

The generation of a collection of human induced pluripotent stem cell (hiPSC) lines expressing endogenously GFP-tagged proteins using CRISPR/Cas9 methods is described. The methods used and the genomic and cell biological data validating the GFP-tagged hiPSC lines are also presented.
Abstract
We present a CRISPR/Cas9 genome-editing strategy to systematically tag endogenous proteins with fluorescent tags in human induced pluripotent stem cells (hiPSC). To date, we have generated multiple hiPSC lines with monoallelic green fluorescent protein tags labeling 10 proteins representing major cellular structures. The tagged proteins include alpha tubulin, beta actin, desmoplakin, fibrillarin, nuclear lamin B1, nonmuscle myosin heavy chain IIB, paxillin, Sec61 beta, tight junction protein ZO1, and Tom20. Our genome-editing methodology using Cas9/crRNA ribonuclear protein and donor plasmid coelectroporation, followed by fluorescence-based enrichment of edited cells, typically resulted in <0.1–4% homology-directed repair (HDR). Twenty-five percent of clones generated from each edited population were precisely edited. Furthermore, 92% (36/39) of expanded clonal lines displayed robust morphology, genomic stability, expression and localization of the tagged protein to the appropriate subcellular structure, pluripotency-marker expression, and multilineage differentiation. It is our conclusion that, if cell lines are confirmed to harbor an appropriate gene edit, pluripotency, differentiation potential, and genomic stability are typically maintained during the clonal line–generation process. The data described here reveal general trends that emerged from this systematic gene-tagging approach. Final clonal lines corresponding to each of the 10 cellular structures are now available to the research community.
INTRODUCTION
The study of cellular processes using new genome-editing strategies, particularly CRISPR/Cas9, is becoming increasingly feasible and powerful (Wood et al., 2011; Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013; Dambournet et al., 2014; Ratz et al., 2015; Hendriks et al., 2016). CRISPR (clustered regularly interspersed short palindromic repeat) RNAs (crRNAs) are commonly used in tandem with the CRISPR-associated 9 (Cas9) nuclease to disrupt genes for loss-of-function analysis, an approach that exploits the error-prone nonhomologous end-joining (NHEJ) pathway of DNA repair (Chiruvella et al., 2013), or to introduce or reverse genomic polymorphisms, often associated with disease, via the much less efficient homology-directed repair (HDR) pathway (Miyaoka et al., 2014). A third powerful application is the introduction (via HDR) of large exogenous sequences into genomic loci that then function as reporters for the activity of that gene (Hockemeyer and Jaenisch, 2016). When this approach is used to fuse a fluorescent protein sequence to an endogenous open reading frame, the subcellular localization and dynamics of the encoded fusion protein can be visualized under endogenous regulatory control (Doyon et al., 2011; Dambournet et al., 2014; Ratz et al., 2015; Cho et al., 2016; White et al., 2017). However, inserting a large tag sequence requires HDR with a correspondingly large, exogenously provided repair template, an inefficient process in human cells. Nevertheless, the ability to endogenously tag proteins in cells offers a major improvement over conventional overexpression systems, especially for live imaging and functional studies.
While a growing number of studies illustrate the power of endogenous gene tagging, it has been used comprehensively in only a few systems (Gavin et al., 2002; Huh et al., 2003). Strategies are now emerging to tag multiple genes in standard human cell line models (Merkle et al., 2015; Kamiyama et al., 2016; Leonetti et al., 2016). They all employ different approaches, using either selection or extensive screening to overcome the inherent inefficiency of HDR (Miyaoka et al., 2014; Merkle et al., 2015). Because most of these previous studies have targeted only a limited number of loci, this study addresses whether HDR-mediated tagging is sufficiently efficient, reproducible, and precise across targets and cellular contexts to enable its systematic use, particularly in human stem cell models.
We generated a collection of human induced pluripotent stem cell (hiPSC) lines in which each cell line harbors a green fluorescent protein (GFP) tag inserted into 1 allele of a unique genomic locus. Before initiating our editing efforts, we engaged the cell biology community to identify proteins that localize to key subcellular structures and are not known to disrupt the organization, dynamics, or function of the labeled protein/cellular structure upon tagging. The genomic loci that encode these proteins were then targeted for GFP tagging using CRISPR/Cas9 methodology. To minimize potential biological effects of the tag in edited cells, we made the isolated clones edited at 1 allele of the target locus the highest priority for our downstream analyses and larger project goals, including live-cell imaging and modeling of dynamic spatial and temporal cellular processes. We used cells edited with gene tags in this manner to improve upon conventional overexpression experiments used for imaging studies, which are often hampered by high background and overexpression artifacts (Gibson et al., 2013). We performed several quality-control assays on edited clones to identify possible biological effects caused by the endogenous tag. Live-cell microscopy and modeling experiments based on these cell lines, which have been made openly available (Coriell Medical Institute Biorepository, 2017), are the foundation of the Allen Cell Explorer (Allen Institute for Cell Science, 2017; Horwitz and Johnson, 2017).
