subsequent alcohol dehydration with Martin’s sulfurane, and allylic oxidation with selenium dioxide. Finally, reductions and global deprotections yielded fully synthetic phorbol (203). By incorporating an unactivated methylene oxidation into their retrosynthetic design, the authors did not have to change their starting chiral pool terpene from that used in the previous ingenol work, thus greatly simplifying the overall pathway. The Baran synthesis of phorbol clearly exemplifies the power of remote C–H bond functionalization in influencing the retrosynthesis of complex terpene natural products.125–129,184
3.3.7.4 Inoue’s Synthesis of (+)-Crotophorbolone (2015) (Scheme 22)
Scheme 22.
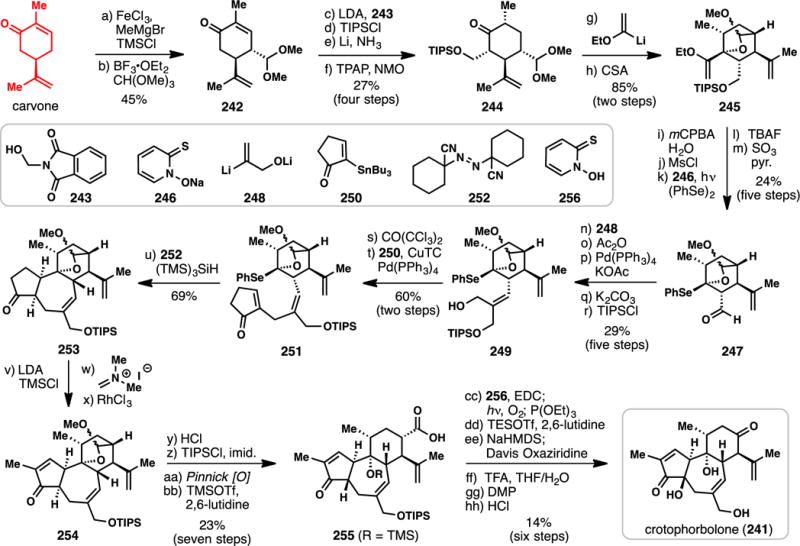
Inoue’s Synthesis of (+)-Crotophorbolone (214) from (−)-Carvone (2015)
Crotophorbolone (241) was first isolated in 1934 as a degradation product of phorbol,296 and was subsequently found to occur naturally in the dried plant roots of Euphorbia fischeriana.297 Though the specific biological activity of this diterpene is unknown, Wender has demonstrated that 241 can be converted in three steps into prostratin (204, Figure 4), a C-12 deoxytigliane that has significant potential in the treatment of HIV.298,299 A structural analysis of 241 identifies a monocyclic monoterpene substructure embedded within its carbon skeleton and in 2015, Inoue and coworkers disclosed the inaugural total synthesis of crotophorbolone starting from (+)-carvone.300
Carvone was first subjected to conditions promoting γ-selective deprotonation and silyl enol ether formation (FeCl3, TMSCl, MeMgBr).301 This sequence was followed by a Lewis acid-mediated Mukaiyama aldol reaction with trimethyl orthoformate (Scheme 22). The acetal formed (242) was then taken through a four-step sequence consisting of: i) stereoselective hydroxymethylation employing formaldehyde equivalent 243, ii) TIPS protection of the resulting primary alcohol, iii) dissolving metal reduction of the enone, and iv) reoxidation of the resulting allylic alcohol to form 244. Addition of the lithiate derived from ethyl vinyl ether followed by acid-mediated cyclization furnished caged compound 245. The Inoue team then began to assemble selenide 251 which was to function as a radical precursor. This was accomplished by epoxidation and Baeyer-Villiger oxidation of the enol ether, mesylation, and selenation under decarboxylative Barton-McCombie-type conditions. After oxidation of the TIPS-protected primary alcohol, selenide 247 was formed. Through a sequence including addition of vinyl lithiate 248 and acetylation, Pd-catalyzed allylic transposition, and protecting group interconversion, alcohol 249 was constructed. This material was converted to its corresponding allylic chloride, thus allowing for a Stille coupling with stannane 250. In the key step of the synthesis, a bridgehead radical was formed from selenide 251 using radical initiator 252. This reactive species then underwent smooth 7-endo radical cyclization onto the pendant enone, and after hydrogen atom abstraction from Tris(trimethylsilyl)silane, complex 5,7,6-fused tricyclic intermediate 253 was produced in an impressive 69% yield. It is of note that cis-stereochemistry was observed at the 5,7-ring junction in the product which would later need to be corrected. Next, the full crotophorbolone core was constructed by silyl enol ether formation, α-methylenation with Eschenmoser’s salt, and thermodynamic olefin isomerization with rhodium(III)chloride. After acidic opening of the bicyclic ketal unit in 254, hydroxyl group protection, and Pinnick oxidation of the resulting aldehyde, the desired trans-configuration at the 5,7-ring fusion could be attained via silyl enol ether formation (TMSOTf, base) and reprotonation. To complete the synthesis, six additional transformations were required. First, the carboxylic acid of 255 was transformed to a Barton ester (256, EDCI), which was subsequently irradiated under aerobic conditions leading to decarboxylation and peroxide formation. Following the addition of a reducing agent (P(OEt)3), a secondary alcohol group was formed which was subsequently protected (TESOTf). α-Oxidation of the cyclopentenone unit (NaHMDS, Davis’ oxaziradine), forged the critical tertiary alcohol moiety. Through various protecting group and redox manipulations, the synthesis of crotophorbolone (241) was then completed. Like the Overman synthesis of aplyviolene (Scheme 12), this work showcases the power of radical-based synthetic methods in the construction of complex, densely functionalized terpene natural products.302–305
3.3.8. Corey’s Synthesis of (+)-Pseudopteroxazole (2003) (Scheme 23)
Scheme 23.
Corey’s (−)-Limonene-derived Synthesis of (+)-Pseudopteroxazole (2003)
The pseudopterosins are marine natural products noted for their diverse biological activities and tricyclic core structures containing a fully-substituted aromatic ring.306 Accordingly, members of this family have attracted considerable synthetic attention over the past several decades, culminating in a variety of completed total syntheses.307–323 Pseudopteroxazole (257), isolated by Rodriguez and co-workers in 1999 from the gorgian sea whip Pseudopteragorgia elisabethae,324 presents unique challenges and opportunities (Scheme 23). Its demonstrated in vitro inhibition of Mycobacterium tuberculosis and highly-substituted benzoxazole core, combined with previous observations that members of this family have been structurally misassigned,325,326 makes 257 an intriguing synthetic target. To date, total syntheses of 257 have been completed by the groups of Corey,327 Harmata,328 Li,329 and Luo.330 The Corey and Li routes utilized the terpene chiral pool and will be discussed herein. While a chiral monocyclic monoterpene can be easily identified within the structure of these marine metabolites, the two research groups employed quite distinct chemistry to access the hallmark aromatic sector of the target.
Corey and coworkers disclosed the first total synthesis of pseudopteroxazole (257) from (−)-limonene, and in doing so, also managed to demonstrate conclusively the correct stereochemical assignment of the natural product (Scheme 23).327 In this work, (−)-limonene was converted to ketone 259 via a three-step protocol previously developed by Corey.317 Notably, the use of a cyclic hydroboration/oxidation strategy (thexylborane/H2O2) was key in setting the stereocenter alpha to the future ketone selectively.331 Chemoselective oxidation of the secondary alcohol (NaOCl) furnished ketone 258. Owing to lack of diastereoselectivity in the initial hydroboration of the limonene isopropenyl group, a kinetic resolution (isopropenyl acetate/lipase) was required at this stage to separate this mixture and produce 259 in pure form. Double silylation of 259 (TBDPSCl/imid. followed by LDA/TMSCl) produced silyl enol ether 260, which was a suitable substrate for SnCl4-mediated conjugate addition to enone 261, thus affording adduct 262 as an inconsequential mixture of diastereomers. Modified Robinson annulation (KOH/EtOH followed by SOCl2/pyr.) then furnished 263.119 Oxime formation followed by pivaloylation forged 264 setting up the key aromatization reaction, in which treatment with one equivalent of acetyl chloride induced a mild Semmler-Wolff rearrangement leading to arene 265.332 Straightforward protecting group manipulations then gave carbamate 266, which was smoothly converted to diene 268 via deprotection of the primary hydroxyl group, oxidation to an aldehyde, and Wittig-Vedejs E-selective olefination (using phosphonium ylide 267).333 Diene 268 then underwent smooth, cationic cyclization to directly generate 269 in a diastereoselective fashion when treated with methanesulfonic acid. During these studies, it was also found that by altering the carbamate protecting group, another epimer could be prepared, thus assisting in confirming the stereochemistry of the natural product. Finally, removal of the carbamate protecting group and condensation of the resulting aminophenol with triethylorthoformate afforded (+)-pseudopteroxazole (257) in 19 steps thereby completing the first total synthesis of this molecule and confirming its relative and absolute stereochemistry.
3.3.9. Li’s Synthesis of (+)-Ileabethoxazole (Scheme 24), (+)-Pseudopteroxazole (Scheme 25), and (+)-seco-Pseudopteroxazole (Scheme 25) (2016)
Scheme 24.
Li’s Isopulegol-based Synthesis of (+)-Ileabethoxazole (2016)
Scheme 25.

Li’s Syntheses of (+)-Pseudopteroxazole and (+)-seco-Pseudopteroxazole from Intermediate 278 (2016)
Recently Li and coworkers reported a second chiral pool-based total synthesis of pseudopteroxazole (257, Scheme 25) as well as syntheses of seco-pseudopteroxazole (270, Scheme 25) and ileabethoxazole (271, Scheme 24), the latter of which had previously been synthesized by Williams and possesses a fused cyclopentane ring.322,334
Whereas the Corey synthesis utilized limonene as the chiral pool building block, Li and co-workers began their studies with (−)-isopulegol (Scheme 24). Once again, stereoselective hydroboration of the isopropenyl group proved troublesome. By preparing epimer 272 via Mitsunobu inversion, however, it was possible for the subsequent hydroboration (BH3•THF) to be directed by the secondary alcohol, thus favoring the correct face of the alkene.335 Silylation of this material with functionalized chlorosilane 273 afforded alkyne 274. Dess-Martin oxidation and triflation (KHMDS, PhNTf2) gave rise to enol triflate 275. Treating this compound with a catalytic quantity of tetrakis(triphenylphosphine)palladium and stannane 276 elicited an impressive carbopalladation/Stille coupling cascade forging two carbon-carbon bonds and a seven-membered silacycle.336 The product formed (see 277) was primed to undergo a 6π-electrocyclization/aromatization sequence and indeed did so when heated to 140 °C followed by oxidation in the presence of air. Thus in two steps, a relatively simple monoterpene derivative was converted into a densely functionalize aromatic precursor. The benzoxazole product (278) subsequently served as a key precursor to ileabethoxazole (271), pseudopteroxazole (257), and seco-pseudopteroxazole (270).
First, intermediate 278 was advanced to ileabethoxazole (271) through a nine-step sequence (Scheme 24). Deprotections and functional group manipulations produced aldehyde 279 in four steps, and a subsequent Gilbert-Seyferth homologation of this aldehyde produced alkyne 280. Formal alkyne C–H insertion of the carbene derived from ethyl diazoacetate afforded ester 281.337 Subjecting this material to reductive radical cyclization conditions (Bu3SnH/AIBN) promoted a 5-exo cyclization process leading to tricycle 282 in high yield (87%). While the product was formed as mixture of olefin isomers, this proved inconsequential as DBU cleanly isomerized the double bond into conjugation. Finally, addition of methyllithium to this conjugated ester (see 283) succinctly completed the synthesis of (+)-ileabethoxazole (271).
Concurrently, key intermediate 278 was also advanced to both pseudopteroxazole (257) and seco-pseudopteroxazole (270) in seven- and six-step sequences, respectively (Scheme 25). Global desilylation of 278 with hydrofluoric acid pyridine complex followed by oxidation of the primary alcohol (DMP) yielded aldehyde 284. Horner-Wadsworth-Emmons olefination, followed by two-step reduction of the unsaturated ester (H2, Pd/C then DIBAL) then afforded key intermediate 285. A straightforward Wittig olefination converted 285 into (+)-seco-pseudopteroxazole (270). To synthesize pseudopteroxazole (257) itself, a diastereoselective oxidative α-arylation of 285 was employed using MacMillan’s shown chiral imidazolidinone catalyst shown and the iron(III) oxidant [Fe(phen)3][(PF6)3].338 Finally, analogous Wittig olefination completed the synthesis of (+)-pseudopteroxazole (257) in 15 total steps. From a strategic perspective, the ability to use common intermediate 278 in multiple synthesis pathways is a powerful method for accessing diverse natural product family members.
3.3.10. Nicolaou and Chen’s Synthesis of (−)-Platensimycin (2008) (Scheme 26)
Scheme 26.
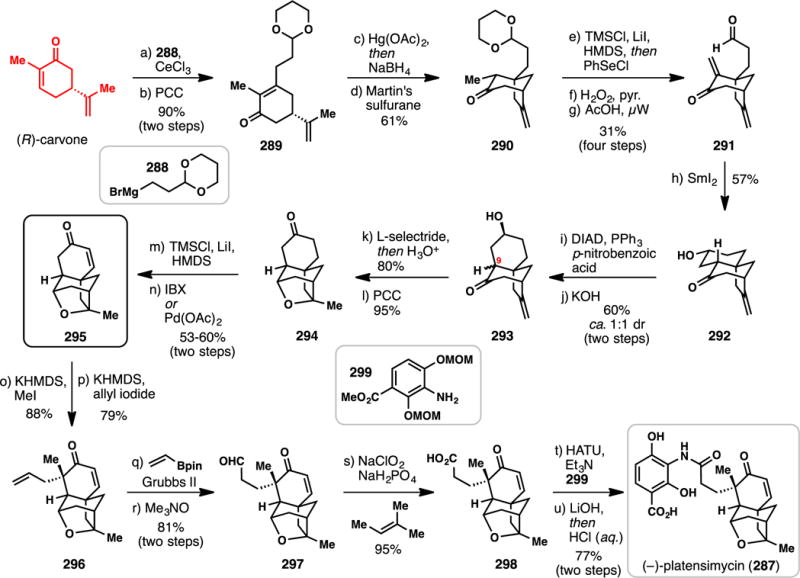
Nicolaou and Chen’s Chiral Pool-based Synthesis of (−)-Platensimycin (2008)
In 2006, scientists at Merck reported the structure of the potent antibiotic platensimycin (287), isolated from South African soil samples (Scheme 26).339,340 Its novel structure, distinctive mechanism of action, and lack of cross-resistance immediately resulted in rarely seen synthetic fervor.341–346 Within the same year of its isolation, the first total synthesis of platensimycin appeared by the group of Nicolaou.347 In approximately ten years since this first report, 287 has already seen total and formal syntheses reported by the groups of Nicolaou,347–351 Mulzer,352 Snider,353 Yamamoto,354 Corey,355 Nicolaou and Chen,356 E. Lee,357 Matsuo,358 Njardarson,359 Ghosh,162,360 D. Lee,361 Canesi,362 Magnus,363 Nakada,364 Ito,365 Wright,366 C.-S. Lee,367 Nagasawa and Kuwahara,368 Zhang and Lee,369 Lear,370,371 and Wang.372 Of these impressive works, the chiral pool of terpenes has been utilized by Nicolaou and Chen,356 Ghosh,162,360 and D. Lee.361 The 2012 Gaich and Mulzer review highlighted Ghosh’s chiral pool route from carvone as well as Mulzer’s and Rutjes’s syntheses of the structurally related natural product platencin.165,166 Herein we will focus on the Nicolaou/Chen and Daesung Lee routes, both of which utilize carvone, but feature markedly different chemical transformations.
In their chiral pool-based synthesis of platensimycin (287), the Nicolaou and Chen groups selected carvone as the tactical precursor to the highly substituted cyclohexane core of the natural product (Scheme 26). To begin, cerium-mediated 1,2-addition of Grignard reagent 288 to carvone, and subsequent Dauben oxidation (PCC) of the resulting tertiary alcohol, afforded enone 289 in high yield. In a key sequence, formal allylation of the enone was brought about in two steps. First, oxymercuration (Hg(OAc)2/H2O) of the isopropenyl group provided a primary alkyl mercury compound. Following reductive work up (NaBH4) of this intermediate, a 5-exo cyclization onto the cyclohexenone ensued, presumably through a primary radical intermediate.373 The tertiary alcohol formed during this step was then dehydrated using Martin’s sulfurane to give bicyclic ketone 290. Conversion of this material to an exo-methylene enone was accomplished by silyl enol ether formation followed by a selenation/elimination (TMSI, PhSeCl then H2O2) sequence. Removal of the cyclic acetal with acetic acid then produced aldehyde 291. In another key ring-forming step, 291 underwent intramolecular 6-endo radical cyclization when subjected to the reductant SmI2, affording 292 as a single diastereomer. Two-step Mitsunobu inversion of the secondary alcohol, with concomitant epimerization of the ketone, furnished compound 293 as a separable ~1:1 mixture of C-9 isomers (shown in red). Stereoselective reduction of the ketone (L-selectride) gave a secondary alcohol that cyclized onto the isopropenyl group when treated with acid and a PCC oxidation of the remaining secondary alcohol then provided caged ketone 294. Further processing to enone 295 was achieved by regioselective silyl enol ether formation (TMSI, HMDS), followed by oxidation with either IBX or Pd(OAc)2.374 Of note, compound 295 serves as the endpoint in most formal syntheses of platensimycin including this work by Nicolaou and Chen. Shown in Scheme 26 is the end game previously developed by Nicolaou to deliver the final natural product.350 Sequential alkylation of 295 (KHMDS, MeI then KHMDS, allyl iodide) led to enone 296 in a stereoselective manner. The allyl group underwent efficient cross metathesis with vinyl boronic acid pinacol ester under the action of Grubbs’ second generation catalyst, and following oxidation at boron (Me3NO) aldehyde 297 was obtained. This somewhat unorthodox two-step sequence was vastly superior to more conventional hydroboration/oxidation sequences in terms of isolated yield. 297 underwent Pinnick oxidation to yield platensic acid (see 298), which was then efficiently coupled (HATU, Et3N) with aniline 299. After a deprotection sequence (LiOH then HCl), platensimycin (287) was unveiled in a total of 21 steps. By leveraging carvone, a route to either enantiomer of 287 has been rendered accessible from simple inexpensive sources.
3.3.11 Lee’s Formal Synthesis of Platensimycin (2009) (Scheme 27)
Scheme 27.
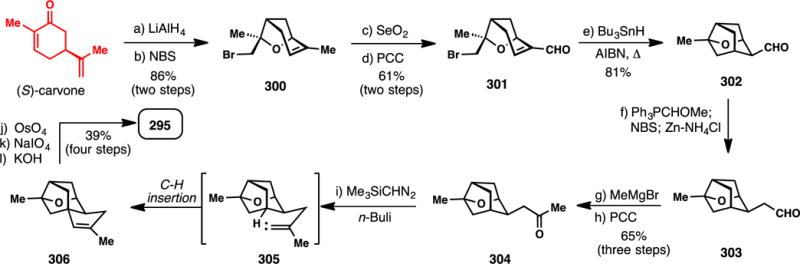
Lee’s Formal Synthesis of Platensimycin from (+)-Carvone (2009)
In 2009, Lee and co-workers reported a distinct route to key intermediate 295 from carvone using an alkylidene carbene C-H insertion strategy (Scheme 27).361 Carvone was first reduced to cis-carveol with lithium aluminum hydride and then subjected to a bromoetherification reaction with NBS, thus forging the tetrahydrofuran ring in the opening sequence. Ether 300 was then converted into enal 301 via two allylic oxidations. In a key C–C bond forming step, a reductive radical cyclization (AIBN, Bu3SnH) was used to construct caged compound 302 via a 5-exo cyclization process.375 One carbon Wittig-homologation afforded aldehyde 303, which was converted into methyl ketone 304 via addition of MeMgBr followed by PCC oxidation. In the key step of the synthesis, 304 was treated with the lithium anion of trimethylsilyl diazomethane, presumably forming vinyl carbene 305 by an addition/Brook rearrangement/elimination/nitrogen extrusion cascade. This reactive species underwent insertion into the neighboring tertiary C-H bond. While there are two tertiary C-H bonds that could react in 305, insertion was observed away from the electronegative oxygen atom due to orbital overlap considerations.361 The C-H insertion product (306) could be advanced efficiently into the platensimycin core (295) via oxidative alkene cleavage (OsO4 then NaIO4) and subsequent aldol condensation (39% over 4 steps). Overall, only 12 steps were needed to access 295, exemplifying the power of alkylidene carbenes in opening up unique and powerful retrosynthetic disconnections.376–379
3.4. Sesterterpene Targets
Twenty-five carbon sesterpenes are relatively uncommon, and far fewer syntheses have been completed in comparison to their sesqui- and diterpene brethren. Nevertheless, this family of terpenes possesses an assortment of diverse chemical architectures that have historically challenged the field of chemical synthesis. An excellent recent review by Trauner specifically covers this area of terpene research from a historic perspective.380
3.4.1. Ma’s Synthesis of (+)-Leucosceptroids A and B (2015) (Scheme 28)
Scheme 28.