We selected hiPSCs for this genome-editing effort for several reasons. When derived from individuals with characterized physiology or pathology, they provide personalized medicine and “disease in a dish” models (Soldner et al., 2011; Young and Goldstein, 2012; Soares et al., 2014). Furthermore, hiPSCs provide the opportunity to study the tagged proteins in a diploid, nontransformed cellular context. Because hiPSCs can be differentiated into multiple cell types, they also offer the opportunity to study tagged gene products in a variety of differentiated cellular contexts. Finally, the propensity of hiPSCs to maintain a stable karyotype over dozens of passages in culture enables the genome-editing and clonal line–generation processes. We chose the Wild Type C (WTC) hiPSC line derived from a healthy, male donor as our parental line for all gene edits for multiple reasons, including its episomal derivation, known genomic stability, availability of genomic sequence, established differentiation into diverse cell fates, and open access to the academic research community (UCSC Genome Browser, n.d.; Kreitzer et al., 2013).
Here we present our methodology for CRISPR/Cas9-mediated genome editing to create a collection of isogenic, clonally derived WTC hiPSC lines labeling 10 distinct intracellular structures and the genetic screening and quality-control data accompanying this effort. With this method, GFP-tag knock-in was successful for 10 targeted genes from diverse loci with monoallelic-editing efficiencies ranging from 0.1% to 4%, with 25% overall recovery of clones with precise monoallelic edits. We also report that 92% (36/39) of the final candidate clonal lines generated from these editing experiments stably expressed and appropriately localized the tagged protein copy, maintained normal overall cellular morphology and a stable karyotype, retained expression of pluripotency markers, and differentiated into multiple germ layers as well as cardiomyocytes. We did in some cases observe diminished abundance of the tagged protein copy, relative to the untagged copy, with no obvious effect on localization or other quality-control criteria used in this study. To our knowledge, this is the first systematic study that introduces, characterizes, validates, and distributes genome-edited hiPSCs expressing complete GFP sequences fused with the endogenous reading frame. Our data illustrate that systematic GFP tagging of diverse loci in hiPSCs is feasible and can result in high-quality stem cell lines for diverse applications.
RESULTS
Genome-editing strategy
We used CRISPR/Cas9 to attempt HDR-mediated incorporation of a full-length monomeric enhanced green fluorescent protein (mEGFP)- or EGFP-tag sequence into 10 genomic loci to label a group of key intracellular structures. Experiments were designed to introduce the GFP tag at the N- or C-terminus along with a short peptide linker between the endogenous protein and the GFP tag (Figure 1A shows an example design strategy for N-terminal tagging). Decisions on the protein terminus and linkers used for each tagging experiment were gathered from the literature and via personal communications from researchers with prior experience with each protein whenever possible (Table 1). We confirmed expression of the transcript isoform(s) designated for tagging by performing RNA-Seq on the parental WTC line before genome editing (Supplemental Figure S1 and unpublished data; Trapnell et al., 2010; Dobin et al., 2013; Martin, 2017).
FIGURE 1:

Genome-editing experimental design and initial comparisons of editing efficiency. (A) Schematic illustrating design features important for genome-editing experiments. An N-terminal GFP-tagging strategy preceding the first exon of the gene of interest is shown as an example. The location of both crRNA binding sequences is indicated in purple. Protospacer-adjacent motif (PAM) sequences are indicated in red. The position of the anticipated double-strand break generated by each crRNA is indicated with a blue arrow. The donor plasmid contained 1-kb HAs on either side of the GFP and linker sequence and a bacterial selection sequence in the backbone. The example in the schematic shows successful N-terminal tagging via HDR, resulting in the tag and linker being inserted after the endogenous start codon (ATG) in frame with the first exon. (B) Schematic depicting the genome-editing process. Transfection included precomplexing of the RNP (Cas9/crRNA/tracrRNA) and coelectroporation with the donor plasmid. FACS was used to enrich for GFP+ cells 3–4 d after transfection. GFP+ cells were collected and expanded as an enriched population for image-based confirmation of tagging before clonal line generation. (C) Flow-cytometry plots displaying GFP intensity (y-axis) 3–4 d after editing. Data shown from several experiments (target locus is indicated), along with control untransfected cells. Gates indicate the population of putatively edited cells, and values reflect the percentage of edited cells within the total population. Forward scatter is shown on the x-axis. (D) Comparison of genome-editing efficiency, as defined by FACS, shown as a percentage of GFP+ cells within the gated cell population in each experiment. (E) Estimated percentage of cells in the FACS-enriched populations expressing GFP, as determined by live microscopy. This analysis was not performed on PXN edited cells. The majority (>50%) of GFP+ cells in each case displayed correctly localized GFP, except where indicated by an asterisk (*); only ∼5% of GFP+ cells in the FBL Cr2 population had correct subcellular localization. (F) Representative image of the LMNB1 Cr1 FACS-enriched population showing an enrichment of GFP+ cells. As expected, the edited population is a mixture of GFP+ and GFP− cells. GFP intensity level was also variable. Scale bars: 10 µm. (G) Schematic overview of the clone isolation, genetic screening, and quality-control workflow. The genetic screening and quality-control assays helped identify 1–2 final clones from each gene-tagging experiment.