Ma’s Syntheses of (+)-leucosceptroids A and B from (−)-Citronellol (2015)
The grandular trichomes of the plant Leucosceptrum canum, which is often avoided by herbivores and many pathogens, were found to contain the novel sesterterpenes leucosceptroids A (307) and B (308).381 These compounds display significant antifeedant activity as well as antifungal activity, and could serve as excellent tool compounds to better understand and develop novel pesticides for crop protection.381 Isolated in 2010, leucosceptroids are relative terpene newcomers, yet several groups have already reported creative solutions to their synthetic construction. In 2013, Liu reported a synthesis of leucosceptroid B (308),382 from 2014–2015 the group of Magauer reported the synthesis of eighteen leucosceptroids (including 307 and 308),383,384 and in 2015, Ma reported a chiral pool approach to both 307 and 308 which will be discussed herein.385
The Ma synthesis utilized (S)-citronellol as starting material and after ozonolysis, Wittig reaction, and DMP oxidation, arrived at allylsilane 309 (Scheme 28). Applying MacMillan’s organo-SOMO oxidative coupling methodology using amine catalyst 310 then furnished key aldehyde coupling partner 311.386,387 Meanwhile, the authors prepared lactone 312 via a seven-step sequence (see inset). The cuprous salt generated from alcohol 313 was coupled with 3-bromopropyne producing a diyne which when treated with a gold catalyst and silver additive cyclized to furan 314 in excellent yield (85%). Addition of the titanium acetylide of 314 to chiral aldehyde 315 (under Felkin-Anh control) yielded an intermediate alcohol which was subjected to alkyne carbomagnesiation (Fe(III), MeMgBr), acetonide deprotection, and selective silylation. Finally, treatment of 316 with PhSeCl facilitated intramolecular selenolactonization producing an intermediate β-phenylseleno alcohol, which was converted to 312 upon treatment with hydrogen peroxide. Key fragments 311 and 312 were then merged via a diastereoselective, boron-mediated aldol reaction which produced 317 in 78% yield. A six-step sequence was then employed to epimerize the resulting alcohol to form trimethylsilyl ether 318, which was subjected to a samarium iodide-mediated 6-exo ketyl olefin cyclization producing 319 after deprotection. The multi-step epimerization sequence proved necessary because, when utilizing the alternative β-disposed alcohol (or TMS ether), the undesired 7-endo cyclization pathway predominated. Oxidation of the primary alcohol of 319 and Wittig olefination completed the synthesis of the pendant prenyl chain of the natural product, and a Swern oxidation of the secondary alcohol accomplished the synthesis of leucosceptroid B (308). Stereo- and regioselective α-oxidation of the ketone moiety then forged leucosceptroid A (307) in a total of 19 steps (longest linear sequence) from alcohol 313. Overall this synthesis further showcases how several powerful radical-based reactions,302–305 in conjunction with chiral pool building blocks and asymmetric catalysis, can lead to creative and convergent solutions to complex problems in terpene synthesis.
3.4.2. Trauner’s Synthesis of (−)-Nitidasin (2014) (Scheme 29)
Scheme 29.
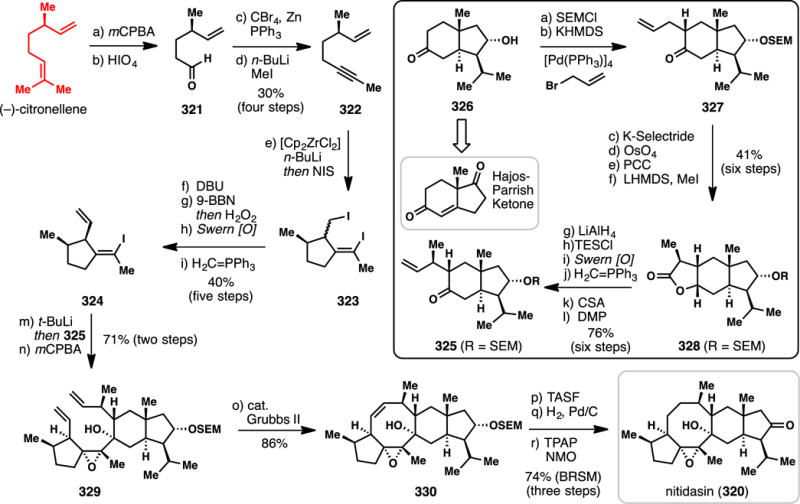
Trauner’s Total Synthesis of (−)-Nitidasin Employing (−)-Citronellene (2014)
The Peruvian infusion “Hercampuri,” which has been used as an ancient remedy against hepatitis, diabetes, and hypertension, was shown in 1997 to contain several complex terpenoids, including the synthetically daunting sesterterpene nitidasin (320).388,389 Comprised of a rare 5,8,6,5-fused tetracyclic architecture possessing ten chiral centers, 320 represents a state-of-the-art challenge in complex terpene synthesis. In 2014, Trauner and coworkers accomplished the first total synthesis of this enigmatic sesterterpene, utilizing chiral pool (−)-citronellene as a key building block (Scheme 29).390
The Trauner synthesis began with a selective oxidative cleavage of the more substituted olefin of (−)-citronellene (mCPBA then HIO4) to produce 321, followed by a methylative Corey-Fuchs reaction sequence affording enyne 322. A zirconium-mediated cycloisomerization of this material,391 utilizing NIS to quench the intermediate zirconocycle, furnished iodide 323.392 The isomeric mixture produced proved inconsequential, as the primary iodide was eliminated with base (DBU) and the resulting olefin subjected to hydroboration/oxidation sequence. A Swern oxidation and Wittig methylenation then afforded key vinyl iodide 324. The “eastern” sector of nitidasin was then constructed beginning from intermediate 326, which is ultimately derived from the Hajos-Parrish ketone (see inset).393–395 Ketone 326 was protected and then subjected to a palladium-catalyzed enolate allylation forming 327. Ketone reduction (K-selectride) and oxidative cleavage of the alkene produced a lactol that was subsequently oxidized with PCC. Deprotonation of the resulting lactone (LHMDS) followed by quenching with methyl iodide produced 328 stereoselectively. Reduction of lactone 328 with lithium aluminum hydride, followed by global silylation and chemoselective oxidation under Swern conditions provided an aldehyde that could be converted into transhydrindanone 325 via a three-step protocol consisting of Wittig olefination, chemoselective deprotection, and subsequent oxidation.
With complex fragments 324 and 325 in hand, a key coupling sequence was developed wherein the vinyl lithiate derived from 324 was added to ketone 325. Following a stereoselective epoxidation of the product, epoxide 329 was constructed. The authors employed a ring closing metathesis reaction using Grubbs’ second-generation catalyst to forge the challenging 8-membered ring of nitidasin. This closure was likely aided by the pre-organization imparted by the geometrically defined epoxide. Completion of the total synthesis from 330 relied on a simple sequence including alkene hydrogenation, deprotection, and alcohol oxidation to furnish nitidasin (320). In addition to showcasing a masterful use of a variety of stereocontrolled transformations, this work also allowed the authors to establish the absolute configuration of the natural product.
3.4.3 Maimone’s Synthesis of (−)-6-epi-Ophiobolin N (2016) (Scheme 30)
Scheme 30.
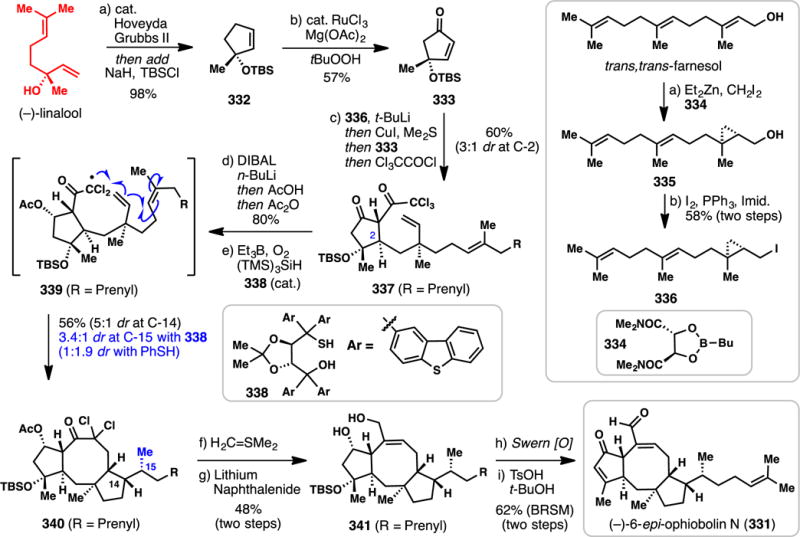
Maimone’s (−)-Linalool-based Synthesis of (−)-6-epi-ophiobolin N (2016)
The ophiobolins are a class of sesterterpenes containing a 5,8,5-fused tricyclic core common to a variety of terpene families including the previously discussed fusiccocanes (see Section 3.3.6).396 Ophiobolins were the first sesterterpenes ever isolated and are well documented phytotoxins.396 More recently a variety of ophiobolins have been found to possess fascinating anticancer activity, including the ability to kill the typically drug resistant brain tumor glioblastoma multiforme cells.397 There have been three total syntheses of members of the ophiobolin family, including the seminal synthesis of ophiobolin C by Kishi in 1989,398 a synthesis of ophiobolin A by Nakada in 2011,399,400 and a recent synthesis of 6-epi-ophiobolin N by Maimone in 2016 (Scheme 30).401
The Maimone synthesis of (−)-6-epi-ophiobolin N (331) utilized (−)-linalool as a chiral pool building block (Scheme 30). Ring-closing metathesis of this material and in situ silylation afforded protected cyclopentenol 332, which was converted smoothly into enone 333 through direct allylic oxidation (RuCl3, tBuOOH). Concurrently, trans,trans-farnesol was cyclopropanated according to Charette’s enantioselective procedure employing tartrate-derived boronate 334.402 The resulting alcohol (see 335) was then converted into highly sensitive iodide 336 via an Appel-type reaction. In a key 3-component coupling sequence, 336 was treated with tert-butyllithium inducing lithium-halogen exchange and anionic cyclopropane opening; after transmetalation with copper iodide, the resulting cuprate formed was added to enone 333, resulting in a diastereoselective conjugate addition reaction. Finally, this mixture was quenched by the addition of trichloroacetyl chloride, resulting in the formation of 337 in 45% isolated yield along with 15% of the C-2 diasteromer. Cyclopentanone 337 could be stereoselectively reduced and acetylated in situ setting up the key step in the synthesis. Exposing this material to reductive radical cyclization conditions (Et3B/O2, (TMS)3SiH) led to a tandem 8-endo/5-exo radical cyclization process (see 339), forming the remaining two rings of the ophiobolins. Crucial to this process was the radical termination step at the C-15 position, which was accomplished diastereoselectively (dr = 3.4:1) under polarity-reversal conditions using the bulky dibenzothiophene derived TADDOL-based monothiol 338.403,404 It is noteworthy that the native selectivity for this reaction, when simple, achiral thiols such as thiophenol were utilized, favored the undesired C-15 diastereomer (dr = 1:1.9). Product 340 then underwent a Corey-Chaykovsky epoxidation and a reductive epoxide opening induced by lithium naphthalenide forging diol 341. Finally, double Swern oxidation of 341 followed by acidic elimination of the linalool-derived tertiary alcohol afforded 331 in nine steps and as the non-natural enantiomer. This work clearly showcases both the power, as well as current practical challenges, of radical cascade processes in the rapid assembly of complex, stereochemically-rich terpene architectures.302–305
3.5. Triterpene-derived Targets
Thirty-carbon triterpenes and their derivatives comprise one of the largest groups of terpenes, with an estimated 20,000 members.405 Given that many of these compounds are frequently modified steroids, semisynthesis from an abundant steroid precursor is often employed.22,23 As noted previously, steroid semisynthesis lies outside the scope of this review. The natural products discussed in this section, while not comprising 30 carbons, are all believed to be biosynthetically derived from triterpenes.
3.5.1 Shing’s Synthesis of (−)-Samaderine Y (2005) (Scheme 31)
Scheme 31.
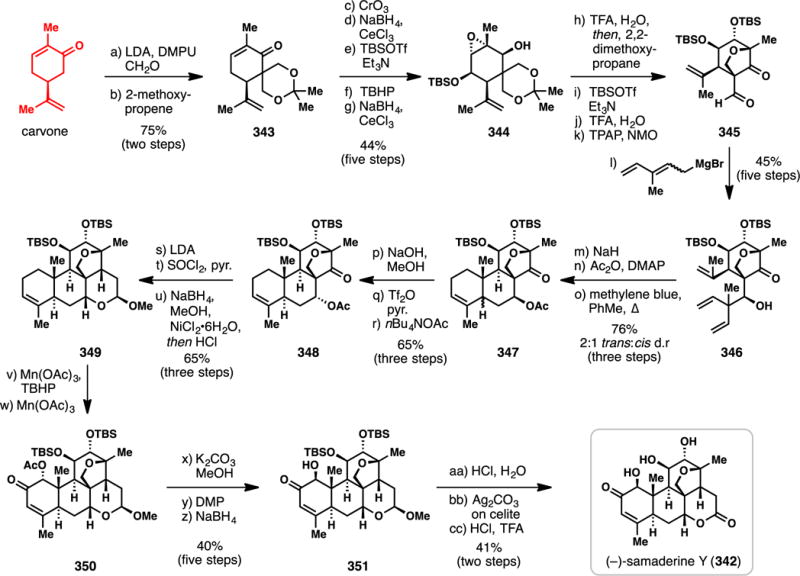
Shing’s (+)-Carvone-based Synthesis of (−)-Samaderine Y (2005)
Samaderine Y (342) belongs to the quassinoid family of natural products and while containing 20 carbon atoms, 342 – like all quassinoids – is more properly described as a degraded triterpene from a biosynthetic perspective.406,407 These compounds are often found in the bitter principles of the Simaroubaceae family of plants and display high levels of oxidation, intricate ring systems, and potent biological activities.408,409 Accordingly, many members of this family have inspired creative syntheses, employing a number of diverse strategies.410,411 Shing’s 2005 synthesis of samaderine Y from carvone exemplifies the chiral pool approach to these natural products.412,413
The synthesis of samaderine Y (342) (Scheme 31) commenced with a double formaldehyde aldol reaction of carvone followed by acetonide formation.414 Product 343 was then processed through a five-step sequence involving a regioselective allylic oxidation, Luche reduction, protection, epoxidation, and a second Luche reduction to give 344. Treatment of this material with TFA removed the acetonide group, triggering an intramolecular epoxide opening from one of the free primary hydroxyl groups. Upon addition of 2,2-dimethoxypropane the resulting diol formed a second acetonide. Secondary alcohol protection (TBSOTf), acetonide removal, and double oxidation furnished keto aldehyde 345. The allylic Grignard reagent shown then added selectively to the aldehyde moiety to furnish secondary alcohol 346. As is common, γ-attack of the allylic nucleophile was observed. Upon treatment with sodium hydride, an anion accelerated 1,3-sigmatropic rearrangement occurred,415,416 which was followed by acetylation of the secondary alcohol. The diene product generated then underwent intramolecular thermal Diels-Alder reaction to forge the 6,6-fused ring system of the quassanoids resulting in 347 with 2:1 trans:cis selectivity at the ring fusion. Next, a three-step sequence was employed to invert the stereochemistry of secondary acetate to provide diastereomer 348. Two-step acetate aldol condensation then delivered the final ring of samaderine Y and a subsequent reduction of this unsaturated lactone (NaBH4, NiII) yielded acetal 349. With the full ring system in place, the concluding steps of the synthesis focused on installing the final requisite oxidations. Allylic oxidation of 349 (Mn(OAc)3, tBuOOH) gave an enone which was α-acetoxylated with additional Mn(OAc)3. Unfortunately, product 350 required another three-step inversion sequence leading to enone 351. With this material in hand, an acetal deprotection (HCl/H2O), lactol oxidation (Fétizon’s reagent), and global deprotection (HCl/TFA), completed the synthesis of (−)-samederine Y (342) in 29 steps, showcasing the power of chiral pool starting materials in concert with pericyclic processes to construct exceedingly compact and stereochemically rich carbocyclic ring systems.
3.5.2 Li’s Synthesis of the Proposed Structure of Rubriflordilactone B (2016) (Scheme 32)
Scheme 32.
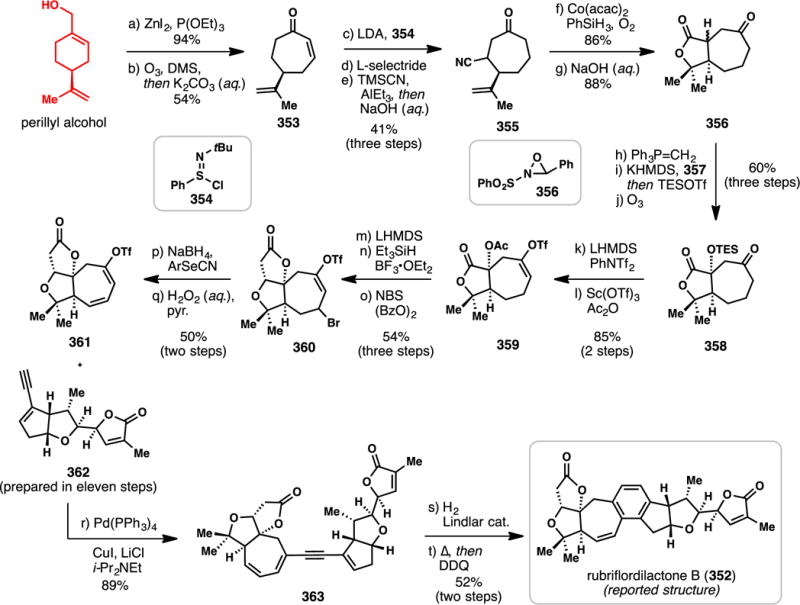
Li’s Total Synthesis of the Reported Structure of Rubriflordilactone B (2016)
Triterpene derivatives from Schisandraceae, like rubriflordilactone B (352, Scheme 32) and schilancitrilactones B and C (364, 365, Scheme 33) have attracted great synthetic attention ever since their isolation and characterization.417–422 Boasting polycyclic structures with a characteristic 7-membered ring and often containing multiple lactone moieties, these natural products are noted for both their structural intricacies and biological potencies – most have strong antiviral properties and many also display antiproliferative effects.423,424 Rubriflordilactone B (352) itself is a bisnortriterpenoid (C-28) that contains a central tetrasubstituted aromatic ring.425 By anticipating the construction of that motif in at the end of their synthesis, Li and coworkers developed a convergent synthetic strategy involving the late-stage union of two complex building blocks.422 Of note, a chiral pool terpene building block is not obviously identified within the gross structure of the final target.
Scheme 33.
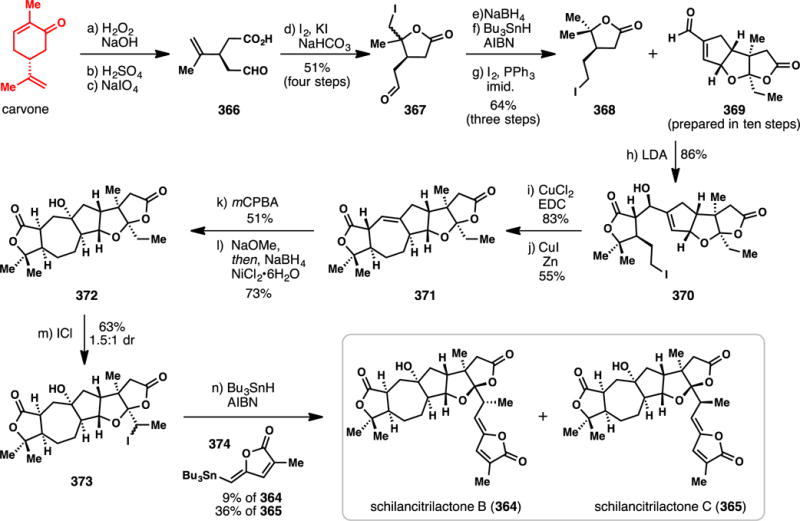
Tang’s Synthesis of Schilancitrilactones B and C from (−)-Carvone (2015)
As part of this plan, the western portion of the molecule, particularly the cycloheptane ring, was traced back to chiral pool starting material perillyl alcohol (Scheme 32). This material was first converted into a phosphonate ester using Zn(II)iodide and triethylphosphite. Ozonolysis then cleaved the more electron-rich, trisubstituted olefin producing an intermediate keto aldehyde that immediately underwent intramolecular Horner-Wadsworth-Emmons olefination in the presence of base. The enone formed (353) was then converted to cyanide 355 via a three-step sequence involving oxidation to a dienone with Mukaiyama’s reagent (354), regioselective hydride conjugate reduction of the less hindered olefin with L-selectride, and conjugate addition of cyanide. An efficient Mukaiyama hydration of the isopropenyl group produced a tertiary alcohol, which cleanly forged a γ-lactone ring (see 356) upon basic hydrolysis of the nitrile. At this point, chemoselective α-hydroxylation of the lactone was required in the presence of the more reactive ketone moiety. To address this challenge, the ketone was protected as an exo-methylene group using the Wittig olefination (Ph3P=CH2), thereby allowing for lactone α-oxygenation (KHMDS, oxaziridine 357, then TESOTf), and finally the ketone was revealed through ozonolysis, yielding lactone 358. Triflation of the ketone (LHMDS, PhNTf2) proceeded smoothly and the triethylsilyl group was exchanged for an acetate, using a procedure particularly suited for hindered tertiary alcohols (Sc(OTf)3, Ac2O).426 Enol triflate 359 was then exposed to LHMDS, promoting an intramolecular acetate aldol condensation which was followed by ionic hydrogenation (Et3SiH, BF3•OEt2). The tricyclic lactone product then underwent regioselective allylic radical bromination (NBS, (BzO)2), producing bromide 360 as an inconsequential mixture of diastereomers. Unfortunately, direct elimination of this bromide proved unsuccessful; therefore, a two-step protocol was developed wherein the bromide was first displaced by an aryl selenide (o-NO2C6H4SeCN, NaBH4) and the resulting intermediate was oxidized and eliminated (H2O2, pyr.), thus delivering key coupling partner 361.
At this juncture, 361 was merged with alkyne 362 – itself prepared in 11 steps from a non-chiral pool starting material – by means of a high yielding Sonogashira coupling. 363 was then semi-hydrogenated (H2, Lindlar’s cat.) forming a triene and setting the stage for Li’s signature electrocyclization/aromatization cascade sequence.329,427,428 Thus, heating the intermediate triene at 80°C brought about the desired 6π-electrocyclization and the crude mixture was then successfully oxidized with DDQ to succinctly produce rubriflordilactone B (352) in a longest linear sequence of 20 steps from perillyl alcohol. Despite X-ray crystallographic data of both synthetic and natural naturally occurring material, the NMR data of synthetic 352 did not match the reported literature values.425 Supported by recent calculations by Kaufman and Sarotti,429 the true structure of rubriflordilactone B is suggested to be a diastereomer of 352. In addition to highlighting the role of total synthesis in structural confirmation, Li’s synthesis of 352, which employs a late-stage coupling of thirteen and fifteen carbon fragments, is a model for convergency in complex terpene assembly.20
3.5.3 Tang’s Synthesis of Schilancitrilactones B and C (2015) (Scheme 33)
Though the schilancitrilactones share some structural similarities to rubriflordilactone B (Section 3.5.2), particularly in the western portion of their structures, their differing carbon skeleton and distinctive oxidation pattern make them unique synthetic challenges.430 In 2015, the group of Tang reported synthetic solutions to the formidable targets schilancitrilactones B (364) and C (365) utilizing carvone as a chiral pool building block (Scheme 33).419
The Tang synthesis began with substantial degradation of carvone utilizing a sequence previously developed by Deslongchamps (Scheme 33).431 Carvone was first epoxidized (H2O2/NaOH) and the resulting epoxide subjected to hydrolytic ring opening with sulfuric acid. Oxidative C–C bond cleavage with sodium periodate not only ruptured the 6-membered ring, but also carved out two carbon atoms furnishing linear acid 366. From this point, iodolactonization proceeded uneventfully, producing lactone 367 as an inconsequential mixture of diastereomers. Reduction of the aldehyde (NaBH4) and radical dehalogenation (Bu3SnH, AIBN) furnished a substrate suitable for an Appel reaction (I2, PPh3, imidazole), leading to iodide 368 in three steps from 367. Concurrently, aldehyde 369 was prepared in ten steps employing a non-chiral pool based route. Straightforward aldol coupling between the lithium enolate of 368 and aldehyde 369 cleanly generated 370 in excellent yield (86%). Alcohol dehydration of 370 (CuCl2, EDC) and subsequent conjugate addition of the alkyl iodide center (CuI/Zn)432 to the resulting unsaturated system zipped up the seven-membered ring, affording pentacycle 371 as the major product. Notably, conventional tin-based and modern photoredox-mediated radical cyclizations failed to elicit this seven membered ring-forming process. Treatment of 371 with mCPBA gave an epoxide, which underwent facile β-elimination when treated with sodium methoxide. The α, β-unsaturated ester product was then chemoselectively reduced (NaBH4, NiCl2•6H2O), yielding tertiary alcohol 372. To complete the synthesis it was necessary to append a butenolide motif onto this advanced core structure, yet 372 did not possess obvious “synthetic handles” to do so. Boldly, Tang and co-workers found that treating 372 with iodine monochloride induced formal oxidative C–H iodination delivering secondary iodide 373 as a 1.5:1 mixture of diastereomers. With this material in hand, a daring radical coupling was saved for the very last step. Under alkyl radical generating conditions (Bu3SnH, AIBN), 373 was found to undergo formal coupling with stannane 374,433 affording a diastereomeric mixtures of schilancitrilactones B (364) (9%) and C (365) (36%). Overall, this concise, 17-step total synthesis elegantly leveraged the known cleavage chemistry of carvone. It is also noteworthy that by utilizing a late stage formal C–H halogenation, the authors did not need to carry a sensitive, and potentially incompatible, secondary halide (or protected precursor) throughout the synthesis.
4. Conclusion
As this review hopefully has made clear, the field of synthetic terpene chemistry is still vibrant with many creative players distributed widely across the globe. Even in an age of advanced analytical techniques, the use of total synthesis in the structural confirmation of terpene natural products is still relevant. Moreover, natural products that several decades earlier seemed nearly impossible to synthesize, many of which ultimately required 30–50 operations to do so, have now been synthesized in a fraction of synthetic steps.434 Furthermore, some of the syntheses discussed herein are quite efficient in terms not only of step count, but yield and material throughput as well.46,49,136,435 Some have overall yields approaching 20%,64,95 and one has produced gram quantities of the target in a single synthetic pass.114 However, much work remains to be able to accomplish this on a consistent basis, particularly owing to the highly variable structures of terpenes and the lack of a universal synthetic strategy. In addition, modular medicinal chemistry-type routes, while becoming increasinlgy feasible with other highly complex natural product scaffolds,436,437 are very much still challenging in the terpenoid arena.438 Moreover, is a chiral pool strategy the best bet for this line of research? And if suitable clinical candidates were to emerge from such studies could they be prepared in kilogram (or more) scales?
Are chiral pool syntheses more efficient than non-chiral pool (i.e. asymmetric synthesis-based) strategies? This question was not addressed in this work and has no simple answer. Many of the syntheses covered in this review are the shortest routes to the given structure. However many chiral pool syntheses have historically been quite lengthy.10,11,21 In many of the latter cases, extensive protecting group and redox manipulations were often required, thereby nullifying the benefits of starting with a substantial portion of the carbon atoms in place. The synthetic practitioner must always be mindful of these drawbacks when considering a chiral pool-based approach to a complex target (of any type). Finally, as documented numerous times in this review, many convergent terpene syntheses employ both a chiral pool component and a fragment prepared by asymmetric synthesis thus complicating this direct evaluation.20
What might the future of this area look like? It is clear from many of the works showcased that truly efficient syntheses are possible when powerful synthetic methodology is leveraged on a judiciously chosen chiral pool scaffold using an overall sound synthetic strategy. Thus this field evolves with the synthetic methods of the period.434,439 It is of note that radical chemistry is as alive as ever, as is clear from the many radical-based key steps in the discussed works. Current activities in the methodological areas of both C–H and C–C activation will also feature prominently for the foreseeable future as these methods not only alter the oxidation state of chiral pool materials but can also rearrange their carbon frameworks.440–446 The methodologic aspect of chiral pool terpene synthesis is highlighted by Reisman’s pulegone-based, and Deslongchamps’ carvone-based syntheses of ryanodol (the latter of which was not discussed due to the period of the review). While both laboratories likely had access to either of these chiral pool materials in house, the Pauson-Khand reaction447 was only just starting to “come online” during the 1970’s when Deslongchamps’ historic campaign toward this target began.227 Thus, chiral pool terpene syntheses are in a way a product of their times, and the terpene starting material simply represents a blank canvas from which the synthetic practitioner can create from. However, with a greater assortment of starting materials, a wider sampling of finished pieces can be expected. Consider Baran’s route to phorbol (Scheme 21). What if modified (+)-3-carene was available with the cyclopropane hydroxyl group already in place? Would these researchers have had to expend the time and steps to deal with breaking and reforming the cyclopropane? How concise could this route become? Would their key remote C–H activation reaction even work with this electronegative substituent already present? What if carene was commercially available with hydroxyl groups at every position? While these questions are left unanswered for now, it’s clear that expanding the chiral pool of terpenes fundamentally alters the disconnections and possibilites available to the synthetic practitioner, aiding in both natural and non-natural synthetic designs.448 Although the field of synthetic biology is actively engaged in this line of research,449–454 substantial engineering (and fundamental research) hurdles are still present in nearly all cases to translate proof-of-concept experiments to portfolios of products available in kilogram quantities for cents/gram. Nevertheless, these goals, in conjunction with advances in synthetic methodology and strategy, could fundamentally alter how chemists synthesize complex terpenes and their diverse analogs well into the 21st century.
Acknowledgments
Funding
Financial support for this work was provided by the NIH (GM116952). T. J. M. is an Alfred P. Sloan Fellow and Cottrell Scholar. Z. G. B, M. L. C., and C. P. T. thank the NSF for pre-doctoral fellowships (DGE-1106400). Z. G. B. and M. L. C. acknowledge UC-Berkeley for Berkeley graduate fellowships. C. P. T. acknowledges Bristol-Myers Squibb for a pre-doctoral fellowship.
Biographies
Tom Maimone received a B.S. degree in chemistry from UC-Berkeley (2004) where he was introduced to organic chemistry research in the laboratory of Prof. Dirk Trauner. From 2005–2009 he was a member of Prof. Phil Baran’s research group at The Scripps Research Institute, and from 2009–2012, an NIH postdoctoral fellow in Prof. Steve Buchwald’s lab at MIT. In 2012, he returned to UC-Berkeley as an assistant professor in the area of natural products total synthesis.
Zachary Brill received his B.A. in chemistry (summa cum laude) from Columbia University in 2012, where he conducted research in the lab of Prof. Scott Snyder. As an undergraduate, he completed the total synthesis and structural revision of the resveratrol dimer caraphenol B and pursued the synthesis of the unusually strained halogenated sesquiterpenoid aplydactone. In 2012, he began his graduate studies at University of California, Berkeley with Prof. Tom Maimone. Continuing his pursuit of total synthesis, his work at Berkeley on complex polycyclic terpenes has explored the use of tandem radical cyclization cascades in the construction of ophiobolin sesterterpenes.
Matthew Condakes received his AB and AM degrees in chemistry summa cum laude from Harvard University in 2014. While an undergraduate there, he conducted research on the synthesis of novel macrolide antibiotic analogs under the guidance of Prof. Andrew Myers. His passion for complex molecules then led him to pursue graduate work with Prof. Tom Maimone at the University of California, Berkeley as an NSF predoctoral fellow. At Berkeley, his work has focused on developing innovative synthetic strategies and methodologies–efforts that recently culminated in an oxidative synthetic strategy toward the Illicium sesquiterpenes.
Chi P. Ting was born in Hong Kong, and grew up in Chicago, Illinois. He received a B.S. degree in Chemistry at the University of Illinois at Urbana-Champaign, where he worked under the direction of Prof. Steven Zimmerman. In 2012, he began his PhD studies at the University of California, Berkeley as a founding member of the Maimone research group. His graduate work has explored concise total synthetic routes to podophyllotoxin, hyperforin, berkeleyone A, and garsubellin A.
Footnotes
Notes
The authors declare no competing financial interests.
References
- 1.Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Wiley-VCH; Weinheim, Germany: 2006. [Google Scholar]
- 2.Rude MA, Schirmer A. New microbial fuels: a biotech perspective. Curr Opin Microbiol. 2009;12:274–281. doi: 10.1016/j.mib.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Wilbon PA, Chu F, Tang C. Progress in Renewable Polymers from Natural Terpenes, Terpenoids, and Rosin. Macromol Rapid Commun. 2013;34:8–37. doi: 10.1002/marc.201200513. [DOI] [PubMed] [Google Scholar]
- 4.Blaser H-U. The chiral pool as a source of enantioselective catalysts and auxiliaries. Chem Rev. 1992;92:935–952. [Google Scholar]
- 5.Ager D, editor. Handbook of Chiral Chemicals. 2nd. CRC Press, Taylor & Francis; Boca Raton, FL: 2005. [Google Scholar]
- 6.Hanessian S. Total Synthesis of Natural Products: The Chiron Approach. Pergamon Press; Oxford: 1983. [Google Scholar]
- 7.Nakata M. Monosaccharides as Chiral Pools for the Synthesis of Complex Natural Compounds. In: Fraser-Reid BO, Tatsuta K, Thiem J, editors. Glycoscience. 2nd. Vol. 2. Springer-Verlag; Berlin: 2008. pp. 957–994. Chapter 4.4. [Google Scholar]
- 8.Chida N, Sato T. Comprehensive Chirality. Vol. 2. Elsevier; 2012. Chiral Pool Synthesis: Chiral Pool Syntheses Starting from Carbohydrates; pp. 207–239. [Google Scholar]
- 9.Paek S-M, Jeong M, Jo J, Heo YM, Han YT, Yun H. Recent Advances in Substrate-Controlled Asymmetric Induction Derived from Chiral Pool α-Amino Acids for Natural Product Synthesis. Molecules. 2016;21:951. doi: 10.3390/molecules21070951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho TL. Enantioselective Synthesis Natural Products from Chiral Terpenes. Wiley; New York: 1992. [Google Scholar]
- 11.Money T, Wong MKC. The Use of Cyclic Monoterpenoids as Enantiopure Starting Materials in Natural Product Synthesis. Stud Nat Prod Chem. 1995;16:123–288. [Google Scholar]
- 12.Heusler F, Pond FJ. The Chemistry of the Terpenes. P. Blakiston’s Son & Co; Philadelphia: 1902. [Google Scholar]
- 13.Ruzicka L. The isoprene rule and the biogenesis of terpenic compounds. Experientia. 1953;9:357–367. doi: 10.1007/BF02167631. [DOI] [PubMed] [Google Scholar]
- 14.Eschenmoser A, Arigoni D. Revisited after 50 Years: The ‘Stereochemical Interpretation of the Biogenetic Isoprene Rule for the Triterpenes,’. Helv Chim Acta. 2005;88:3011–3050. [Google Scholar]