TABLE 1:
Summary of tagged structures.
| Gene | Protein | Cellular structure | Terminus tagged | Linker | Linker-specific reference |
|---|---|---|---|---|---|
| PXN | Paxillin | Matrix adhesions | C-terminus | GTSGGS | Commonly used linker sequence |
| TUBA1B | Alpha tubulin | Microtubules | N-terminus | GGSGGS | Gan et al., 2016 |
| DSP | Desmoplakin | Desmosomes | C-terminus | HDPPVAT | Godsel et al., 2005 |
| LMNB1 | Nuclear lamin B1 | Nuclear envelope | N-terminus | SGLRSRAQAS | Michael Davidson Fluorescent Protein Collection |
| TOMM20 | Tom20 | Mitochondria | C-terminus | GGSGDPPVAT | Michael Davidson Fluorescent Protein Collection |
| ACTB | Beta actin | Actin filaments | N-terminus | AGSGT | Grassart et al., 2014 |
| SEC61B | Sec61 beta | ER | N-terminus | SGLRS | Shibata et al., 2008 |
| FBL | Fibrillarin | Nucleolus | C-terminus | KPNSAVDGTAGPG | Dundr et al., 2000 |
| MYH10 | Nonmuscle myosin heavy chain IIB | Actomyosin bundles | N-terminus | YSDELELKLRIP | Wei and Adelstein, 2000 |
| TJP1 | Tight junction protein ZO1 | Tight junctions | N-terminus | SGLRSRALERDK | Riesen et al., 2002 |
The 10 genes and corresponding proteins successfully targeted in genome-editing experiments are listed. The peptide terminus chosen for introduction of the GFP tag and the peptide linker chosen to fuse the GFP tag with the endogenous protein are indicated. References guiding linker design choices are provided where appropriate.
While plasmid-based guide RNA (gRNA) and Cas9 systems are commonly used for gene editing, we used the ribonuclear protein (RNP) approach, because it has been reported to reduce off-target effects due to more abbreviated editing kinetics (Lin et al., 2014). We also reasoned that preferred monoallelic edits would more likely result from the shorter predicted window of editing activity. We used commercially produced gRNA to ensure quality and consistency across our editing experiments. We precomplexed wild-type Cas9 protein with a synthetic CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) duplex (Kim et al., 2014; Lin et al., 2014). This RNP complex was coelectroporated into the WTC cells along with a GFP donor plasmid specific to the target locus (Figure 1B). The donor plasmids contained 1-kb homology arms (HAs) and a target-specific linker sequence (Figure 1A and Table 1) (Dundr et al., 2000; Wei and Adelstein, 2000; Riesen et al., 2002; Godsel et al., 2005; Shibata et al., 2008; Grassart et al., 2014; Gan et al., 2016). Other design features specific to each target locus were included in the donor plasmid HAs, including single-nucleotide polymorphisms (SNPs) specific to the WTC genome (if homozygous) and mutations to inactivate crRNA binding sites that would otherwise remain intact after the introduction of the linker and GFP sequence (Supplemental Table S1; UCSC Genome Browser, n.d.). At least two independent crRNA sequences were used in each editing experiment to maximize editing success (Figure 1A and Supplemental Table S1). When possible, we only used crRNAs targeting Cas9 to within 50 base pairs of the intended GFP integration site with a strong preference for any crRNAs with binding sites within 10 base pairs (Supplemental Table S1; Elliott et al., 1998). Cas-OFFinder was used to select and rank available crRNA sequences with respect to their genome-wide specificity (Supplemental Figure S2A; Bae et al., 2014). Only crRNAs unique within the human genome were used, with one unavoidable exception (TOMM20, where the locus sequence restricted crRNA choice), and crRNAs whose alternative binding sites include mismatches in the “seed” region and are in nongenic regions were prioritized whenever possible (Supplemental Figure S2; Graham and Root, 2015; Tsai et al., 2015; Fu et al., 2016).
Fluorescence-activated cell sorting (FACS) was used to enrich the population of gene-edited cells after transfection and to evaluate rates of HDR (Figure 1, B and C, and Supplemental Table S1). We observed GFP fluorescence in putatively edited cells at variable rates over a range of signal intensities (Figure 1, C and D, and Supplemental Table S1). We used the percentage of GFP+ cells above the background defined by untransfected, unedited cells as a measure of HDR-mediated knock-in efficiency (Figure 1D and Supplemental Table S1). Successful GFP tagging was observed using at least 1 crRNA at 10 of the 12 loci, even when HDR was inefficient (<1%). Across the 10 targets resulting in successful editing, the efficiency of HDR typically ranged from <0.1% to 4%, with SEC61B a notable exception at 24% (Figure 1, C and D). In many cases, HDR efficiency at a given locus depended on the crRNA used.
As expected for tagging experiments targeting diverse cellular proteins, the observed GFP intensity among edited cell lines varied widely. We observed weak GFP signal in some experiments in which the target gene transcript was relatively scarce (PXN) and/or the protein is known to localize to small foci in cells corresponding to cell junctions (DSP) or substrate adhesion sites (PXN) (Figure 1C and Supplemental Figure S1). These structures may produce insufficient live fluorescence signal for robust flow sorting using this protocol. We were nevertheless able to enrich for cells edited at these loci, despite their minimal expression level relative to background (Figure 1, C and D). We failed to obtain edited cells from GALT, another relatively rare transcript, and TUBG1, whose product is known to localize to small cellular foci (Figure S1and Supplemental Table S1; Gunawardane et al., 2000; Kollman et al., 2011). All GFP+ cells were sorted as a single population (“FACS-enriched population”) for further characterization and clone generation.
Live imaging of the FACS-enriched populations was performed to assess the percentage of GFP+ cells within the enriched population of sorted cells and confirm GFP localization to the appropriate structure. These enriched populations were generally mixed with respect to the fraction of GFP+ cells and levels of GFP intensity (Figure 1, E and F). We observed GFP+ cells in 40–100% of the imaged cells within each population across editing experiments (Figure 1E). In all experiments except for FBL Cr2 (in which only ∼5% of GFP+ cells had the anticipated nucleolar GFP localization), the majority of GFP+ cells displayed GFP localization to the appropriate cellular structure (Figure 1F; unpublished data). Where observed, we hypothesize that variance in the localization (e.g., FBL) and intensity of the GFP signal (e.g., LMNB1) may reflect heterogeneous genome-editing outcomes that include disruption of regulatory sequences in the target gene in a subpopulation of imprecisely edited cells (discussed further in the Genetic analysis of clones section below).
We subsequently generated clonal lines starting from these edited, enriched cell populations to identify and isolate precisely edited cells. Because stem cells are sensitive to single-cell sorting, we followed established methods to passage the enriched population of sorted cells at low density such that colonies would be derived from individual cells in the majority of cases (Woodruff et al., 2013; Dambournet et al., 2014). We isolated ∼102 colonies per target gene using physical detachment with a pipette and distributed colonies to 96-well culture format. Following clonal recovery, we subjected these clones to genetic screening, as described in the following section, followed by a suite of quality-control assays to select preferred clones based on stringent genomic, phenotypic, cell biological, and stem cell criteria (Figure 1G). More than 95% of physically isolated clones survived screening and expansion (unpublished data).
Genetic analysis of clones
We developed an efficient screening strategy to rapidly discriminate between precisely and imprecisely edited clones. Our criteria for precise editing were 1) incorporation of the GFP tag in-frame with the targeted exon, 2) the absence of random or on-target donor plasmid backbone integration, and 3) no unintended mutations in either allele. Toward this aim, we employed a three-step PCR-based screening approach (Figure 2A). Because primers and probes for GFP, the donor plasmid backbone, and the RPP30 reference gene could be used to analyze all gene edits, a droplet digital PCR (ddPCR) assay was used to rapidly interrogate large sets of clones in parallel without having to optimize parameters specifically for each target gene, a significant advantage for our high-throughput platform (Miyaoka et al., 2014, 2016; Findlay et al., 2016).
FIGURE 2:

Genetic assays to screen for precise genome editing in clones. (A) Schematic illustrating the sequential process for identifying precisely tagged clones. In step 1 (left), ddPCR was used to identify clones with GFP insertion (normalized genomic GFP copy number ∼1 or ∼2) and no plasmid integration (normalized genomic plasmid backbone copy number <0.2). Hypothetical example of a typical editing experiment is shown with examples for pass (green) and fail (red) criteria. In step 2 (middle), junctional PCR amplification of the tagged allele was used to determine precise on-target GFP insertion. In step 3 (right), the untagged allele of a clone with monoallelic GFP insertion is amplified. The amplicon was then sequenced to ensure that no mutations have been introduced to this allele. (B) Examples of ddPCR screening data (step 1) from experiments representative of the range of outcomes observed. Each data point represents 1 clone. Clones with GFP genomic copy number of ∼1 to ∼2 and plasmid backbone genomic copy number <0.2 were considered for further analysis. (C) Step1 results: percentage of clones confirmed by ddPCR to have incorporated the GFP tag but not the plasmid backbone. Data are shown across experiments for which ddPCR was performed as the initial screen. (D) Step 2 results: percentage of clones confirmed in step 1 that also had correctly sized junctional PCR amplicons. (E) Step 3 results: percentage of clones confirmed to have wild-type untagged alleles by PCR amplification and Sanger sequencing following steps 1 and 2. (C–E) Number of clones analyzed for each experiment is shown in blue. Percentage of clones across all experiments (combined) that met the screening criteria (C–E) is shown in green (39%, 90%, and 77%). TUBA1B, DSP, and TJP1 analysis is omitted from C and D because junctional screening (step 2) was performed before ddPCR screening (step 1) in these experiments. (F) The percentage of clones in each experiment with KAN/AMP copy number ≥0.2 is displayed on the y-axis. Stacked bars represent three observed subcategories of rejected clones. Clones with one correct and one incorrect or missing junctions were interpreted as plasmid backbone integration at the targeted locus (purple). Clones in which no junctions are amplified (black) were interpreted to contain random integration of the donor plasmid. Clones in which both junctions are correct (pink) were interpreted to contain duplications of the GFP-tag sequence at the targeted locus.