- 15.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 16.Nugent WA, RajanBabu TV, Burk MJ. Beyond Nature’s Chiral Pool: Enantioselective Catalysis in Industry. Science. 1993;259:479–483. doi: 10.1126/science.259.5094.479. [DOI] [PubMed] [Google Scholar]
- 17.Maimone TJ, Baran PS. Modern synthetic efforts toward biologically active terpenes. Nat Chem Biol. 2007;3:396–407. doi: 10.1038/nchembio.2007.1. [DOI] [PubMed] [Google Scholar]
- 18.Jansen DJ, Shenvi RA. Synthesis of medicinally relevant terpenes: reducing the cost and time of drug discovery. Future Med Chem. 2014;6:1127–1148. doi: 10.4155/fmc.14.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiao T, Liang G. Recent advances in terpenoid synthesis from China. Sci China Chem. 2016;59:1142–1174. [Google Scholar]
- 20.Urabe D, Asaba T, Inoue M. Convergent Strategies in Total Syntheses of Complex Terpenoids. Chem Rev. 2015;115:9207–9231. doi: 10.1021/cr500716f. [DOI] [PubMed] [Google Scholar]
- 21.Gaich T, Mulzer J. In: Comprehensive Chirality. Carreira EM, Yamamoto H, editors. Vol. 2. Elsevier Ltd; 2012. pp. 163–206. [Google Scholar]
- 22.Hanson JR. Steroids: partial synthesis in medicinal chemistry. Nat Prod Rep. 2007;24:1342–1349. doi: 10.1039/b705948p. [DOI] [PubMed] [Google Scholar]
- 23.Lednicer D. Steroid Chemistry at a Glance. Wiley; 2011. [Google Scholar]
- 24.Lowest price (USD) in bulk quantities as of October 2016
- 25.Lenardão EJ, Botteselle GV, de Azambuja F, Perin G, Jacob RG. Citronellal as a key compound in organic synthesis. Tetrahedron. 2007;63:6671–6712. [Google Scholar]
- 26.Akutagawa S. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer; New York: 1999. Chapter 41.4. [Google Scholar]
- 27.Wu YK, Liu HJ, Zhu JL. An Efficient Procedure for the 1,3-Transposition of Allylic Alcohols Based on Lithium Naphthalenide Induced Reductive Elimination of Epoxy Mesylates. Synlett. 2008:0621–0623. [Google Scholar]
- 28.Macaev FZ. Bioactive Natural Products from Enantiomeric Carvones. In: Atta-ur-Rahman Ed., editor. Studies in Natural Products Chemistry. Vol. 39. Elsevier; 2013. Chapter 7. [Google Scholar]
- 29.Money T. Camphor: a chiral starting material in natural product synthesis. Nat Prod Rep. 1985:253–289. doi: 10.1039/np9850200253. [DOI] [PubMed] [Google Scholar]
- 30.Taylor MD, Minaskanian G, Winzenberg KN, Santone P, Smith AB., III Preparation, stereochemistry, and nuclear magnetic resonance spectroscopy of 4-hydroxy(acetoxy)bicyclo[5.1.0]octanes. Synthesis of (−)- and (±)-8,8-dimethylbicyclo[5.1.0]oct-2-en-4-one. J Org Chem. 1982;47:3960–3964. [Google Scholar]
- 31.Santana A, Molinillo JMG, Macías FA. Trends in the Synthesis and Functionalization of Guaianolides. Eur J Org Chem. 2015:2093–2110. [Google Scholar]
- 32.Ellerbrock P, Armanino N, Ilg MK, Webster R, Trauner D. An eight-step synthesis of epicolactone reveals its biosynthetic origin. Nat Chem. 2015;7:879–882. doi: 10.1038/nchem.2336. [DOI] [PubMed] [Google Scholar]
- 33.Corey EJ, Pearce HL. Total synthesis of picrotoxinin. J Am Chem Soc. 1979;101:5841–5843. [Google Scholar]
- 34.Schmid G, Hofheinz W. Total synthesis of qinghaosu. J Am Chem Soc. 1983;105:624–625. [Google Scholar]
- 35.Corey EJ, Cheng XM. The Logic of Chemical Synthesis. Wiley; New York: 1995. [Google Scholar]
- 36.Nicolaou KC, van Delft F, Ohshima T, Vourloumis D, Xu J, Hosokawa S, Pfefferkorn J, Kim S, Li T. Total Synthesis of Eleutherobin. Angew Chem Int Ed. 1997;36:2520–2524. [Google Scholar]
- 37.Chen X-T, Gutteridge CE, Bhattacharya SK, Zhou B, Pettus TR, Hascall T, Danishefsky SJ. Total Synthesis of Eleutherobin. Angew Chem Int Ed. 1998;37:185–187. doi: 10.1002/(SICI)1521-3773(19980403)37:6<789::AID-ANIE789>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 38.Wender PA, Holt DA. Macroexpansion methodology. 3. Eight-step synthesis of (−)-(3Z)-cembrene A. J Am Chem Soc. 1985;107:7771–7772. [Google Scholar]
- 39.Smith AB, III, Liverton NJ, Hrib NJ, Sivaramakrishnan H, Winzenberg KN. Total synthesis of (+)-jatropholone A and B. J Org Chem. 1985;50:3239–3241. [Google Scholar]
- 40.Rowley M, Tsukamoto M, Kishi Y. Total synthesis of (+)-ophiobolin C. J Am Chem Soc. 1989;111:2735–2731. [Google Scholar]
- 41.Walji AB, Macmillan DW. Strategies to Bypass the Taxol Problem. Enantioselective cascade catalysis, a New Approach for the Efficient Construction of Molecular Complexity. Synlett. 2007:1477–1489. [Google Scholar]
- 42.Ishihara Y, Baran PS. Two-Phase Terpene Total Synthesis: Historical Perspective and Application to the Taxol® Problem. Synlett. 2010:1733–1745. [Google Scholar]
- 43.Wender PA, Badham NF, Conway SP, Floreancig PE, Glass TE, Houze JB, Krauss NE, Lee D, Marquess DG, McGrane PL, et al. The Pinene Path to Taxanes. 6. A Concise Stereocontrolled Synthesis of Taxol. J Am Chem Soc. 1997;119:2757–2758. [Google Scholar]
- 44.Holton RA, Kim HB, Somoza C, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim S. First total synthesis of taxol. 2. Completion of the C and D rings. J Am Chem Soc. 1994;116:1599–1600. [Google Scholar]
- 45.Burns NZ, Baran PS, Hoffman RW. Redox Economy in Organic Synthesis. Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 46.Wender PA, Verman VA, Paxton TJ, Pillow TH. Function-Oriented Synthesis, Step Economy, and Drug Design. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 47.Hoffman RW. Protecting-Group-Free Synthesis. Synthesis. 2006;21:3531–3541. [Google Scholar]
- 48.Young IS, Baran PS. Protecting-group-free synthesis as an opportunity for invention. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 49.Newhouse T, Baran PS, Hoffmann RW. The economies of synthesis. Chem Soc Rev. 2009;38:3010–3021. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mangion IK, Macmillan DWC. Total Synthesis of Brasoside and Littoralisone. J Am Chem Soc. 2005;127:3696–3697. doi: 10.1021/ja050064f. [DOI] [PubMed] [Google Scholar]
- 51.He D-Y, Dai S-M. Anti-inflammatory and immunomodulatory effects of Paeonia lactiflora Pall., a traditional Chinese herbal medicine. Front Pharmacol. 2011;2:1–5. doi: 10.3389/fphar.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu S-H, Wu D-G, Chen Y-W. Chemical constituents and bioactivities of plants from the genus Paeonia. Chem Biodivers. 2010;7:90–104. doi: 10.1002/cbdv.200800148. [DOI] [PubMed] [Google Scholar]
- 53.Corey EJ, Wu Y-J. Total synthesis of (±)-paeoniflorigenin and paeoniflorin. J Am Chem Soc. 1993;115:8871–8872. [Google Scholar]
- 54.Hatakeyama S, Kawamura M, Takano S. Total Synthesis of (−)-Paeoniflorin. J Am Chem Soc. 1994;116:4081–4082. [Google Scholar]
- 55.Hatakeyama S, Kawamura M, Mukugi Y, Irie H. Total syntheses of (+)-paeonilactone B and (−)-paeonisuffrone. Tetrahedron Lett. 1995;36:267–268. [Google Scholar]
- 56.Martín-Rodríquez M, Galán-Fernández R, Marcos-Escribano A, Bermejo FA. Ti(III)-Promoted Radical Cyclization of Epoxy Enones. Total Synthesis of (+)-Paeonisuffrone. J Org Chem. 2009;74:1798–1801. doi: 10.1021/jo8026033. [DOI] [PubMed] [Google Scholar]
- 57.Bermejo FA, Mateos AF, Escribano AM, Lago RM, Burón LM, López MR, González RR. Ti(III)-promoted cyclizations. Application to the synthesis of (E)-endo-bergamoten-12-oic acids Moth oviposition stimulants isolated from Lycopersicon hirsutum. Tetrahedron. 2006;62:8933–8942. [Google Scholar]
- 58.Gansäuer A, Fleckhaus A. Biology and Materials, Online. Wiley; 2012. Epoxides in Titanocene-Mediated and –Catalyzed Radical Reactions In Encyclopedia of Radicals in Chemistry. [Google Scholar]
- 59.Morcillo SP, Miguel D, Campaña AG, Álvarez de Cienfuegos L, Justicia J, Cuerva JM. Recent applications of Cp2TiCl in natural product synthesis. Org Chem Front. 2014;1:15–33. [Google Scholar]
- 60.Kamchonwongpaisan S, Nilanonta C, Tarnchompoo B, Thebtaranonth C, Thebtaranonth Y, Yuthavong Y, Kongsaeree P, Clardy J. An antimalarial peroxide from Amomum krervanh Pierre. Tetrahedron Lett. 1995;36:1821–1824. [Google Scholar]
- 61.Slack RD, Jacobine AM, Posner GH. Antimalarial peroxides: advances in drug discovery and design. Med Chem Commun. 2012;3:281–297. [Google Scholar]
- 62.Tang Y, Dong Y, Vennerstrom JL. Synthetic peroxides as antimalarials. Med Res Rev. 2004;24:425–448. doi: 10.1002/med.10066. [DOI] [PubMed] [Google Scholar]
- 63.Muraleedharan KM, Avery MA. Progress in the development of peroxide-based anti-parasitic agents. Drug Discov Today. 2009;14:793–803. doi: 10.1016/j.drudis.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 64.Hu X, Maimone TJ. Four-Step Synthesis of the Antimalarial Cardamom Peroxide via an Oxygen Stitching Strategy. J Am Chem Soc. 2014;136:5287–5290. doi: 10.1021/ja502208z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghogare AA, Greer A. Using Singlet Oxygen to Synthesize Natural Products and Drugs. Chem Rev. 2016;116:9994–10034. doi: 10.1021/acs.chemrev.5b00726. [DOI] [PubMed] [Google Scholar]
- 66.Isayama S, Mukaiyama T. A new method for preparation of alcohols from olefins with molecular oxygen and phenylsilane by the use of bis(acetylacetonato)cobalt(II) Chem Lett. 1989:1071–1074. [Google Scholar]
- 67.Magnus P, Payne AH, Waring MJ, Scott DA, Lynch V. Conversion of α,β-unsaturated ketones into α-hydroxy ketones using an Mn(III) catalyst, phenylsilane and dioxygen: acceleration of conjugate hydride reduction by dioxygen. Tetrahedron Lett. 2000;41:9725–9730. [Google Scholar]
- 68.Korshin EE, Bachi MD. Synthesis of Cyclic Peroxides, Patai’s Chemistry of Functional Groups, Online. Wiley; 2009. pp. 1–117. [Google Scholar]
- 69.Hu X, Maimone TJ. In: Peroxy Radical Additions, in Science of Synthesis, Applications of Domino Transformations in Organic Synthesis. Snyder SA, editor. Vol. 1. Georg Thieme Verlag; Stuttgart, Germany: 2016. pp. 157–186. [Google Scholar]
- 70.Crossley SWM, Martinez RM, Obradors C, Shenvi RA. Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hugelshofer CL, Magauer T. Bioinspired Total Synthesis of Terpenoids. Org Biomol Chem. 2017;15:12–16. doi: 10.1039/c6ob02488b. [DOI] [PubMed] [Google Scholar]
- 72.Angeles AR, Waters SP, Danishefsky SJ. Total Syntheses of (+)- and (–)-Peribysin E. J Am Chem Soc. 2008;130:13765–13770. doi: 10.1021/ja8048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pardeshi SG, Ward DE. Enantiospecific Total Synthesis of Lairdinol A. J Org Chem. 2008;73:1071–1076. doi: 10.1021/jo7024465. [DOI] [PubMed] [Google Scholar]
- 74.Nicolaou KC, Sarlah D, Shaw DM. Total Synthesis and Revised Structure of Biyouyanagin A. Angew Chem Int Ed. 2007;46:4708–4711. doi: 10.1002/anie.200701552. [DOI] [PubMed] [Google Scholar]
- 75.Fürstner A, Hannen P. Platinum- and Gold-Catalyzed Rearrangement Reactions of Propargyl Acetates: Total Syntheses of (-)-α-Cubebene, (-)-Cubebol, Sesquicarene and Related Terpenes. Chem Eur J. 2006;12:3006–3019. doi: 10.1002/chem.200501299. [DOI] [PubMed] [Google Scholar]
- 76.Oliver SF, Högenauer K, Simic O, Antonello A, Smith MD, Ley SV. A route to the thapsigargins from (S)-carvone providing a substrate-controlled total synthesis of trilobolide, nortrilobolide, and thapsivillosin F. Angew Chem Int Ed. 2003;42:5996–6000. doi: 10.1002/anie.200353140. [DOI] [PubMed] [Google Scholar]
- 77.Yang H, Qiao X, Li F, Ma H, Xie L, Xu X. Diastereoselective total synthesis of 8-epigrosheimin. Tetrahedron Lett. 2009;50:1110–1112. [Google Scholar]
- 78.Kopp S, Schweizer WB, Altmann KH. Total Synthesis of Valerenic Acid. Synlett. 2009:1769–1772. [Google Scholar]
- 79.Zhang W, Luo S, Chen Q, Hu H, Jia X, Zhai H. Total Synthesis of Absinthin. J Am Chem Soc. 2005;127:18–19. doi: 10.1021/ja0439219. [DOI] [PubMed] [Google Scholar]
- 80.Liang XT, Yu DQ, Wu WL, Deng HC. The Structure of Yingzhaosu. Acta Chim Sin. 1979;37:215–230. [Google Scholar]
- 81.Xu X-X, Zhu J, Huang D-Z, Zhou W-S. Total synthesis of (+)-Yingzhaosu A. Tetrahedron Lett. 1991;32:5785–5788. [Google Scholar]
- 82.Szpilman AM, Korshin EE, Rozenberg H, Bachi MD. Total Syntheses of Yingzhaosu A and of Its C(14)- Epimer Including the First Evaluation of Their Antimalarial and Cytotoxic Activities. J Org Chem. 2005;70:3618–3632. doi: 10.1021/jo050074z. [DOI] [PubMed] [Google Scholar]
- 83.Kan SBJ, Ng KK-H, Paterson I. The Impact of the Mukaiyama Aldol Reaction in Total Synthesis. Angew Chem Int Ed. 2013;52:9097–9108. doi: 10.1002/anie.201303914. [DOI] [PubMed] [Google Scholar]
- 84.Corey EJ, Helal CJ. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew Chem Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 85.Dussault P. The Peroxide Changes Everything: New Methodology for the Synthesis of Peroxide-Containing Natural Products. Synlett. 1995:997–1003. [Google Scholar]
- 86.Wan KK, Evans-Klock CD, Fielder BC, Vosburg D. A Synthesis of cis- and trans-Davanoids: Artemone, Hydroxydavanone, Isodavanone, and Nordavanone. Synthesis. 2013;45:1541–1545. [Google Scholar]
- 87.Nacsa ED, Fiedler BC, Wetzler SP, Srisuknimit V, Litz JP, Van Vleet MJ, Quach K, Vosburg D. A Direct, Biomimetic Synthesis of (+)-Artemone via a Stereoselective, Organocatalytic Cyclization. Synthesis. 2015;47:2599–2602. [Google Scholar]
- 88.Naegeli P, Klimes J, Weber G. Structure and synthesis of Artemone. Tetrahedron Lett. 1970:5021–5024. [Google Scholar]
- 89.Honda Y, Ori A, Tsuchihashi GI. Stereoselective Syntheses of (+)-Davanone and (+)-Artemone via Anti-Selective Epoxidation and Iodo-cyclization. Chem Lett. 1987:1259–1262. [Google Scholar]
- 90.Jensen KL, Dickmeiss G, Jiang HAlbrecht Ł, Jørgensen KA. The Diarylprolinol Silyl Ether System: A General Organocatalyst. Acc Chem Res. 2012;45:248–264. doi: 10.1021/ar200149w. [DOI] [PubMed] [Google Scholar]
- 91.Fleury LM, Ashfeld BL. Organozinc Generation via the Titanium-Catalyzed Activation of Alkyl Halides. Org Lett. 2009;11:5670–5673. doi: 10.1021/ol902374v. [DOI] [PubMed] [Google Scholar]
- 92.McMorris TC, Lira R, Gantzel PK, Kelner MJ, Dawe R. Sesquiterpenes from the Basidiomycete Omphalotus illudens. J Nat Prod. 2000;63:1557–1559. doi: 10.1021/np9904760. [DOI] [PubMed] [Google Scholar]
- 93.Zheng Y-B, Lu C-H, Zheng Z-H, Lin X-J, Su W-J, Shen Y-M. New sesquiterpenes from Edible Fungus Clavicorona pyxidata. Helv Chim Acta. 2008;91:2174–2180. [Google Scholar]
- 94.Fraga BM. Natural sesquiterpenoids. Nat Prod Rep. 2013;30:1226–1264. doi: 10.1039/c3np70047j. [DOI] [PubMed] [Google Scholar]
- 95.Liu G, Romo D. Total Synthesis of (+)-Omphadiol. Angew Chem Int Ed. 2011;50:7537–7540. doi: 10.1002/anie.201102289. [DOI] [PubMed] [Google Scholar]
- 96.Henry-Riyad H, Lee C, Purohit VC, Romo D. Bicyclic- and Tricyclic-β-lactones via Organonucleophile-Promoted Bis-Cyclizations of Keto Acids: Enantioselective Synthesis of (+)-Dihydroplakevulin. Org Lett. 2006;8:4363. doi: 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]
- 97.Vougioukalakis GC, Grubbs RH. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 98.Acebey L, Sauvain M, Beck S, Moulis C, Gimenez A, Jullian V. Bolivianine, a New Sesterpene with an Unusual Skeleton from Hedyosmum angustifolium, and Its Isomer, Isobolivianine. Org Lett. 2007;9:4693–4696. doi: 10.1021/ol7015725. [DOI] [PubMed] [Google Scholar]
- 99.Du B, Yuan C, Yu T, Yang L, Yang Y, Liu B, Qin S. Asymmetric Total Synthesis of Onoseriolide, Bolivianine, and Isobolivianine. Chem Eur J. 2014;20:2613–2622. doi: 10.1002/chem.201304378. [DOI] [PubMed] [Google Scholar]
- 100.Yuan C, Du B, Yang L, Liu B. Bioinspired Total Synthesis of Bolivianine: A Diels− Alder/Intramolecular Hetero-Diels−Alder Cascade Approach. J Am Chem Soc. 2013;135:9291–9294. doi: 10.1021/ja4040335. [DOI] [PubMed] [Google Scholar]
- 101.Watanabe M, Awen BZ, Kato M. Efficient Synthesis of (lS,5S)-4-Alkyl-6,6-dimethylbicyclo[3.1.1]hept-3-en-2-ones from (1R,5S)-(+)-Nopinone and Preparation of Some Chiral Building Blocks Suitable for the Asymmetric Synthesis. J Org Chem. 1993;58:3923–3927. [Google Scholar]
- 102.Fulton JR, Aggarwal VK, de Vicente J. The Use of Tosylhydrazone Salts as a Safe Alternative for Handling Diazo Compounds and Their Applications in Organic Synthesis. Eur J Org Chem. 2005:1479–1492. [Google Scholar]
- 103.Doyle MP. Catalytic methods for metal carbene transformations. Chem Rev. 1986;86:919–939. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- 104.Oikawa H, Tokiwano T. Enzymatic catalysis of the Diels-Alder reaction in the biosynthesis of natural products. Nat Prod Rep. 2004;21:321–352. doi: 10.1039/b305068h. [DOI] [PubMed] [Google Scholar]
- 105.Stocking EM, Williams RM. Chemistry and Biology of Biosynthetic Diels-Alder Reactions. Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]
- 106.Klas K, Tsukamoto S, Sherman DH, Williams RM. Natural Diels-Alderases: Elusive and Irresistible. J Org Chem. 2015;80:11672–11685. doi: 10.1021/acs.joc.5b01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. The Diels-Alder Reaction in Total Synthesis. Angew Chem Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 108.Fukuyama Y, Huang J-M. Chemistry and neurotrophic activity of seco-prezizaane-and anislactone-type sesquiterpenes from Illicium species. In: Atta-ur-Rahman Ed., editor. Studies in Natural Products Chemistry. Vol. 32. Elsevier; Amsterdam; 2005. pp. 395–429. [Google Scholar]
- 109.Kubo M, Okada C, Huang J-M, Harada K, Hioki H, Fukuyama Y. Novel Pentacyclic seco-Prezizaane-Type Sesquiterpenoids with Neurotrophic Properties from Illicium jiadifengpi. Org Lett. 2009;11:5190–5193. doi: 10.1021/ol9021029. [DOI] [PubMed] [Google Scholar]
- 110.Xu J, Trzoss L, Chang WK, Theodorakis EA. Enantioselective Total Synthesis of (−)-Jiadifenolide. Angew Chem Int Ed. 2011;50:3672–3676. doi: 10.1002/anie.201100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trzoss L, Xu J, Lacoske MH, Mobley WC, Theodorakis EA. Illicium Sesquiterpenes: Divergent Synthetic Strategy and Neurotrophic Activity Studies. Chem Eur J. 2013;19:6398–6408. doi: 10.1002/chem.201300198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paterson I, X M, Dalby SM. Total Synthesis of Jiadifenolide. Angew Chem Int Ed. 2014;53:7286–7289. doi: 10.1002/anie.201404224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Siler DA, Mighion JD, Sorensen EJ. An Enantiospecific Synthesis of Jiadifenolide. Angew Chem Int Ed. 2014;53:5332–5335. doi: 10.1002/anie.201402335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu H-H, Martinez MD, Shenvi RA. An eight-step gram-scale synthesis of (−)-jiadifenolide. Nat Chem. 2015;7:604–607. doi: 10.1038/nchem.2283. [DOI] [PubMed] [Google Scholar]
- 115.Shen Y, Li L, Pan Z, Wang Y, Li J, Wang K, Wang X, Zhang Y, Hu T, Zhang Y. Protecting-Group-Free Total Synthesis of (−)-Jiadifenolide: Development of a [4 + 1] Annulation toward Multisubstituted Tetrahydrofurans. Org Lett. 2015;17:5480–5483. doi: 10.1021/acs.orglett.5b02845. [DOI] [PubMed] [Google Scholar]
- 116.Gomes J, Daeppen C, Liffert R, Roesselein J, Kaufman E, Heikinheimo A, Neuburger M, Gademann KJ. Formal Total Synthesis of (−)-Jiadifenolide and Synthetic Studies toward seco-Prezizaane-Type Sesquiterpenes. J Org Chem. 2016;81:11017–11034. doi: 10.1021/acs.joc.6b02039. [DOI] [PubMed] [Google Scholar]
- 117.Wolinsky J, Chan D. The Favorskii Rearrangement of Pulegone Dibromide. J Org Chem. 1965;30:41–43. [Google Scholar]
- 118.Vettel PR, Coates RM. Total Synthesis of (−)-Prezizaene and (−)-Prezizanol. J Org Chem. 1980;45:5430–5432. [Google Scholar]
- 119.Gawley RE. The Robinson Annelation and Related Reactions. Synthesis. 1976:777–794. [Google Scholar]
- 120.Niwa H, Nisiwaki M, Tsukada I, Ishigaki T, Ito S, Wakamatsu K, Mori T, Ikagawa M, Yamada K. Stereocontrolled Total Synthesis of (−)-Anisatin: A Neurotoxic Sesquiterpenoid Possessing a Novel Spiro β-Lactone. J Am Chem Soc. 1990;112:9001–9003. [Google Scholar]
- 121.Leusen DV, Leusen AMV. Synthetic Uses of Tosylmethyl Isocyanide (TosMIC) Organic Reactions. 2004;57:3, 417–666. [Google Scholar]
- 122.Desai LV, Hull KL, Sanford MS. Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 123.Neufeldt SR, Sanford MS. O-Acetyl Oximes as Transformable Directing Groups for Pd-Catalyzed C-H Bond Functionalization. Org Lett. 2010;12:532–535. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gutekunst WR, Baran PS. C–H functionalization logic in total synthesis. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 126.Chen DY-K, Youn SW. C–H Activation: A Complementary Tool in the Total Synthesis of Complex Natural Products. Chem Eur J. 2012;18:9452–9474. doi: 10.1002/chem.201201329. [DOI] [PubMed] [Google Scholar]
- 127.Brückl T, Baxter RD, Ishihara Y, Baran PS. Innate and Guided C–H Functionalization Logic. Acc Chem Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yamaguchi J, Yamaguchi AD, Itami K. C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 129.Hartwig JF. Evolution of C–H Bond Functionalization from Methane to Methodology. J Am Chem Soc. 2016;138:2–24. doi: 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tao P, Jia Y. C–H bond activation in the total syntheses of natural products. Sci China Chem. 2016;59:1109–1125. [Google Scholar]
- 131.Molander GA. Sequencing Reactions with Samarium(II) Iodide. Chem Rev. 1996;96:307–338. doi: 10.1021/cr950019y. [DOI] [PubMed] [Google Scholar]
- 132.Edmonds DJ, Johnston D, Procter DJ. Samarium(II)-Iodide-Mediated Cyclizations in Natural Product Synthesis. Chem Rev. 2004;104:3371–3404. doi: 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]
- 133.Nicolaou KC, Ellery SP, Chen JS. Samarium diiodide mediated reactions in total synthesis. Angew Chem Int Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lyapkalo IM, Vogel MAK, Boltukhina EV, Vavříka J. A general one-step synthesis of alkynes from enolisable carbonyl compounds. Synlett. 2009:558–561. [Google Scholar]
- 135.Peixoto PA, Boulangé A, Leleu S, Franck X. Versatile synthesis of acylfuranones by reaction of acylketenes with α-hydroxy ketones: application to the one-step multi-component synthesis of cadiolide B and its analogues. Eur J Org Chem. 2013:3316–3327. [Google Scholar]
- 136.Kuttruff CA, Eastgate MD, Baran PS. Natural product synthesis in the age of scalability. Nat Prod Rep. 2014;31:419–432. doi: 10.1039/c3np70090a. [DOI] [PubMed] [Google Scholar]
- 137.Ratnayake R, Covell D, Ransom TT, Gustafson KR, Beutler JA. Englerin A, a Selective Inhibitor of Renal Cancer Cell Growth, from Phyllanthus engleri. Org Lett. 2009;11:57–60. doi: 10.1021/ol802339w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pouwer RH, Richard J-A, Tseng C-C, Chen DY-K. Chemical Synthesis of the Englerins. Chem Asian J. 2011;7:22–35. doi: 10.1002/asia.201100780. [DOI] [PubMed] [Google Scholar]
- 139.Willot M, Radtke L, Könnig D, Fröhlich R, Gessner VH, Strohmann C, Christmann M. Total Synthesis and Absolute Configuration of the Guaiane Sesquiterpene Englerin A. Angew Chem Int Ed. 2009;48:9105–9108. doi: 10.1002/anie.200905032. [DOI] [PubMed] [Google Scholar]
- 140.Nicolaou KC, Kang Q, Ng SY, Chen DY-K. Total Synthesis of Englerin A. J Am Chem Soc. 2010;132:8219–8222. doi: 10.1021/ja102927n. [DOI] [PubMed] [Google Scholar]
- 141.Xu J, Caro-Diaz EJE, Theodorakis EA. Enantioselective Formal Synthesis of (−)-Englerin A via a Rh-Catalyzed [4+3] Cycloaddition Reaction. Org Lett. 2010;12:3708–3711. doi: 10.1021/ol1015652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou Q, Chen X, Ma D. Asymmetric, Protecting-Group-Free Total Synthesis of (−)-Englerin A. Angew Chem Int Ed. 2010;49:3513–3516. doi: 10.1002/anie.201000888. [DOI] [PubMed] [Google Scholar]
- 143.Molawi K, Delpont N, Echavarren AM. Enantioselective Synthesis of (−)-Englerins A and B. Angew Chem Int Ed. 2010;49:3517–3519. doi: 10.1002/anie.201000890. [DOI] [PubMed] [Google Scholar]
- 144.Li Z, Nakashige M, Chain WJ. A Brief Synthesis of (−)-Englerin A. J Am Chem Soc. 2011;133:6553–6556. doi: 10.1021/ja201921j. [DOI] [PubMed] [Google Scholar]
- 145.Takahashi K, Komine K, Yokoi Y, Ishihara J, Hatakeyama S. Stereocontrolled Total Synthesis of (−)-Englerin A. J Org Chem. 2012;77:7364–7370. doi: 10.1021/jo301145r. [DOI] [PubMed] [Google Scholar]
- 146.Lee J, Parker KA. A Formal Synthesis of (−)-Englerin A by Relay Ring Closing Metathesis and Transannular Etherification. Org Lett. 2012;14:2682–2685. doi: 10.1021/ol3007524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gao P, Cook SP. A Reductive-Heck Approach to the Hydroazulene Ring System: A Formal Synthesis of the Englerins. Org Lett. 2012;14:3340–3343. doi: 10.1021/ol3013167. [DOI] [PubMed] [Google Scholar]
- 148.Zahel M, Keβberg A, Metz P. A Short Enantioselective Total Synthesis of (−)-Englerin A. Angew Chem Int Ed. 2013;52:5390–5392. doi: 10.1002/anie.201301247. [DOI] [PubMed] [Google Scholar]
- 149.Wang J, Chen S-G, Sun B-F, Lin G-Q, Shang Y-J. Collective Total Synthesis of Englerin A and B, Orientalol E and F, and Oxyphyllol: Application of the Organocatalytic [4+3] Cycloaddition Reaction. Chem Eur J. 2013;19:2539–2547. doi: 10.1002/chem.201203467. [DOI] [PubMed] [Google Scholar]
- 150.Zhang J, Zheng S, Peng W, Shen Z. Total synthesis of (−)-Englerin A. Tetrahedron Lett. 2014;55:1339–1341. [Google Scholar]
- 151.Hanari T, Shimada N, Kurosaki Y, Thrimurtulu N, Nambu H, Anada M, Hashimoto S. Asymmetric Total Synthesis of (−)-Englerin A through Catalytic Diastereo- and Enantioselective Carbonyl Ylide Cycloaddition. Chem Eur J. 2015;21:11671–11676. doi: 10.1002/chem.201502009. [DOI] [PubMed] [Google Scholar]
- 152.Kusama H, Tazawa A, Ishida K, Iwasawa N. Total Synthesis of (±)-Englerin A Using An Intermolecular [3+2] Cycloaddition Reaction of Platinum-Containing Carbonyl Ylide. Chem Asian J. 2016;11:64–67. doi: 10.1002/asia.201500935. [DOI] [PubMed] [Google Scholar]
- 153.Nelson R, Gulías M, Mascareñas JL, López F. Concise, Enantioselective, and Versatile Synthesis of (−)-Englerin A Based on a Platinum-Catalyzed [4C+3C] Cycloaddition of Allenedienes. Angew Chem Int Ed. 2016;55:14359–14363. doi: 10.1002/anie.201607348. [DOI] [PubMed] [Google Scholar]
- 154.Takano S, Inomata K, Samizu K, Tomita S, Yanase M, Suzuki M, Iwabuchi Y, Sugihara T, Ogasawara KA. Convenient One-flask Synthesis of α-Methylenealdehydes from Primary Alcohols. Chem Lett. 1989:1283–1284. [Google Scholar]
- 155.Chavez DE, Jacobsen EN. Catalyst-Controlled Inverse-Electron-Demand Hetero-Diels−Alder Reactions in the Enantio- and Diastereoselective Synthesis of Iridoid Natural Products. Org Lett. 2003;5:2563–2565. doi: 10.1021/ol034883l. [DOI] [PubMed] [Google Scholar]
- 156.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 157.Vulovic B, Bihelovic F, Matovic R, Saicic RN. Organocatalyzed Tsuji–Trost reaction: a new method for the closure of five- and six-membered rings. Tetrahedron. 2009;65:10485–10494. [Google Scholar]
- 158.Busch T, Kirschning A. Recent advances in the total synthesis of pharmaceutically relevant diterpenes. Nat Prod Rep. 2008;25:318–341. doi: 10.1039/b705652b. [DOI] [PubMed] [Google Scholar]
- 159.Soucy P, L’ Heureux A, Toró A, Deslongchamps P. Pyranophane Transannular Diels−Alder Approach to (+)-Chatancin: A Biomimetic Asymmetric Total Synthesis. J Org Chem. 2003;68:9983–9987. doi: 10.1021/jo035193y. [DOI] [PubMed] [Google Scholar]
- 160.Corminboeuf O, Overman LE, Pennington LD. Enantioselective Total Synthesis of Briarellins E and F: The First Total Syntheses of Briarellin Diterpenes. J Am Chem Soc. 2003;125:6650–6652. doi: 10.1021/ja035445c. [DOI] [PubMed] [Google Scholar]
- 161.Shipe WD, Sorensen EJ. Convergent, Enantioselective Syntheses of Guanacastepenes A and E Featuring a Selective Cyclobutane Fragmentation. J Am Chem Soc. 2006;128:7025–7035. doi: 10.1021/ja060847g. [DOI] [PubMed] [Google Scholar]
- 162.Ghosh AK, Xi K. Total Synthesis of (−)-Platensimycin, a Novel Antibacterial Agent. J Org Chem. 2009;74:1163–1170. doi: 10.1021/jo802261f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harrowven DC, Pascoe DD, Demurtas D, Bourne HO. Total Synthesis of (−)-Colombiasin A and (−)-Elisapterosin B. Angew Chem Int Ed. 2005;44:1221–1222. doi: 10.1002/anie.200462268. [DOI] [PubMed] [Google Scholar]
- 164.Mohr PJ, Halcomb RL. Total Synthesis of (+)-Phomactin A Using a B-Alkyl Suzuki Macrocyclization. J Am Chem Soc. 2003;125:1712–1713. doi: 10.1021/ja0296531. [DOI] [PubMed] [Google Scholar]
- 165.Tiefenbacher K, Mulzer J. Short Formal Synthesis of (−)-Platencin. Angew Chem Int Ed. 2008;47:6199–6200. doi: 10.1002/anie.200801441. [DOI] [PubMed] [Google Scholar]
- 166.Waalboer DCJ, Schaapman MC, van Delft FL, Rutjes FPJT. High-Pressure Entry into Platencin. Angew Chem Int Ed. 2008;47:6576–6578. doi: 10.1002/anie.200802912. [DOI] [PubMed] [Google Scholar]
- 167.Molander GA, St Jean DJ, Jr, Haas J. Toward a General Route to the Eunicellin Diterpenes: The Asymmetric Total Synthesis of Deacetoxyalcyonin Acetate. J Am Chem Soc. 2004;126:1642–1643. doi: 10.1021/ja0398464. [DOI] [PubMed] [Google Scholar]
- 168.Cheng HM, Tian W, Peixoto PA, Dhudshia B, Chen DY-K. Synthesis of ent-Nanolobatolide. Angew Chem Int Ed. 2011;50:4165–4168. doi: 10.1002/anie.201100926. [DOI] [PubMed] [Google Scholar]
- 169.González MA. Total Syntheses and Synthetic Studies of Spongiane Diterpenes. Tetrahedron. 2008;64:445–467. [Google Scholar]
- 170.Hambley TW, Poiner A, Taylor WC. Diterpene Metabolites of the Marine Sponge Chelonaplysilla violacea: Aplyviolene and Aplyviolacene. Tetrahedron Lett. 1986;27:3281–3282. [Google Scholar]
- 171.Overman LE, Pennington LD. A new strategy for synthesis of attached rings. Can J Chem. 2000;78:732–738. [Google Scholar]
- 172.Schnermann MJ, Overman LE. Enantioselective Total Synthesis of Aplyviolene. J Am Chem Soc. 2011;133:16425–16427. doi: 10.1021/ja208018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schnermann MJ, Overman LE. A Concise Synthesis of (−)-Aplyviolene Facilitated by a Strategic Tertiary Radical Conjugate Addition. Angew Chem Int Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kreiser W, Below P. Erste Synthese von (+)-Homofenchon (1,4,4-Trimethylbicyclo[3.2.1] octan-3-on) Liebigs Ann Chem. 1985:203–209. [Google Scholar]
- 175.Jamison CR, Overman LE. Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photocatalysis. Acc Chem Res. 2016;49:1578–1586. doi: 10.1021/acs.accounts.6b00284. [DOI] [PubMed] [Google Scholar]
- 176.Singh M, Pal M, Sharma RP. Biological Activity of the Labdane Diterpenes. Planta Med. 1999;65:2–8. doi: 10.1055/s-1999-13952. [DOI] [PubMed] [Google Scholar]
- 177.Demetzos C, Dimas KS. Labdane-type diterpenes: Chemistry and biological activity. Studies in Natural Products Chemistry. 2001;25:235–292. [Google Scholar]
- 178.Malochet-Grivois C, Cotelle P, Biard JF, Hénichart JP, Debitus C, Roussakis C, Verbist J-F. Dichlorolissoclimide, a New Cytotoxic Labdane Derivative from Lissoclinum voeltzkowi Michaelson (Urochordata) Tetrahedron Lett. 1991;32:6701–6702. [Google Scholar]
- 179.Malochet-Grivois C, Roussakis C, Robillard N, Biard JF, Riou D, Debitus C, Verbist J-F. Effects in vitro of two marine substances, chlorolissoclimide and dichlorolissoclimide, on a non-small-cell bronchopulmonary carcinoma line (NSCLC-N6) Anti-Cancer Drug Des. 1992;7:493–502. [PubMed] [Google Scholar]
- 180.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. Site-Selective Aliphatic C−H Chlorination Using N‑Chloroamides Enables a Synthesis of Chlorolissoclimide. J Am Chem Soc. 2015;138:696–702. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fokin AA, Schreiner PR. Metal-Free, Selective Alkane Functionalizations. Adv Synth Catal. 2003;345:1035–1052. [Google Scholar]
- 182.Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. Site-Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J Am Chem Soc. 2014;136:14389–14392. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- 183.Chen K, Eschenmoser A, Baran PS. Strain Release in C–H Bond Activation? Angew Chem Int Ed. 2009;48:9705–9708. doi: 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Newhouse T, Baran PS. If C–H bonds could talk: selective C–H bond oxidation. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen MS, White MC. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 186.Zhang X, Yang H, Tang P. Transition-Metal-Free Oxidative Aliphatic C−H Azidation. Org Lett. 2015;17:5828–5831. doi: 10.1021/acs.orglett.5b03001. [DOI] [PubMed] [Google Scholar]
- 187.Huang X, Bergsten TM, Groves JT. Manganese-Catalyzed Late-Stage Aliphatic C−H Azidation. J Am Chem Soc. 2015;137:5300–5303. doi: 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- 188.Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 189.Xia J-B, Ma Y, Chen C. Vanadium-Catalyzed C(sp3)-H Fluorination Reactions. Org Chem Front. 2014;1:468–472. doi: 10.1039/C4QO00057A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Halperin SD, Fan H, Chang S, Martin RE, Britton RA. Convenient Photocatalytic Fluorination of Unactivated C–H Bonds. Angew Chem, Int Ed. 2014;53:4690–4693. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- 191.Bloom S, Knippel JL, Lectka T. A photocatalyzed aliphatic fluorination. Chem Sci. 2014;5:1175–1178. [Google Scholar]
- 192.Kee CW, Chan KM, Wong MW, Tan C-H. Selective Bromination of sp3 C–H Bonds by Organophotoredox Catalysis. Asian J Org Chem. 2014;3:536–544. [Google Scholar]
- 193.Liu W, Groves JT. Manganese Porphyrins Catalyze Selective C−H Bond Halogenations. J Am Chem Soc. 2010;132:12847–12849. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- 194.Kee CW, Chin KF, Wong MW, Tan C-H. Selective fluorination of alkyl C–H bonds via photocatalysis. Chem Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 195.Zhang X, Guo S, Tang P. Transition-metal free oxidative aliphatic C–H fluorination. Org Chem Front. 2015;2:806–810. [Google Scholar]
- 196.Nguyen TM, Vu NQ, Youte J-J, Lau J, Cheong A, Ho YS, Tan BSW, Yoganathan K, Butler MS, Chai CLL. A fast and straightforward route towards the synthesis of the lissoclimide class of anti-tumour agents. Tetrahedron. 2010;66:9270–9276. [Google Scholar]
- 197.Tius MA. Synthesis of cembranes and cembranolides. Chem Rev. 1988;88:719–732. [Google Scholar]
- 198.Prestwich GD, Wiemer DF, Meinwald J, Clardy J. Cubitene: An Irregular Twelve-Membered-Ring Diterpene from a Termite Soldier. J Am Chem Soc. 1978;100:2560–2561. [Google Scholar]