First, GFP-tagged clones lacking plasmid backbone integration were identified using ddPCR, with equivalently amplifying primer sets and probes corresponding both to the GFP tag and the donor plasmid backbone (Figure 2A, left panel, step 1, and Supplemental Figure S3B). To accomplish this, we quantified the abundance of the GFP-tag sequence (x-axis in Figure 2, A, left panel, and B, and Supplemental Figure S3A) and normalized this value to a known two-copy genomic reference gene (RPP30) in order to calculate genomic GFP copy number in the sample. We also calculated the copy number of a marker sequence in the donor plasmid (AMP or KAN resistance genes) in each clone (y-axis in Figure 2, A, left panel, and B, and Supplemental Figure S3A). Clones with a GFP copy number of ∼1.0 (monoallelic) or ∼2.0 (biallelic) and AMP/KAN <0.2 were putatively identified as correctly edited clones. Combining data across all 10 successful editing experiments, 39% of clones were retained as candidates using this assay (Figure 2C). Clones with GFP copy number 0.2–1 were considered possible mosaics of edited and unedited cells and were typically rejected (Figure 2, A, left panel, and B, and Supplemental Figure S3A). The abundance of unedited and mosaic clones observed for target genes such as PXN may have reflected the relative difficulty of enriching for endogenously tagged proteins with low expression (Figures 1C and 2B and Supplemental Figure S1). The relative rates of putative clonal confirmation and rejection in this assay varied widely based both on the locus and the crRNA used (Figure 2C). Putatively confirmed clones were almost exclusively tagged at 1 allele (Figure 2B and Supplemental Figure S3A). Clones with putative biallelic edits with no plasmid incorporation were rare (Figure 2B and Supplemental Figure S3A). Therefore we further screened clones with a GFP copy number between ∼1 and ∼2 to potentially identify biallelic clones from mixed cultures. However, the majority of these clones (six of eight) showed evidence of faulty DNA repair in the subsequent analysis step, as discussed later in this section (Figure 2B and Supplemental Figure S3A).
As a second step in our screening, we performed junctional PCR by amplifying 2 overlapping PCR amplicons that spanned the 5′ and 3′ junctions between the GFP tag and the host cell genome distal to the 1-kb donor plasmid HA sequences. This allowed us to confirm GFP-tag incorporation without large insertions or deletions (Figure 2A, middle panel, step 2) (Jasin and Rothstein, 2013; Oceguera-Yanez et al., 2016). While a high fraction of clones had correct junctional products in most experiments, certain loci with relatively few clones putatively confirmed by ddPCR were more error prone (e.g., PXN Cr1 and TOMM20 Cr1) (Figure 2D). The small number of clones confirmed by ddPCR (passing step 1) in these cases may also indicate locus-specific challenges for precise editing. Nevertheless, 90% (n = 231) of the overall clones tested in this assay contained expected junctional PCR products after initial confirmation by ddPCR (Figure 2D). Sanger sequencing of the junctional amplicons from a subset of these clones (n = 107) confirmed correct sequences in all cases (unpublished data).
In a third step, the untagged allele (for monoallelic GFP-tagged clones) was amplified and sequenced to ensure that no mutations had been introduced via the NHEJ repair pathway at the binding site of the crRNA used for editing (Figure 2A, right panel, step 3, and E). We chose a subset of clones confirmed by ddPCR and junctional PCR from each gene edit and performed Sanger sequencing on the amplicon corresponding to the untagged allele. Clones with mutations caused by NHEJ in the untagged allele were rejected. Seventy-seven percent (n = 177) of the clones analyzed from all experiments contained a wild-type untagged allele (Figure 2E), and a subset of these clones was chosen for further analysis in additional quality-control assays (Figure 1G). To rule out the possibility of misleading junctional PCR outcomes in our final clones, such as rearrangements and duplications, we performed a single PCR designed to amplify both the tagged and untagged allele across both HA junctions (Supplemental Figure S4). In nine out of 10 cases, we confirmed the presence of the expected products for both the tagged and untagged alleles, with the exception of TOMM20 (Supplemental Figure S4C).
Because clones were frequently rejected (45% of clones in all experiments) due to stable integration of plasmid backbone sequence, we further characterized these rejected clones in hopes of understanding and eliminating this inefficiency. In many cases, clones were derived from FACS-enriched populations in which most cells displayed the correct anticipated subcellular GFP-tag localization but nevertheless harbored the GFP tag and donor plasmid backbone at equivalent copy number. We hypothesized that nonrandom HDR-mediated incorporation of both the tag and the donor plasmid backbone at the targeted locus might explain this pattern. Such an outcome would result in a tagged protein but also unintended insertions of exogenous sequence into the locus (Rouet et al., 1994; Hockemeyer et al., 2009). We evaluated this possibility by performing the tiled junctional PCR assay (step 2) on clones rejected by ddPCR due to integrated plasmid backbone, in the same manner as clones putatively confirmed by ddPCR (Figure 2F). The large majority of clones gave rise to at least 1 junctional PCR amplicon, suggesting that plasmid integration occurs at the target locus. Clones with no amplified junctions, as expected in the case of donor plasmid integration at random genomic locations, were uncommon (4% of failed clones). Much more frequently (51% of failed clones), junctions from rejected clones failed to amplify or were aberrantly large on one side of the tag but intact on the other side (Figure 2F). Forty-five percent of the plasmid-integrated clones rejected by ddPCR (which were 45% of all clones) had correct junctions on both sides of the tag (Figure 2F, “combined”). We hypothesize that these categories of clones harbor insertions and/or duplications derived from the donor cassette sequence delivered by HDR to noncoding regions flanking the GFP tag at the target locus. The prevalence of clones with this flawed editing outcome may underlie heterogeneity in the GFP signal intensity observed in some experiments (Figure 1F). The fact that ddPCR results largely correlated with the presence or absence of appropriate junctions (Figure 2D) validated the use of ddPCR as an efficient screening assay. However, we caution that the reverse is not true. Confirmation of clones with amplification of both junctions does not, on its own, exclude the possibility of incorrect repair at the targeted locus (Figure 2F).
Faulty editing was particularly evident in several experiments that were locus and crRNA specific. For example, TOMM20 editing yielded GFP+ cells from only 1 crRNA (Cr1), all of which contained integrated plasmid (80/83) and/or faulty junctions (3/83) (Figure 2, B–D, and Supplemental Figures S3A and S4). In the absence of precise editing at this locus, we chose several TOMM20 clones with evidence of plasmid backbone insertion in the noncoding sequences at the TOMM20 locus for expansion and downstream quality-control analysis. The large majority of TUBA1B clones edited with Cr2 contained integrated plasmid, while most clones from Cr1 were unaffected (Figure 2B). Similarly, the frequency and type of mutations found in the unedited allele were also target and locus specific, with ACTB Cr1 a notable outlier case in which NHEJ-mediated mutations in the untagged allele occurred in all analyzed clones (n = 24) unlike ACTB Cr2 (Figure 2E).
Clones with ddPCR signatures consistent with biallelic editing (GFP copy number ∼2) were observed at low frequency across all experiments (total n = 8) (Figure 2B and Supplemental Figure S3A). Only one clone (PXN Cr2 cl. 53) was confirmed as a biallelic edit with predicted junctional products (Figure 2A, step 2) and absence of the untagged allele (Figure 2A, step 3; unpublished data), but was later rejected due to poor morphology (see later discussion of Figure 5A). Other suspected biallelic clones were rejected due to incorrect junctional products (Figure 2A, step 2) and/or presence of the untagged allele (Figure 2A, step 3; unpublished data) indicating that these clones did not precisely incorporate the GFP tag in both alleles. The frequency of faulty HDR demonstrated by these data underscores the importance of multistep genomic screening to identify precisely edited clones and confirm monoallelic editing.
FIGURE 5:

Assessment of stem cell quality after genome editing. (A) Representative phase-contrast images depicting cell and colony morphology of the unedited WTC line and several GFP-tagged clones (LMNB1, ACTB, TOMM20, and PXN). Images are of mature stem cell colonies after expansion. PXN cl. 53, rejected due to poor stem cell morphology, is shown as a counterexample. Scale bars: 100 µm. (B) Representative flow-cytometry plots of gene-edited LMNB1 cl. 210 cells (blue), and unedited WTC cells (red) immunostained for indicated pluripotency markers (Nanog, Oct3/4, Sox2, SSEA-3, and TRA-1-60) and a marker of differentiation (SSEA-1). FMO controls (gray) defined the positive staining threshold. (C) Representative flow-cytometry plots of differentiated unedited WTC cells or gene-edited LMNB1 cl. 210 cells immunostained for markers of ectoderm (Pax6), mesoderm (Brachyury), and endoderm (Sox17) lineages. Differentiated cells stained with isotype control antibody (red) were used to define the positive staining threshold. Undifferentiated, gene-edited or WTC unedited cells (yellow) and their respective isotype controls (green) are overlaid. (D) Cardiomyocytes differentiated from unedited WTC cells and stained with cTnT antibody to label cardiac myofibrils. Scale bars: 50 µm. (E) Representative flow-cytometry plots showing cTnT expression (blue) in unedited WTC control cells and several gene-edited cell lines (LMNB1 cl. 210, ACTB cl. 184, and TOMM20 cl. 27). Isotype controls (gray) defined the positive staining threshold. (B–E) Antibody information available in Supplemental Table S3.