- 199.Wahlberg I, Eklund AM. Cembranoids, Pseudopteranoids, and Cubitanoids of Natural Occurrence. In: Herz W, Kirby GW, Moore RE, Steglich W, Tamm C, editors. Prog Chem Nat Prod. Springer; New York: 1992. pp. 142–294. [Google Scholar]
- 200.Snyder SA, Wespe DA, von Hof JMA. Concise, Stereocontrolled Total Synthesis of Rippertenol. J Am Chem Soc. 2011;133:8850–8853. doi: 10.1021/ja202859f. [DOI] [PubMed] [Google Scholar]
- 201.Vig OP, Bari SS, Trehan IR, Vig R. Indian J Chem Sect B. 1980:446–449. [Google Scholar]
- 202.Kodama M, Takahashi T, Kojima T, Itô S. Synthesis of Macrocyclic Terpenoids by Intramolecular Cyclization VII. Total Synthesis of (±)-cubitene. Tetrahedron Lett. 1982;23:3397–3400. [Google Scholar]
- 203.Kodama M, Maeda H, Hioki H. Enantioselective Synthesis and Absolute Configuration of (+)-Cubitene. Chem Lett. 1996:809–810. [Google Scholar]
- 204.Simon K, Wefer J, Schöttner E, Lindel T. Enantioselective Total Synthesis of the Diterpene (+)-Cubitene. Angew Chem Int Ed. 2012;51:10889–10892. doi: 10.1002/anie.201205143. [DOI] [PubMed] [Google Scholar]
- 205.Schöttner E, Wiechoczek M, Jones PG, Lindel T. Enantiospecific Synthesis of the Cubitane Skeleton. Org Lett. 2010;12:784–787. doi: 10.1021/ol902854t. [DOI] [PubMed] [Google Scholar]
- 206.Hoffmann RW. Elements of Synthesis Planning. Springer; Berlin: 2009. pp. 106–117. [Google Scholar]
- 207.Su J-H, Huang H-C, Chao C-H, Yan L-Y, Wu Y-C, Sheu J-H. Vigulariol, a New Metabolite from the Sea Pen Vigularia juncea. Bull ChemSoc Jpn. 2005;78:877–879. [Google Scholar]
- 208.Sung P-J, Chen M-C. THE HETEROCYCLIC NATURAL PRODUCTS OF GORGONIAN CORALS OF GENUS BRIAREUM EXCLUSIVE OF BRIARANE-TYPE DITERPENOIDS. Heterocycles. 2002;57:1705–1715. [Google Scholar]
- 209.Welford AJ, Collins I. The 2,11-Cyclized Cembranoids: Cladiellins, Asbestinins, and Briarellins (Period 1998−2010) J Nat Prod. 2011;74:2318–2328. doi: 10.1021/np200125v. [DOI] [PubMed] [Google Scholar]
- 210.Ellis JM, Crimmins MT. Strategies for the Total Synthesis of C2-C11 Cyclized Cembranoids. Chem Rev. 2008;108:5278–5298. doi: 10.1021/cr078117u. [DOI] [PubMed] [Google Scholar]
- 211.Bernardelli P, Moradei OM, Friedrich D, Yang J, Gallou F, Dyck BP, Doskotch RW, Lange T, Paquette LA. Total Asymmetric Synthesis of the Putative Structure of the Cytotoxic Diterpenoid (−)-Sclerophytin A and of the Authentic Natural Sclerophytins A and B. J Am Chem Soc. 2001;123:9021–9032. doi: 10.1021/ja011285y. [DOI] [PubMed] [Google Scholar]
- 212.Clark JS, Hayes ST, Wilson C, Gobbi L. A Concise Total Synthesis of (±)-Vigulariol. Angew Chem Int Ed. 2007;46:437–440. doi: 10.1002/anie.200603880. [DOI] [PubMed] [Google Scholar]
- 213.Becker J, Bergander K, Fröhlich R, Hoppe D. Asymmetric Total Synthesis and X-Ray Crystal Structure of the Cytotoxic Marine Diterpene (+)-Vigulariol. Angew Chem Int Ed. 2008;47:1654–1657. doi: 10.1002/anie.200704678. [DOI] [PubMed] [Google Scholar]
- 214.Crimmins MT, Stauffer CS, Mans MC. Total Syntheses of (+)-Vigulariol and (−)-Sclerophytin A. Org Lett. 2011;13:4890–4893. doi: 10.1021/ol201981j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chen K, Baran PS. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 216.Ünaldi S, Özlügedik M, Fröhlich R, Hoppe D. Diastereoselective Synthesis of Enantioenriched, Annulated Tetrahydrofurans by Simultaneous Formation of the O-1–C-5 and the C-5–C-4 bonds. Adv Synth Catal. 2005;347:1621–1626. [Google Scholar]
- 217.Hoppe D, Marr F, Brüggemann M. In: Organolithiums in Enantioselective Synthesis. Hodgson DM, editor. Springer; Heidelberg: 2003. pp. 61–137. [Google Scholar]
- 218.Hoppe D, Christoph G. In: The Chemistry of Organolithium Compunds. Rappoport Z, Marek I, editors. Wiley; Chichester: 2004. p. 1055. [Google Scholar]
- 219.Rogers EF, Koniuszy FR, Shavel J, Folkers K. Plant insecticides I. Ryanodine, a new alkaloid from Ryania speciosa Vahl. J Am Chem Soc. 1948;70:3086–3088. doi: 10.1021/ja01189a074. [DOI] [PubMed] [Google Scholar]
- 220.Wiesner K, Valenta Z, Findlay JA. The structure of ryanodine. Tetrahedron Lett. 1967;8:221–223. [Google Scholar]
- 221.Srivastava SN, Przybylska M. The molecular structure of ryanodol-p-bromo benzyl ether. Can J Chem. 1968;46:795–797. [Google Scholar]
- 222.Pessah IN, Waterhouse AL, Casida JE. The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem Biophys Res Commun. 1985;128:449–456. doi: 10.1016/0006-291x(85)91699-7. [DOI] [PubMed] [Google Scholar]
- 223.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Waterhouse AL, Pessah IN, Francini AO, Casida JE. Structural aspects of ryanodine action and selectivity. J Med Chem. 1987;30:710–716. doi: 10.1021/jm00387a022. [DOI] [PubMed] [Google Scholar]
- 225.Welch W, Ahmad S, Airey JA, Geron K, Humerickhouse RA, Besch HR, Ruest L, Deslongchamps P, Sutko JL. Structural determinants of high-affinity binding of ryanoids to the vertebrate skeletal muscle ryanodine receptor: A comparative molecular field analysis. Biochemistry. 1994;33:6074–6085. doi: 10.1021/bi00186a006. [DOI] [PubMed] [Google Scholar]
- 226.Sutko JL, Airey JA, Welch W, Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol Rev. 1997;49:53–98. [PubMed] [Google Scholar]
- 227.Bélanger A, Berney DJF, Borschberg H-J, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao C-C, et al. Total synthesis of ryanodol. Can J Chem. 1979;57:3348–3354. [Google Scholar]
- 228.Deslongchamps P, Bélanger A, Berney DJF, Borschberg H-J, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, et al. The total synthesis of (+)-ryanodol. Part I. General strategy and search for a convenient diene for the construction of a key tricyclic intermediate. Can J Chem. 1990;68:115–126. [Google Scholar]
- 229.Deslongchamps P, Bélanger A, Berney DJF, Borschberg H-J, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, et al. The total synthesis of (+)-ryanodol. Part II. Model studies for rings B and C and of (+)-anhydroryanodol. Preparation of a key pentacyclic intermediate. Can J Chem. 1990;68:127–152. [Google Scholar]
- 230.Deslongchamps P, Bélanger A, Berney DJF, Borschberg H-J, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, et al. The total synthesis of (+)-ryanodol. Part III. Preparation of (+)-anhydroryanodol from a key pentacyclic intermediate. Can J Chem. 1990;68:153–185. [Google Scholar]
- 231.Deslongchamps P, Bélanger A, Berney DJF, Borschberg H-J, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, et al. The total synthesis of (+)-ryanodol. Part IV. Preparation of (+)-ryanodol from (+)-anhydroryanodol. Can J Chem. 1990;68:186–192. [Google Scholar]
- 232.Nagamoto M, Koshimizu M, Masuda K, Tabuchi T, Urabe D, Inoue M. Total synthesis of ryanodol. J Am Chem Soc. 2014;136:5916–5919. doi: 10.1021/ja502770n. [DOI] [PubMed] [Google Scholar]
- 233.Chuang KV, Xu C, Reisman S. E A 15-step synthesis of (+)-ryanodol. Science. 2016;353:912–915. doi: 10.1126/science.aag1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Nagamoto M, Hagiwara K, Masuda K, Koshimizu M, Kawamata T, Matsui Y, Urabe D, Inoue M. Symmetry-driven strategy for the assembly of the core tetracycle of (+)-ryanodine: Synthetic utility of a cobalt-catalyzed olefin oxidation and α-alkoxy bridgehead radical reaction. Chem Eur J. 2016;22:222–229. doi: 10.1002/chem.201503640. [DOI] [PubMed] [Google Scholar]
- 235.Masuda K, Koshimizu M, Nagatomo M, Inoue M. Asymmetric total synthesis of (+)-ryanodol and (+)-ryanodine. Chem Eur J. 2016;22:230–236. doi: 10.1002/chem.201503641. [DOI] [PubMed] [Google Scholar]
- 236.Egi M, Ota Y, Nishimura Y, Shimizu K, Azechi K, Akai S. Efficient intramolecular cyclizations of phenoxyethynyl diols into multisubstituted α,β-unsaturated lactones. Org Lett. 2013;15:4150–4153. doi: 10.1021/ol401824v. [DOI] [PubMed] [Google Scholar]
- 237.Koga Y, Kobayashi T, Narasaka K. Rhodium-catalyzed intramolecular Pauson-Khand reaction. Chem Lett. 1998;27:249–250. [Google Scholar]
- 238.Petasis NA, Patane M. A The synthesis of carbocyclic eight-membered rings. Tetrahedron. 1992;48:5757–5821. [Google Scholar]
- 239.DeBoer B. Fusicoccin – a key to multiple 14-3-3 locks? Trends Plant Sci. 1997;2:60–66. [Google Scholar]
- 240.Asahi K, Honma Y, Hazeki K, Sassa T, Kubohara Y, Sakurai A, Takahashi N. Cotylenin A, a Plant-Growth Regulator, Induces the Differentiation in Murine and Human Myeloid Leukemia Cells. Biochem Biophys Res Commun. 1997;238:758–763. doi: 10.1006/bbrc.1997.7385. [DOI] [PubMed] [Google Scholar]
- 241.De Boer AH, de Vries-Van Leeuwen IJ. Fusicoccanes: diterpenes with surprising biological functions. Trends Plant Sci. 2012;17:360–368. doi: 10.1016/j.tplants.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 242.Okamoto H, Arita H, Kato N, Takeshita H. Total Synthesis of (−)-Cotylenol, a Fungal Metabolite Having a Leaf Growth Activity. Chem Lett. 1994:2335–2338. [Google Scholar]
- 243.Kato N, Okamoto H, Takeshita H. Total Synthesis of (−)-Cotylenol, a Fungal Metabolite Having a Leaf Growth Activity. Tetrahedron. 1996;52:3921–3932. [Google Scholar]
- 244.Paquette LA, Sun L-Q, Friedrich D, Savage PB. Total Synthesis of (+)-Epoxydictymene. Application of Alkoxy-Directed Cyclization to Diterpenoid Construction. J Am Chem Soc. 1997;119:8438–8450. [Google Scholar]
- 245.Jamison TF, Shambayati S, Crowe WE, Schreiber SL. Cobalt-Mediated Total Synthesis of (+)-Epoxydictymene. J Am Chem Soc. 1994;116:5505–5506. [Google Scholar]
- 246.Williams DR, Robinson LA, Nevill CR, Reddy JP. Strategies for the Synthesis of Fusicoccanes by Nazarov Reaction of Dolabelladienones: Total Synthesis of (+)-Fusicoauritone. Angew Chem Int Ed. 2007;46:915–918. doi: 10.1002/anie.200603853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Zapp J, Burkhardt G, Becker H. Sphenolobane and fusicoccane diterpenoids from the liverword Anastrophyllum auritum. Phytochemistry. 1994;37:787–793. [Google Scholar]
- 248.Chiba R, Minami A, Gomi K, Oikawa H. Identification of Ophiobolin F Synthase by a Genome Mining Approach: A Sesterterpene Synthase from Aspergillus clavatus. Org Lett. 2013;15:594–597. doi: 10.1021/ol303408a. [DOI] [PubMed] [Google Scholar]
- 249.White JD, Ruppert JF, Avery A, Torii S, Nokami J. Spiroannelation via intramolecular ketocarbenoid addition. Stereocontrolled synthesis of (−)-acorenone B and (±)-alpha-chamigrene. J Am Chem Soc. 1981;103:1813–1821. [Google Scholar]
- 250.Williams DR, Coleman PJ. Studies on the Dolabellanes: Stereoselective Transannular Cyclizations of Dolabelladiene Macrocycles. Tetrahedron Lett. 1995;36:39–42. [Google Scholar]
- 251.Poupon E, Nay B. Biomimetic Organic Synthesis. Wiley-VCH; Weinheim: 2011. pp. 1–2. [Google Scholar]
- 252.Razzak M, De Brabander JK. Lessons and revelations from biomimetic syntheses. Nat Chem Biol. 2011;7:865–875. doi: 10.1038/nchembio.709. [DOI] [PubMed] [Google Scholar]
- 253.Bulger PG, Bagal SK, Marquez R. Recent advances in biomimetic natural product synthesis. Nat Prod Rep. 2008;25:254–297. doi: 10.1039/b705909b. [DOI] [PubMed] [Google Scholar]
- 254.Brunoldi E, Luparia M, Porta A, Zanoni G, Vidari G. Biomimetic cyclizations of functionalized isoprenoid polyenes: a cornucopia of synthetic opportunities. Curr Org Chem. 2006;10:2259–2282. [Google Scholar]
- 255.de la Torre MC, Sierra MA. Comments on recent achievements in biomimetic organic synthesis. Angew Chem Int Ed. 2004;43:160–181. doi: 10.1002/anie.200200545. [DOI] [PubMed] [Google Scholar]
- 256.Ilardi EA, Stivala CE, Zakarian A. [3,3]-Sigmatropic rearrangements: recent applications in the total synthesis of natural products. Chem Soc Rev. 2009;38:3133–3148. doi: 10.1039/b901177n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Majumdar KC, Nandi RK. The Claisen rearrangement in the syntheses of bioactive natural products. Tetrahedron. 2013;69:6921–6957. [Google Scholar]
- 258.Fernandes RA, Chowdhury AK, Kattanguru P. The Orthoester Johnson–Claisen Rearrangement in the Synthesis of Bioactive Molecules, Natural Products, and Synthetic Intermediates – Recent Advances. Eur J Org Chem. 2014:2833–2871. [Google Scholar]
- 259.Shi Q-W, Su X-H, Kiyota H. Chemical and Pharmacological Research of the Plants in Genus Euphorbia. Chem Rev. 2008;108:4295–4327. doi: 10.1021/cr078350s. [DOI] [PubMed] [Google Scholar]
- 260.Vasas A, Hohmann J. Euphorbia Diterpenes: Isolation, Structure, Biological Activity, and Synthesis (2008–2012) Chem Rev. 2014;114:8579–8612. doi: 10.1021/cr400541j. [DOI] [PubMed] [Google Scholar]
- 261.Adolf W, Hecker E. Diterpenoid Irritants and Cocarcinogens in Euphorbiaceae and Thymelacaceae: Structural Relationships in View of their Biogenesis. Isr J Chem. 1977;16:75–83. [Google Scholar]
- 262.Liao S-G, Chen H-D, Yue J-M. Plant Orthoesters. Chem Rev. 2009;109:1092–1140. doi: 10.1021/cr0782832. [DOI] [PubMed] [Google Scholar]
- 263.Newton AC. Protein Kinase C: Structural and Spatial Regulation by Phosphorylation, Cofactors, and Macromolecular Interactions. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 264.Cromer BA, Mclntyre P. Painful toxins acting at TRPV1. Toxicon. 2008;57:163–173. doi: 10.1016/j.toxicon.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 265.Hecker E. Cocarcinogenic Principles from the Seed Oil of Croton tiglium and from Other Euphorbiaceae. Cancer Res. 1968;28:2338–2349. [PubMed] [Google Scholar]
- 266.Ogbourne SM, Suhrbier A, Jones B, Cozzi SJ, Boyle GM, Morris M, McAlpine D, Johns J, Scott TM, Sutherland KP, et al. Antitumor activity of 3-ingenyl angelate: Plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004;64:2833–2839. doi: 10.1158/0008-5472.can-03-2837. [DOI] [PubMed] [Google Scholar]
- 267.Fujiwara M, Ijichi K, Tokuhisa K, Katsuura K, Shigeta S, Konno K, Wang GY, Uemura D, Yokota T, Baba M. Mechanism of selective inhibition of human immunodeficiency virus by ingenol triacetate. Antimicrob Agents Chemother. 1996;40:271–273. doi: 10.1128/aac.40.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Hasler CM, Acs G, Blumberg PM. Specific binding to protein kinase C by ingenol and its induction of biological responses. Cancer Res. 1992;52:202–208. [PubMed] [Google Scholar]
- 269.U.S. Food Drug Administration. 2012 Novel New Drugs Summary. FDA; Washington, DC: 2013. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM337830.pdf. [Google Scholar]
- 270.Alder RW, East SP. In/out Isomerism. Chem Rev. 1996;96:2097–2112. doi: 10.1021/cr940246k. [DOI] [PubMed] [Google Scholar]
- 271.Kuwajima I, Tanino K. Total Syntheses of Ingenol. Chem Rev. 2005;105:4661–4670. doi: 10.1021/cr040636z. [DOI] [PubMed] [Google Scholar]
- 272.Winkler JD, Rouse MB, Greaney MF, Harrison SJ, Jeon YT. The First Total Synthesis of (±)-Ingenol. J Am Chem Soc. 2002;124:9726–9728. doi: 10.1021/ja026600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Tanino K, Onuki K, Asano K, Miyashita M, Nakamura T, Takahashi Y, Kuwajima I. Total Synthesis of Ingenol. J Am Chem Soc. 2003;125:1498–1500. doi: 10.1021/ja029226n. [DOI] [PubMed] [Google Scholar]
- 274.Watanabe K, Suzuki Y, Aoki K, Sakakura A, Suenaga K, Kigoshi H. Formal Synthesis of Optically Active Ingenol via Ring-Closing Olefin Metathesis. J Org Chem. 2004;69:7802–7808. doi: 10.1021/jo048833l. [DOI] [PubMed] [Google Scholar]
- 275.Nickel A, Maruyama T, Tang H, Murphy PD, Greene B, Yusuff N, Wood JL. Total Synthesis of Ingenol. J Am Chem Soc. 2004;126:16300–16301. doi: 10.1021/ja044123l. [DOI] [PubMed] [Google Scholar]
- 276.Jørgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J, Baran PS. 14-Step Synthesis of (+)-Ingenol from (+)-3-Carene. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 277.Funk RL, Olmstead TA, Parvez MA. Solution to the in,out-Bicyclo[4.4.1]undecan-7-one Problem Inherent in Ingenane Total Synthesis. J Am Chem Soc. 1988;110:3298–3300. [Google Scholar]
- 278.Tang H, Yusuff N, Wood JL. Progress toward the Total Synthesis of Ingenol: Construction of the Complete Carbocyclic Skeleton. Org Lett. 2001;3:1563–1566. doi: 10.1021/ol015855a. [DOI] [PubMed] [Google Scholar]
- 279.Nicolaou KC, Bulger PG, Sarlah D. Metathesis Reactions in Total Synthesis. Angew Chem Int. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]
- 280.McKerrall SJ, Jørgensen L, Kuttruff CA, Ungeheuer F, Baran PS. Development of a Concise Synthesis of (+)-Ingenol. J Am Chem Soc. 2014;136:5799–5810. doi: 10.1021/ja501881p. [DOI] [PubMed] [Google Scholar]
- 281.Epstein OL, Cha JK. Rapid Access to the “in,out”-Tetracyclic Core of Ingenol. Angew Chem Int Ed. 2004;44:121–123. doi: 10.1002/anie.200461807. [DOI] [PubMed] [Google Scholar]
- 282.Brummond KM, Chen H, Fisher KD, Kerekes AD, Rickards B, Sill PC, Geib SJ. An Allenic Pauson-Khand-Type Reaction: A Reversal in π-Bond Selectivity and the Formation of Seven-Membered Rings. Org Lett. 2002;4:1931–1934. doi: 10.1021/ol025955w. [DOI] [PubMed] [Google Scholar]
- 283.Bohm R, Flaschentrager B, Lendle L. Über die Wirksamkeit von Substanzen aus dem Krotonöl. Arch Exp Pathol Pharmakol. 1935;177:212–220. [Google Scholar]