We observed overall confirmation rates of 39% (GFP incorporation with no plasmid), 90% (correct junctions), and 77% (wild-type untagged allele) in each of the three screening steps across all gene-targeting experiments (Figure 2, C–E). Thus ∼25% of the clones screened in this manner met all three of these precise editing criteria. Donor plasmid integration was the most common category of imprecise editing, affecting 45% of all clones (Figure 2F). Our data suggest that this frequently occurs at the edited locus as a faulty by-product of the editing process and that screening by junctional PCR alone, without a method to directly detect the plasmid backbone, leads to misidentification of clones with imprecise editing, despite appropriate localization of the tagged protein resulting from the edit (Figure 2F and Supplemental Figure S4; Jasin and Rothstein, 2013; Oceguera-Yanez et al., 2016).
To assess whether the clones that met the above gene-editing criteria contained off-target mutations due to nonspecific CRISPR/Cas9 activity, we analyzed several final candidate clones from each experiment for mutations at off-target sites predicted by Cas-OFFinder (Supplemental Figure S2; Bae et al., 2014). Potential off-target sites for each crRNA were prioritized for screening based both on their similarity to the on-target site used for editing and their proximity to genic regions (see Materials and Methods). PCR amplification of these regions followed by Sanger sequencing was performed to identify potential mutations in three to five final candidate clones for all 10 genome-editing experiments (6–12 sequenced sites per clone) across 142 unique sites. We were unable to identify any off-target editing events among a total of 406 sequenced loci (Supplemental Figure S2).
Establishing clonal hiPSC lines and culturing them long term is known to carry the risk of fixing somatic mutations and introducing genomic instability (Weissbein et al., 2014). To address this concern, we karyotyped each candidate clone during our clonal line–generation and expansion process (Figure 1G). Of the 39 final candidate clonal lines tested, we only detected a karyotypic abnormality in one candidate clone (TUBA1B) that was then rejected (unpublished data). Therefore our data indicate that chromosomal abnormalities arise at a permissively low rate for high-throughput editing in the WTC line using our methodology.
The assays described in this section allowed us to identify and expand a refined set of 5–10 candidate clones for further analysis of genomic, cell biological, and stem cell integrity as described in the Cell biological analyses to validate genome editing and Stem cell quality-control analyses sections, below (Figure 1G). These quality-control assays were often performed in sequence, with only several clones being tested in all assays (Figure 1G; see also the later discussion of Table 4). Although multiple crRNAs were tested in parallel in each editing experiment, only 1 crRNA per target locus generated clones under consideration for the final clonal line in eight out of 10 experiments (DSP and SEC61B were the exceptions). The remaining crRNAs resulted in inefficient or imprecise HDR (TOMM20, TJP1, TUBA1B, ACTB, and PXN), altered morphology and/or survival (MYH10 and LMNB1), or aberrant tag localization (FBL) (Figures 1D and 2, C–E). One of these clones for each target gene, chosen based on the aggregate result from all quality-control assays (Figure 1G), was chosen for internal imaging studies and public distribution through the Allen Cell Collection (Coriell Medical Institute Biorepository, 2017).
TABLE 4:
Summary of clones screened and analyzed.
| Experiment | Clones isolated (n) | Clones analyzed (n) | Clones passed all other quality-control (passed/analyzed) | Final clone chosen for distribution (crRNA used) |
|---|---|---|---|---|
| SEC61B | 118 | 58 | 3/3 | cl. 55 (Cr2) |
| FBL | 58 | 20 | 2/2 | cl. 6 (Cr1) |
| ACTB | 192 | 181 | 5/5 | cl. 184 (Cr2) |
| TJP1 | 93 | 91 | 4/4 | cl. 20 (Cr3) |
| PXN | 79 | 68 | 4/5 | cl. 50 (Cr2) |
| LMNB1 | 202 | 93 | 3/3 | cl. 210 (Cr1) |
| TOMM20 | 96 | 83 | 4/4 | cl. 27 (Cr1) |
| DSP | 173 | 164 | 2/3 | cl. 65 (Cr2) |
| TUBA1B | 144 | 138 | 4/5 | cl. 105 (Cr1) |
| MYH10 | 102 | 93 | 5/5 | cl. 80 (Cr4) |
| Total | 1257 | 989 | 36/39 |
Screen results for all 10 gene-tagging experiments. The total number of clones isolated from the FACS-enriched populations, clones screened for GFP insertion, clones that satisfied all quality-control assays, and the final clone chosen for distribution are shown. The crRNA used to derive the final clone is indicated in parentheses.
Live-cell imaging characterization
Live-cell imaging was performed at multiple steps throughout our quality-control process starting with the FACS-enriched population of gene-edited cells (Figure 1, B, E, and F) and then again during the clonal line–generation process to ensure proper subcellular localization of the endogenously GFP-tagged proteins. Cells were imaged using spinning-disk confocal microscopy at low (10× or 20×) and high (100×) magnification. Healthy, undifferentiated WTC hiPSCs ranged from 5 to 20 µm in diameter and 10 to 20 µm in height and grew in tightly packed colonies (Figure 3, A and B). The observations and subcellular features (z-stacks and time-lapse movies) can be seen in greater detail in the cell catalogue available through the Allen Cell Explorer Web portal (Allen Institute for Cell Science, 2017).