- 284.Wang H-B, Wang X-Y, Liu L-P, Qin G-W, Kang T-G. Tigliane Diterpenoids from the Euphorbiaceae and Thymelaeaceae Families. Chem Rev. 2015;115:2975–3011. doi: 10.1021/cr200397n. [DOI] [PubMed] [Google Scholar]
- 285.Isakov N, Altman A. Regulation of immune system cell functions by protein kinase C. Front Immunol. 2013;4:384. doi: 10.3389/fimmu.2013.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.McKernan LN, Momjian D, Kulkosky J, Protein Kinase C. One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv Virol. 2012;805347 doi: 10.1155/2012/805347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nature Rev Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 288.Wender PA, Lee HY, Wilhelm RS, Williams PD. Studies on tumor promoters. 7. The synthesis of a potentially general precursor of the tiglianes, daphnanes, and ingenanes. J Am Chem Soc. 1989;111:8954–8957. [Google Scholar]
- 289.Wender PA, Kogen H, Lee HY, Munger JD, Wilhelm RS, Williams PD. Studies on tumor promoters. 8. The synthesis of phorbol. J Am Chem Soc. 1989;111:8957–8958. [Google Scholar]
- 290.Wender PA, McDonald FE. Studies on tumor promoters. 9. A second-generation synthesis of phorbol. J Am Chem Soc. 1990;112:4956–4958. [Google Scholar]
- 291.Wender PA, Rice KD, Schnute ME. The first formal asymmetric synthesis of phorbol. J Am Chem Soc. 1997;119:7897–7898. [Google Scholar]
- 292.Lee K, Cha JK. Formal Synthesis of (+)-Phorbol. J Am Chem Soc. 2001;123:5590–5591. doi: 10.1021/ja010643u. [DOI] [PubMed] [Google Scholar]
- 293.Kawamura S, Chu H, Felding J, Baran PS. Nineteen-step total synthesis of (+)-phorbol. Nature. 2016;532:90–93. doi: 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zou L, Paton RS, Eschenmoser A, Newhouse TR, Baran PS, Houk KN. Enhanced Reactivity in Dioxirane C−H Oxidations via Strain Release: A Computational and Experimental Study. J Org Chem. 2013;78:4037–4048. doi: 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Waldemar A, Curci R, Edwards JO. Dioxiranes: a new class of powerful oxidants. Acc Chem Res. 1989;22:205–211. [Google Scholar]
- 296.Flaschenträger B, von Falkenhausen F. Frhr. Über den Giftstoff im Krotonöl. II. Zur Konstitution von Krotophorbolon. Justus Liebigs Ann Chem. 1934;514:252–260. [Google Scholar]
- 297.Wang HB, Chu W-J, Wang Y, Ji P, Wang Y-B, Yu Q, Qin G-W. Diterpenoids from the roots of Euphorbia fischeriana. J Asian Nat Prod Res. 2010;12:1038–1043. doi: 10.1080/10286020.2010.532490. [DOI] [PubMed] [Google Scholar]
- 298.Wender PA, Kee J-M, Warrington JM. Practical Synthesis of Prostratin, DPP, and Their Analogs, Adjuvant Leads Against Latent HIV. Science. 2008;320:649–652. doi: 10.1126/science.1154690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Beans EJ, Fournogerakis D, Gauntlett C, Haumann LV, Kramer R, Marsden MD, Murray D, Chun T-W, Zack JA, Wender PA. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci USA. 2013;110:11698–11703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Asaba T, Katoh Y, Urabe D, Inoue M. Total Synthesis of Crotophorbolone. Angew Chem Int Ed. 2015;54:14457–14461. doi: 10.1002/anie.201509160. [DOI] [PubMed] [Google Scholar]
- 301.Krafft ME, Holton RA. The Kharasch Reagent. Regioselective Generation of Dienol Ethers from Enones. J Am Chem Soc. 1984;106:7619–7621. [Google Scholar]
- 302.Jasperse CP, Curran DP, Fevig TL. Radical reactions in natural product synthesis. Chem Rev. 1991;91:1237–1286. [Google Scholar]
- 303.Chatgilialoglu C, Studer A. Encyclopedia of Radicals in Chemistry, Biology and Materials. Wiley-Interscience. 2012 [Google Scholar]
- 304.Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Wiley-Verlag; 2008. [Google Scholar]
- 305.Yan M, Lo JC, Edwards JT, Baran PS. Radicals: Reactive Intermediates with Translational Potential. J Am Chem Soc. 2016;138:12692–12714. doi: 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Look SA, Fenical W, Jacobs RS, Clardy J. The pseudopterosins: Anti-inflammatory and analgesic natural products from the sea whip Pseudoptergorgia elisabethae. Proc Natl Acad Sci. 1986;83:6238–6240. doi: 10.1073/pnas.83.17.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Broka CA, Chan S, Peterson B. Total synthesis of (−)-pseudopterosin A. J Org Chem. 1988;53:1584–1586. [Google Scholar]
- 308.Corey EJ, Carpino P. Enantiospecific total synthesis of pseudopterosins A and E. J Am Chem Soc. 1989;111:5472–5474. [Google Scholar]
- 309.Corey EJ, Carpino P. A new enantiospecific route to the pseudopterosins. Tetrahedron Lett. 1990;31:3857–3858. [Google Scholar]
- 310.McCombie SW, Cox B, Ling SI, Ganguly AK, McPhail AT. Controlling benzylic functionality and stereochemistry: 1. Synthesis of the secopseudopterosin aglycone. Tetrahedron Lett. 1991:2083–2086. [Google Scholar]
- 311.McCombie SW, Ortiz C, Cox B, Ganguly AK. Controlling Benzylic and Anomeric Functionality and Stereochemistry: Methodology and Syntheses Utilizing Intramolecular Ionic Hydrogenation. Synlett. 1993:541–547. [Google Scholar]
- 312.Buszek KR, Bixby DL. Total synthesis of pseudopterosin A and E aglycon. Tetrahedron Lett. 1995;36:9129–9132. [Google Scholar]
- 313.Gill S, Kocienski P, Kohler A, Pontiroli A, Qun L. A synthetic approach to the pseudopterosins. Chem Comm. 1996:1743–1744. [Google Scholar]
- 314.Majdalani A, Schmalz H-G. Chiral η6-Arene-Cr(CO)3 complexes in organic synthesis: A short and highly selective synthesis of the 18-nor-seco-pseudopterosin aglycone. Tetrahedron Lett. 1997;38:4545–4548. [Google Scholar]
- 315.Majdalani A, Schmalz HG. Enantioselective Synthesis of the Aglycones of Pseudopterosin and seco-Pseudopterosin via a Common Synthetic Intermediate. Synlett. 1997:1303–1305. [Google Scholar]
- 316.LeBrazidec JY, Kocienski PJ, Connolly JD, Muir KW. Synthetic approaches to pseudopterosin G aglycone dimethyl ether. J Chem Soc Perkin Trans. 1:1998, 2475–2478. [Google Scholar]
- 317.Corey EJ, Lazerwith SE. A Direct and Efficient Stereocontrolled Synthetic Route to the Pseudopterosins, Potent Marine Antiinflamatory Agents. J Am Chem Soc. 1998;120:12777–12782. [Google Scholar]
- 318.Kocienski PJ, Pontiroli A, Qun L. Enantiospecific syntheses of pseudopterosin aglycones. Part 2. Synthesis of pseudopterosin K–L aglycone and pseudopterosin A–F aglycone via a B→BA→BAC annulation strategy. J Chem Soc Perkin Trans 1. 2001:2356–2366. [Google Scholar]
- 319.Harrowven DC, Tyte MJ. Total synthesis of (±)-pseudopterosin A–F and K–L aglycone. Tetrahedron Lett. 2004;45:2089–2091. [Google Scholar]
- 320.Mans DJ, Cox GA, RajanBabu TV. Ethylene in organic synthesis. Repetitive hydrovinylation of alkenes for highly enantioselective syntheses of pseudopterosins. J Am Chem Soc. 2011;133:5776–5779. doi: 10.1021/ja201321v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Cooksey JP, Kocieński PJ, Schmidt AW, Snaddon TN, Kilner CA. A synthesis of the pseudopterosin A–F aglycone. Synthesis. 2012;44:2779–2785. [Google Scholar]
- 322.Williams DR, Shah AA. Total Synthesis of (+)-Ileabethoxazole via an Iron-Mediated Pauson-Khand [2+2+1] Carbocyclization. J Am Chem Soc. 2014;136:8829–8836. doi: 10.1021/ja5043462. [DOI] [PubMed] [Google Scholar]
- 323.Newton CG, Drew SL, Lawrence AL, Willis AC, Paddon-Row MN, Sherburn MS. Pseudopterosin synthesis from a chiral cross-conjugated hydrocarbon through a series of cycloadditions. Nat Chem. 2015;7:82–86. doi: 10.1038/nchem.2112. [DOI] [PubMed] [Google Scholar]
- 324.Rodríguez AD, Ramírez C, Rodríguez II, González E. Novel Antimycobacterial Benzoxazole Alkaloids, from the West Indian Sea Whip Pseudopterogorgia elisabethae. Org Lett. 1999;1:527–530. doi: 10.1021/ol9907116. [DOI] [PubMed] [Google Scholar]
- 325.Lazerwith SE, Johnson TW, Corey EJ. Syntheses and Stereochemical Revision of Pseudopterosin G−J Aglycon and Helioporin E. Org Lett. 2000;2:2389–2392. doi: 10.1021/ol006192k. [DOI] [PubMed] [Google Scholar]
- 326.Nicolaou KC, Snyder SA. Chasing Molecules That Were Never There: Misassigned Natural Products and the Role of Chemical Synthesis in Modern Structure Elucidation. Angew Chem Int Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- 327.Davidson JP, Corey EJ. First Enantiospecific Total Synthesis of the Antitubercular Marine Natural Product Pseudopteroxazole. Revision of Assigned Stereochemistry. J Am Chem Soc. 2003;125:13486–13489. doi: 10.1021/ja0378916. [DOI] [PubMed] [Google Scholar]
- 328.Harmata M, Hong X. Benzothiazines in Synthesis. A Total Synthesis of Pseudopteroxazole. Org Lett. 2005;7:3581–3583. doi: 10.1021/ol0515412. [DOI] [PubMed] [Google Scholar]
- 329.Yang M, Yang X, Sun H, Li A. Total Synthesis of Ileabethoxazole, Pseudopteroxazole and seco-Pseudopteroxazole. Angew Chem Int Ed. 2016;55:2851–2855. doi: 10.1002/anie.201510568. [DOI] [PubMed] [Google Scholar]
- 330.Yu X, Su F, Liu C, Yuan H, Zhao S, Zhou Z, Quan T, Luo T. Enantioselective Total Syntheses of Various Amphilectane and Serrulatane Diterpenoids via Cope Rearrangements. J Am Chem Soc. 2016;138:6261–6270. doi: 10.1021/jacs.6b02624. [DOI] [PubMed] [Google Scholar]
- 331.Brown HC, Pfaffenberger CD. Hydroboration of terpenes – VIII: Cyclic hydroboration of D-(+)-limonene A stereoselective synthesis of D-(−)-(1R, 2R, 4R)-limonene-2,9-diol and D-(−)-(1R, 2R, 4R)-carvomenthol. Tetrahedron. 1975;31:925–928. [Google Scholar]
- 332.Tabolin AA, Ioffe SL. Rearrangement of N-Oxyenamines and Related Reactions. Chem Rev. 2014;114:5426–5476. doi: 10.1021/cr400196x. [DOI] [PubMed] [Google Scholar]
- 333.Vedejs E, Fang HW. An E-selective 1,3-diene synthesis from moderated ylides and aldehydes. J Org Chem. 1984;49:210–212. [Google Scholar]
- 334.Heckrodt TJ, Mulzer J. Marine Natural Products from Pseudopterogorgia elisabethae: Structures, Biosynthesis, Pharmacology, and Total Synthesis. Top Curr Chem. 2005;244:1–41. [Google Scholar]
- 335.Yadav JS, Bhasker EV, Srihari P. Synthesis of a key intermediate for the total synthesis of pseudopteroxazole. Tetrahedron. 2010;66:1997–2004. [Google Scholar]
- 336.Negishi E, Noda Y, Lamaty F, Vawter EJ. Effects of organometals on the palladium-catalyzed carbopalladation-cross coupling for preparing stereodefined exocyclic alkenes. Tetrahedron Lett. 1990;31:4393–4396. [Google Scholar]
- 337.Suárez A, Fu GC. A straightforward and mild synthesis of functionalized 3-alkynoates. Angew Chem Int Ed. 2004;43:3580–3582. doi: 10.1002/anie.200454070. [DOI] [PubMed] [Google Scholar]
- 338.Conrad JC, Kong J, Laforteza BN, MacMillan DWC. Enantioselective α-Arylation of Aldehydes via Organo-SOMO Catalysis. An Ortho-Selective Arylation Reaction Based on an Open-Shell Pathway. J Am Chem Soc. 2009;131:11640–11641. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 340.Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, et al. Isolation, Structure, and Absolute Stereochemistry of Platensimycin, A Broad Spectrum Antibiotic Discovered Using an Antisense Differential Sensitivity Strategy. J Am Chem Soc. 2006;128:11916–11920. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]
- 341.Palanichamy K, Kaliappan KP. Discovery and syntheses of “superbug challengers” –platensimycin and platencin. Chem Asian J. 2010;5:668–703. doi: 10.1002/asia.200900423. [DOI] [PubMed] [Google Scholar]
- 342.Saleem M, Hussain H, Ahmed I, van Ree T, Krohn K. Platensimycin and its relatives: A recent story in the struggle to develop new naturally derived antibiotics. 2011;28:1534–1579. doi: 10.1039/c1np00010a. [DOI] [PubMed] [Google Scholar]
- 343.Martens E, Demain AL. Platensimycin and platencin: Promising antibiotics for future application in human medicine. J Antibiot. 2011;64:705–710. doi: 10.1038/ja.2011.80. [DOI] [PubMed] [Google Scholar]
- 344.Allahverdiyev AM, Bagirova M, Abamor ES, Ates SC, Koc RC, Miraloglu M, Elcicek S, Yaman S, Unal G. The use of platensimycin and platencin to fight antibiotic resistance. Infect Drug Resist. 2013;6:99–114. doi: 10.2147/IDR.S25076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Tiefenbacher K, Mulzer J. Synthesis of Platensimycin. Angew Chem Int Ed. 2008;47:2548–2555. doi: 10.1002/anie.200705303. [DOI] [PubMed] [Google Scholar]
- 346.Das M, Ghosh PS, Manna KA. Review on Platensimycin: A Selective FabF Inhibitor. Int J Med Chem. 2016:1–16. doi: 10.1155/2016/9706753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Nicolaou KC, Li A, Edmonds DJ. Total Synthesis of Platensimycin. Angew Chem Int Ed. 2006;45:7086–7090. doi: 10.1002/anie.200603892. [DOI] [PubMed] [Google Scholar]
- 348.Nicolaou KC, Tang Y, Wang J. Formal synthesis of (±)-platensimycin. Chem Commun. 2007:1922–1923. doi: 10.1039/b704589a. [DOI] [PubMed] [Google Scholar]
- 349.Nicolaou KC, Edmonds DJ, Li A, Tria GS. Asymmetric Total Syntheses of Platensimycin. Angew Chem Int Ed. 2008;46:3942–3945. doi: 10.1002/anie.200700586. [DOI] [PubMed] [Google Scholar]
- 350.Nicolaou KC, Li A, Edmonds DJ, Tria GS, Ellery SP. Total Synthesis of Platensimycin and Related Natural Products. J Am Chem Soc. 2009;131:16905–16918. doi: 10.1021/ja9068003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Nicolaou KC, Li A, Ellery SP, Edmonds DJ. Rhodium-Catalyzed Asymmetric Enyne Cycloisomerization of Terminal Alkynes and Formal Total Synthesis of (−)-Platensimycin. Angew Chem Int Ed. 2009;48:6293–6295. doi: 10.1002/anie.200901992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Tiefenbacher K, Mulzer J. Protecting-Group-Free Formal Synthesis of Platensimycin. Angew Chem Int Ed. 2007;46:8074–8075. doi: 10.1002/anie.200702852. [DOI] [PubMed] [Google Scholar]
- 353.Zou Y, Chen C-H, Taylor CD, Foxman BM, Snider BB. Formal Synthesis of (±)-Platensimycin. Org Lett. 2007;9:1825–1828. doi: 10.1021/ol070563g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Li P, Payette JN, Yamamoto H. Enantioselective Route to Platensimycin: An Intramolecular Robinson Annulation Approach. J Am Chem Soc. 2007;129:9534–9535. doi: 10.1021/ja073547n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Lalic G, Corey EJ. An Effective Enantioselective Route to the Platensimycin Core. Org Lett. 2007;9:4921–4923. doi: 10.1021/ol702323s. [DOI] [PubMed] [Google Scholar]
- 356.Nicolaou KC, Pappo D, Tsang KY, Gibe R, Chen DY-K. A Chiral Pool Based Synthesis of Platensimycin. Angew Chem Int Ed. 2008;47:944–946. doi: 10.1002/anie.200705080. [DOI] [PubMed] [Google Scholar]
- 357.Kim CH, Jang KP, Choi SY, Chung YK, Lee E. A Carbonyl Ylide Cycloaddition Approach to Platensimycin. Angew Chem Int Ed. 2008;47:4009–4011. doi: 10.1002/anie.200800568. [DOI] [PubMed] [Google Scholar]
- 358.Matsuo J-I, Takeuchi K, Ishibashi H. Stereocontrolled Formal Synthesis of (±)-Platensimycin. Org Lett. 2008;10:4049–4052. doi: 10.1021/ol801584r. [DOI] [PubMed] [Google Scholar]
- 359.McGrath NA, Bartlett ES, Sittihan S, Njarsarson JTA. Concise Ring-Expansion Route to the Compact Core of Platensimycin. Angew Chem Int Ed. 2009;48:8543–8546. doi: 10.1002/anie.200903347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ghosh AK, Xi K. Enantioselective Synthesis of (−)-Platensimycin Oxatetracyclic Core by Using an Intramolecular Diels−Alder Reaction. Org Lett. 2007;9:4013–4016. doi: 10.1021/ol701783z. [DOI] [PubMed] [Google Scholar]
- 361.Yun SY, Zheng J-C, Lee D. Stereoelectronic Effect for the Selectivity in C-H Insertion of Alkylidene Carbenes and Its Application to the Synthesis of Platensimycin. J Am Chem Soc. 2009;131:8413–8415. doi: 10.1021/ja903526g. [DOI] [PubMed] [Google Scholar]
- 362.Beaulieu M-A, Sabot C, Achache N, Guérard KC, Canesi S. An Oxidative Prins–Pinacol Tandem Process and its Application to the synthesis of (−)-Platensimycin. Chem Eur J. 2010;16:11224–11228. doi: 10.1002/chem.201001813. [DOI] [PubMed] [Google Scholar]
- 363.Magnus P, Rivera H, Lynch V. Concise Formal Total Synthesis of Platensimycin Mediated by a Stereoselective Autoxidation and Hydroxyl Group Directed Conjugative Reduction. Org Lett. 2010;12:5677–5679. doi: 10.1021/ol102557k. [DOI] [PubMed] [Google Scholar]
- 364.Hirai S, Nakada M. Enantioselective divergent approaches to both (−)-platensimycin and (−)-platencin. Tetrahedron. 2011;67:518–530. [Google Scholar]
- 365.Ueda Y, Iwahashi K, Iguchi K, Ito H. Enantioselective Synthesis of the Tetracyclic Core of Platensimycin. Synthesis. 2011:1532–1536. [Google Scholar]
- 366.Oblak EZ, Wright DL. Highly Substituted Oxabicyclic Derivatives from Furan: Synthesis of (±)-Platensimycin. Org Lett. 2011;13:2263–2265. doi: 10.1021/ol2005775. [DOI] [PubMed] [Google Scholar]
- 367.Zhu L, Han Y, Du G, Lee C-S. A Bifunctional Lewis Acid Induced Cascade Cyclization to the Tricyclic Core of ent-Kaurenoids and Its Application to the Formal Synthesis of (±)-Platensimycin. Org Lett. 2013;15:524–527. doi: 10.1021/ol3033412. [DOI] [PubMed] [Google Scholar]
- 368.Horii S, Torihata M, Nagasawa T, Kuwahara S. Stereoselective Approach to the Racemic Oxatetracyclic Core of Platensimycin. J Org Chem. 2013;78:2798–2801. doi: 10.1021/jo302813y. [DOI] [PubMed] [Google Scholar]
- 369.Zhu L, Zhou C, Yang W, He S, Cheng G-J, Zhang X, Lee C-S. Formal Syntheses of (±)-Platensimycin and (±)-Platencin via a Dual-Mode Lewis Acid Induced Cascade Cyclization Approach. J Org Chem. 2013;78:7912–7929. doi: 10.1021/jo401105q. [DOI] [PubMed] [Google Scholar]
- 370.Eey STC, Lear MJ. Total Synthesis of (−)-Platensimycin by Advancing Oxocarbenium- and Iminium-Mediated Catalytic Methods. Chem Eur J. 2014;20:11556–11573. doi: 10.1002/chem.201400131. [DOI] [PubMed] [Google Scholar]
- 371.Eey STC, Lear MJA. Bismuth(III)-Catalyzed Friedel−Crafts Cyclization and Stereocontrolled Organocatalytic Approach to (−)-Platensimycin. Org Lett. 2010;12:5510–5513. doi: 10.1021/ol102390t. [DOI] [PubMed] [Google Scholar]
- 372.Jiao Z-W, Tu Y-Q, Zhang Q, Liu W-X, Wang S-H, Wang M. Formal synthesis of (−)-platensimycin. Org Chem Front. 2015;2:913–916. [Google Scholar]
- 373.Weinges K, Reichert H. Radical cyclisation of dienes; part IV. Enantiomerically pure building blocks from (R)- (−)- and (S)-(+)-carvone for the synthesis of naturally occurring triquinanes. Synlett. 1991:785–786. [Google Scholar]
- 374.Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Oxidation of silyl enol ethers by using IBX and IBX•N-oxide complexes: A mild and selective reaction for the synthesis of enones. Angew Chem Int Ed. 2002;41:996–1000. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 375.McCarroll AJ, Walton JC. Programming Organic Molecules: Design and Management of Organic Syntheses through Free-Radical Cascade Processes. Angew Chem Int Ed. 2001;40:2224–2248. doi: 10.1002/1521-3773(20010618)40:12<2224::aid-anie2224>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 376.Stang PJ. Unsaturated Carbenes. Chem Rev. 1978;78:383–405. [Google Scholar]
- 377.Taber DF. In: Methods of Organic Chemistry. 4th. Helmchen G, editor. E21. Georg Thieme Verlag; New York: 1995. p. 1127. [Google Scholar]
- 378.Kirmse W. Alkenylidenes in Organic Synthesis. Angew Chem Int Ed. 1997;36:1164–1170. [Google Scholar]
- 379.Knorr R. Alkylidenecarbenes, Alkylidenecarbenoids, and Competing Species: Which Is Responsible for Vinylic Nucleophilic Substitution, [1 + 2] Cycloadditions, 1,5-CH Insertions, and the Fritsch−Buttenberg−Wiechell Rearrangement? Chem Rev. 2004;104:3795–2850. doi: 10.1021/cr030616h. [DOI] [PubMed] [Google Scholar]
- 380.Hog DT, Webster R, Trauner D. Synthetic approaches toward sesterterpenoids. Nat Prod Rep. 2012;29:752–779. doi: 10.1039/c2np20005h. [DOI] [PubMed] [Google Scholar]
- 381.Luo S-H, Luo Q, Niu X-M, Xie M-J, Zhao X, Schneider B, Gershenzon J, Li S-H. Glandular Trichomes of Leucosceptrum canum Harbor Defensive Sesterterpenoids. Angew Chem Int Ed. 2010;49:4471–4475. doi: 10.1002/anie.201000449. [DOI] [PubMed] [Google Scholar]
- 382.Huang X, Song L, Xu J, Zhu G, Liu B. Asymmetric Total Synthesis of Leucosceptroid B. Angew Chem Int Ed. 2013;52:952–955. doi: 10.1002/anie.201208687. [DOI] [PubMed] [Google Scholar]
- 383.Hugelshofer CL, Magauer TA. General Entry to Antifeedant Sesterterpenoids: Total Synthesis of (+)-Norleucosceptroid A, (−)-Norleucosceptroid B, and (−)-Leucosceptroid K. Angew Chem Int Ed. 2014;53:11351–11355. doi: 10.1002/anie.201407788. [DOI] [PubMed] [Google Scholar]
- 384.Hugelshofer CL, Magauer T. Total Synthesis of the Leucosceptroid Family of Natural Products. J Am Chem Soc. 2015;137:3807–3810. doi: 10.1021/jacs.5b02021. [DOI] [PubMed] [Google Scholar]
- 385.Guo S, Liu J, Ma D. Total Synthesis of Leucosceptroids A and B. Angew Chem Int Ed. 2015;54:1298–1301. doi: 10.1002/anie.201410134. [DOI] [PubMed] [Google Scholar]
- 386.Comito RJ, Finelli FG, MacMillan DWC. Enantioselective Intramolecular Aldehyde α-Alkylation with Simple Olefins: Direct Access to Homo-Ene Products. J Am Chem Soc. 2013;135:9358–9361. doi: 10.1021/ja4047312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Pham PV, Ashton K, MacMillan DWC. The intramolecular asymmetric allylation of aldehydes via organo-SOMO catalysis: A novel approach to ring construction. Chem Sci. 2011;2:1470–1473. doi: 10.1039/C1SC00176K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Senatore F, Feo VD, Zhou ZL. Chemical constituents of Gentianella alborosea (Gilg) Fabris. Ann Chem. 1991;81:269–274. [Google Scholar]
- 389.Kawahara N, Nozawa M, Flores D, Bonilla P, Sekita S, Satake M, Kawai K-I. Nitidasin, a novel sesterterpenoid, from the Peruvian folk medicine “Hercampuri” (Gentianella nitida) Chem Pharm Bull. 1997;45:1717–1719. [Google Scholar]
- 390.Hog DT, Huber FME, Mayer P, Trauner D. The Total Synthesis of (−)-Nitidasin. Angew Chem Int Ed. 2014;53:8513–8517. doi: 10.1002/anie.201403605. [DOI] [PubMed] [Google Scholar]
- 391.Agnel G, Owczarczyk Z, Negishi E-I. Diastereoselective zirconocene-promoted bicyclization-carbonylation of allylically methyl-substituted enynes. Synthesis of (+)-iridomyrmecin. Tetrahedron Lett. 1992;33:1543–1546. [Google Scholar]
- 392.García AM, Mascereñas JL, Castedo A, Mouriño A. Alkenylzinc-Mediated Approach to the Vitamin D Skeleton. Application to the Synthesis of 6-Methyl Analogs of Vitamin and Previtamin D. J Org Chem. 1997;62:6353–6358. [Google Scholar]
- 393.Hajos ZG, Parrish DR. “(+)-(7aS)-7a-methyl-2,3,7,7a-tetrahydro-1 H-indene-1,5-(6H-Dione) Org Synth. 1985;63:26–31. [Google Scholar]
- 394.Micheli RA, Hajos ZG, Cohen N, Parrish DR, Portland LA, Sciamanna W, Scott MA, Wehrli PA. Total syntheses of optically active 19-nor steroids. (+)-Estr-4-ene-3,17-dione and (+)-13β-ethylgon-4-ene-3,17-dione. J Org Chem. 1975;40:675–681. doi: 10.1021/jo00894a003. [DOI] [PubMed] [Google Scholar]
- 395.Hog DT, Mayer P, Trauner DA. Unified Approach to trans-Hydrindane Sesterterpenoids. J Org Chem. 2012;77:5838–5843. doi: 10.1021/jo300726z. [DOI] [PubMed] [Google Scholar]
- 396.Au TK, Chick WS, Leung PC. The biology of ophiobolins. Life Sci. 2000;67:733–742. doi: 10.1016/s0024-3205(00)00668-8. [DOI] [PubMed] [Google Scholar]
- 397.Bury M, Girault G, Mégalizzi V, Spiegl-Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A, Gailly P, Vandier C, et al. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis. 2013;4:e561. doi: 10.1038/cddis.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Rowley M, Tsukamoto M, Kishi Y. Total synthesis of (+)-ophiobolin C. J Am Chem Soc. 1989;111:2735–2737. [Google Scholar]
- 399.Tsuna K, Noguchi N, Nakada M. Convergent Total Synthesis of (+)-Opiobolin A. Angew Chem Int Ed. 2011;50:9452–9455. doi: 10.1002/anie.201104447. [DOI] [PubMed] [Google Scholar]
- 400.Tsuna K, Noguchi N, Nakada M. Enantioselective Total Synthesis of (+)-Ophiobolin A. Chem Eur J. 2013;19:5476–5486. doi: 10.1002/chem.201204119. [DOI] [PubMed] [Google Scholar]
- 401.Brill ZG, Grover HK, Maimone T. J Enantioselective synthesis of an ophiobolin sesterterpene via a programmed radical cascade. Science. 2016;352:1078–1082. doi: 10.1126/science.aaf6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Charette AB, Juteau H, Lebel H, Molinaro C. Enantioselective Cyclopropanation of Allylic Alcohols with Dioxaborolane Ligands: Scope and Synthetic Applications. J Am Chem Soc. 1998;120:11943–11952. [Google Scholar]
- 403.Roberts BP. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: Concepts and applications in organic chemistry. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 404.Cai Y, Roberts BP, Tocher DA. Carbohydrate-derived thiols as protic polarity-reversal catalysts for enantioselective radical-chain reactions. J Chem Soc Perkin Trans. 1:2002, 1376–1386. [Google Scholar]
- 405.Sheng H, Sun H. Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat Prod Rep. 2011;28:543–593. doi: 10.1039/c0np00059k. [DOI] [PubMed] [Google Scholar]
- 406.Moron J, Polonsky J. Sur l’origine triterpenique des constituants amers des simarubacees. Tetrahedron Lett. 1968;9:385–390. [Google Scholar]
- 407.Moron J, Merrien M-A, Polonsky J. Sur la biosynthèse des quassinoïdes de Simaruba glauca (Simarubaceae) Phytochemistry. 1971;10:585–592. [Google Scholar]
- 408.Guo Z, Vangapandu S, Sindelar RW, Walker LA, Sindelar RD. Biologically active quassinoids and their chemistry: Potential leads for drug design. Curr Med Chem. 2005;12:173–190. doi: 10.2174/0929867053363351. [DOI] [PubMed] [Google Scholar]
- 409.Fiaschetti G, Grotzer MA, Shalaby T, Castelletti D, Arcaro A. Quassinoids: From traditional drugs to new cancer therapeutics. Curr Med Chem. 2011;18:316–328. doi: 10.2174/092986711794839205. [DOI] [PubMed] [Google Scholar]
- 410.Kawada K, Kim M, Watt DS. Synthesis of quassinoids: A review. Org Prep and Proc Int. 1989;21:521–618. [Google Scholar]
- 411.Vieira IJC, Braz-Filho R. Quassinoids: Structural diversity, biological activity and synthetic studies. Studies in Nat Prod Chem. 2006;33:433–492. [Google Scholar]
- 412.Shing TKM, Jiang Q. Total Synthesis of (+)-quassin. J Org Chem. 2000;65:7059–7069. doi: 10.1021/jo000877g. [DOI] [PubMed] [Google Scholar]
- 413.Shing TKM, Yeung YY. Total Synthesis of (−)-Samaderine Y from (S)-(+)-carvone. Angew Chem Int Ed. 2005;44:7981–7984. doi: 10.1002/anie.200502763. [DOI] [PubMed] [Google Scholar]
- 414.Shing TKM, Zhu XY, Mak TCW. Synthetic studies towards pentacyclic quassinoids: Facile and stereocontrolled construction of the E ring. Tetrahedron Asymm. 1996;7:673–676. [Google Scholar]
- 415.Wilson SR, Mao DT, Jernberg KM, Ezmirly ST. [1,3]-sigmatropic shifts of alkoxides. Tetrahedron Lett. 1977;18:2559–2562. [Google Scholar]
- 416.Spangler CW. Thermal [1,j] sigmatropic rearrangements. Chem Rev. 1976;76:187–217. [Google Scholar]
- 417.Xiao Q, Ren W-W, Chen Z-X, Sun T-W, Li Y, Ye Q-D, Gong J-X, Meng F-K, You L, Liu Y-F, et al. Diastereoselective Total Synthesis of (±)-Schindilactone A. Angew Chem Int Ed. 2011;50:7373–7377. doi: 10.1002/anie.201103088. [DOI] [PubMed] [Google Scholar]
- 418.Li J, Yang P, Yao M, Deng J, Li A. Total Synthesis of Rubriflordilactone A. J Am Chem Soc. 2014;136:16477–16480. doi: 10.1021/ja5092563. [DOI] [PubMed] [Google Scholar]
- 419.Wang L, Wang H, Li Y, Tang P. Total Synthesis of Schilancitrilactones B and C. Angew Chem Int Ed. 2015;54:5732–5735. doi: 10.1002/anie.201501169. [DOI] [PubMed] [Google Scholar]
- 420.You L, Liang X-T, Xu L-M, Wang Y-F, Zhang J-J, Su Q, Li Y-H, Zhang B, Yang S-L, Chen J-H, et al. Asymmetric Total Synthesis of Propindilactone G. J Am Chem Soc. 2015;137:10120–10123. doi: 10.1021/jacs.5b06480. [DOI] [PubMed] [Google Scholar]
- 421.Goh S-S, Chaubet G, Gockel B, Cordonnier M-CA, Baars H, Phillips AW, Anderson EA. Total Synthesis of (+)-Rubriflordilactone A. Angew Chem Int Ed. 2015;54:12618–12621. doi: 10.1002/anie.201506366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Yang P, Yao M, Li J, Li Y, Li A. Total Synthesis of Rubriflordilactone B. Angew Chem Int Ed. 2016;55:6964–6968. doi: 10.1002/anie.201601915. [DOI] [PubMed] [Google Scholar]
- 423.Xiao W-L, Li R-T, Huang S-X, Pu J-X, Sun H-D. Triterpenoids from the Schisandraceae family. Nat Prod Rep. 2008;25:871–891. doi: 10.1039/b719905h. [DOI] [PubMed] [Google Scholar]
- 424.Shi Y-M, Xiao W-L, Pu J-X, Sun HD. Triterpenoids from the Schisandraceae family: An update. Nat Prod Rep. 2015;32:367–410. doi: 10.1039/c4np00117f. [DOI] [PubMed] [Google Scholar]
- 425.Xiao W-L, Yang L-M, Gong N-B, Wu L, Wang R-R, Pu J-X, Li X-L, Huang S-X, Zheng Y-T, Li R-T, et al. Rubriflordilactones A and B, Two Novel Bisnortriterpenoids from Schisandra rubriflora and Their Biological Activities. Org Lett. 2006;8:991–994. doi: 10.1021/ol060062f. [DOI] [PubMed] [Google Scholar]
- 426.Ishihara K, Kubota M, Kurihara H, Yamamoto H. Scandium Trifluoromethanesulfonate as an Extremely Active Acylation Catalyst. J Am Chem Soc. 1995;117:4413–4414. doi: 10.1021/jo952237x. [DOI] [PubMed] [Google Scholar]
- 427.Lu Z, Li Y, Deng J, Li A. Total synthesis of the Daphniphyllum alkaloid daphenylline. Nat Chem. 2013;5:679–684. doi: 10.1038/nchem.1694. [DOI] [PubMed] [Google Scholar]
- 428.Yang M, Li J, Li A. Total synthesis of clostrubin. Nat Commun. 2015;6:6445. doi: 10.1038/ncomms7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Grimblat N, Kaufman TS, Sarotti AM. Computational Chemistry Driven Solution to Rubriflordilactone B. Org Lett. 2016;18:6420–6423. doi: 10.1021/acs.orglett.6b03318. [DOI] [PubMed] [Google Scholar]
- 430.Luo X, Shi Y-M, Luo R-H, Luo S-H, Li X-N, Wang R-R, Li S-H, Zheng Y-T, Du X, Xiao W-L, et al. Schilancitrilactones A–C: Three Unique Nortriterpenoids from Schisandra lancifolia. Org Lett. 2012;14:1286–1289. doi: 10.1021/ol300099e. [DOI] [PubMed] [Google Scholar]
- 431.Lavallée Spino C, Ruel R, Hogan TK, Deslongchamps P. Stereoselective synthesis of cis-decalins via Diels-Alder and double Michael addition of substituted Nazarov reagents. Can J Chem. 1992;70:1406–1426. [Google Scholar]
- 432.Luche JL, Allavena C. Ultrasound in organic synthesis 16. Optimisation of the conjugate additions to α,β-unsaturated carbonyl compounds in aqueous media. Tetrahedron Lett. 1988;29:5369–5372. [Google Scholar]
- 433.Benneche T, Hussain Z. Synthesis of 5-(bromomethylene)furan-2-(5H)-ones and 3-(bromomethylene)isobenzofuran-1(3H)-ones as inhibitors of microbial quorum sensing. New J Chem. 2008;32:1567–1572. [Google Scholar]
- 434.Li J, Eastgate MD. Current complexity: a tool for assessing the complexity of organic molecules. Org Biomol Chem. 2015;13:7164–7176. doi: 10.1039/c5ob00709g. [DOI] [PubMed] [Google Scholar]
- 435.Gaich T, Baran PS. Aiming for the Ideal Synthesis. J Org Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- 436.Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AGA. Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- 437.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, et al. A Platform for the Discovery of New Macrolide Antibiotics. Nature. 2016;533:338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Feng J, Noack F, Krische MJ. Modular Terpenoid Construction via Catalytic Enantioselective Formation of All-Carbon Quaternary Centers: Total Synthesis of Oridamycin A, Triptoquinones B and C, and Isoiresin. J Am Chem Soc. 2016;138:12364–12367. doi: 10.1021/jacs.6b08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Armaly AM, DePorre YC, Groso EJ, Riehl PS, Schindler CS. Discovery of Novel Synthetic Methodologies and Reagents during Natural Product Synthesis in the Post-Palytoxin Era. Chem Rev. 2015;115:9232–9276. doi: 10.1021/acs.chemrev.5b00034. [DOI] [PubMed] [Google Scholar]
- 440.Dong G, editor. C−C Bond activation. Topics in Current Chemistry. Vol. 346 Springer; Berlin: p. 2014. [Google Scholar]
- 441.Murakami M, Ito Y. Cleavage of Carbon−Carbon Single Bonds by Transition Metals. In: Murai S, editor. Topics in Organometallic Chemistry. Vol. 3. Springer; Berlin: 1999. p. 97. [Google Scholar]
- 442.Rybtchinski B, Milstein D. Metal Insertion into C–C Bonds in Solution. Angew Chem Int Ed. 1999;38:870–883. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 443.Jun CH. Transition metal-catalyzed carbon-carbon bond activation. Chem Soc Rev. 2004;33:610–618. doi: 10.1039/b308864m. [DOI] [PubMed] [Google Scholar]
- 444.Masarwa A, Weber M, Sarpong R. Selective C–C and C–H Bond Activation/Cleavage of Pinene Derivatives: Synthesis of Enantiopure Cyclohexenone Scaffolds and Mechanistic Insights. J Am Chem Soc. 2015;137:6327–6334. doi: 10.1021/jacs.5b02254. [DOI] [PubMed] [Google Scholar]
- 445.Weber M, Owens K, Masarwa A, Sarpong R. Construction of Enantiopure Taxoid and Natural Product-like Scaffolds Using a C−C Bond Cleavage/Arylation Reaction. Org Lett. 2015;17:5432–5435. doi: 10.1021/acs.orglett.5b02797. [DOI] [PubMed] [Google Scholar]
- 446.Hung K, Condakes ML, Morikawa T, Maimone TJ. Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Synthesis of (+)-Pseudoanisatin. J Am Chem Soc. 2016;138:16616–16619. doi: 10.1021/jacs.6b11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Brummond KM, Kent JL. Recent Advances in the Pauson-Khand Reaction and Related [2+2+1] Cycloadditions. Tetrahedron. 2000;56:3263–3283. [Google Scholar]
- 448.Morrison KC, Hergenrother P. Natural Products as Starting Points for the Synthesis of Complex and Diverse Compounds. J Nat Prod Rep. 2014;31:6. doi: 10.1039/c3np70063a. [DOI] [PubMed] [Google Scholar]
- 449.Lewis JC, Coelho PS, Arnold FH. Enzymatic Functionalization of Carbon–hydrogen Bonds. Chem Soc Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Fasan R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012;2:647–666. [Google Scholar]
- 451.Agudo R, Roiban GD, Reetz MT. Achieving regio- and enantioselectivity of P450-catalyzed oxidative C–H activation of small functionalized molecules by structure-guided directed evolution. ChemBioChem. 2012;13:1465–1473. doi: 10.1002/cbic.201200244. [DOI] [PubMed] [Google Scholar]
- 452.Schrader J, Bohlmann J, editors. Biotechnology of Isoprenoids. Springer; International Publishing Switzerland: 2015. [Google Scholar]
- 453.Reetz MT. Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology. Wiley-VCH; 2017. [Google Scholar]
- 454.Gotor V, Alfonso I, García-Urdiales E, editors. Asymmetric Organic Synthesis with Enzymes. Wiley-VCH; 2008. [Google Scholar]