FIGURE 3:

Live-cell imaging of final 10 edited clonal lines. Additional images and movies, displaying additional biological features of each cell line, can be found at the Allen Cell Explorer (Allen Institute for Cell Science, 2017). All images are single images taken from a z-stack unless otherwise noted. (A) WTC hiPSC stem cell colony (transmitted light image; TL) depicting normal morphology when cells were plated on Matrigel-coated glass. (B) mEGFP-tagged nuclear lamin B1 localized to the nuclear envelope (nuclear periphery) in nondividing cells and to an extended nuclear lamina within the cytoplasm during mitosis. Image is a maximum-intensity projection of the entire colony. (C) EGFP-tagged paxillin localized to puncta at the bottom surface of the cell and larger patches near the dynamic edges of the cell colony, consistent with the localization to matrix adhesions. Some diffuse signal throughout the cytosol was also observed. Images are from the bottoms of the cells. Right, fluorescence channel overlaid onto the TL channel to indicate colony edges. (D) mEGFP-tagged beta actin localized to stress fibers and lamellipodia at the bottom of the cells (left), to a junctional band at the top of cells (right), and to regions of cell–cell contact in the center of cells (unpublished data). Some diffuse signal throughout the cytosol is consistent with depolymerized actin. (E) mEGFP-tagged nonmuscle myosin heavy chain IIB localized basolaterally to stress fibers (left), to an apical actin band (right), and to regions of cell–cell contact in the centers of cells (unpublished data). (F) mEGFP-tagged desmoplakin localized to puncta at apical cell–cell boundaries, consistent with desmosomes. Puncta are not visible in all cells; however, when present, there were varying numbers per cell (left, a maximum-intensity projection of the upper half of the volume of the cells; right, fluorescence channel overlaid onto the TL channel to indicate cell–cell boundaries). (G) mEGFP-tagged tight junction protein ZO1 localized to an apical tight junction band. Weak signal is detectable at cell–cell boundaries in the apicobasal middle of cells (unpublished data). Image is a maximum-intensity projection. (H) mEGFP-tagged alpha tubulin localized to microtubules, mitotic spindles, primary cilia, and midbodies; some diffuse signal was also observed throughout the cytosol, consistent with depolymerized tubulin. (I) mEGFP-tagged Sec61 beta was detected in ER sheets and ER tubules throughout the cytoplasm (right, image from near the middles of the cells) and in the nuclear periphery (left, image from near the bottoms of the cells). (J) mEGFP-tagged Tom20 localized to mitochondrial networks throughout the cytoplasm. Image is a maximum-intensity projection of 5 z-slices near the bottom of the cells. (K) mEGFP-tagged fibrillarin was observed in intranuclear structures. Image is a maximum-intensity projection. (A–K) Scale bars in all panels are as indicated. All imaging was performed in 3D on live cells using spinning-disk confocal microscopy with a 100× objective, except A and B, which were obtained with a 10× objective. (B–K) Representative images of final gene-edited cell lines.
The GFP-tagged proteins in the genome-edited cells served as effective markers of several cell structures (see Table 1) that exhibited localization patterns reminiscent of the apicobasal polarity in epithelial cells. For example, we observed paxillin in the matrix adhesions formed between substrate contact points and the basal surface of cells, as well as at the dynamic edges of colonies (Figure 3C). Beta actin localized to the basal surface of colonies both in prominent filaments (stress fibers) and at the periphery of cell protrusions (lamellipodia), as well as in an apical actin band at cell–cell contacts, a feature common in epithelial cells (Figure 3D). Non-muscle myosin heavy chain IIB had similar localization in actomyosin bundles, including at basal stress fibers and in an apical band (Figure 3, D and E). Desmoplakin localized to distinct puncta at apical cell–cell boundaries, as expected of desmosomes, which form junctional complexes in epithelial cells (Figure 3F). Tight junction protein ZO1 also localized apically to cell–cell contacts where tight junctions are formed (Figure 3G). These observations suggest the presence of multiple distinct epithelial junction complexes and an overall apical junction zone in edited hiPSC colonies. In addition, alpha tubulin was both diffuse, as unpolymerized tubulin, and localized to microtubules, which exhibited apicobasal polarity in nondividing cells with many microtubules extending parallel to the z-direction as reported for some epithelial cell types (Figure 3H; Musch, 2004; Toya and Takeichi, 2016).
Sec61 beta localized to endoplasmic reticulum (ER; Figure 3I), and Tom20 localized to mitochondria (Figure 3J) and was distributed throughout the cytoplasm, often with greatest density in a cytoplasmic “pocket” near the top of the cell and at lowest density in the central periphery of the cell. The center region of the cell was almost entirely occupied by the nucleus, which we observed outlined by nuclear lamin B1 (Figure 3B). Fibrillarin was localized to nucleoli within the center of the nuclei (Figure 3K).
In summary, these observations are consistent with the epithelial nature of tightly packed undifferentiated WTC hiPSCs grown on two-dimensional surfaces. All final candidate clones, spanning 10 editing experiments, exhibited predicted subcellular localization of their tagged proteins (Figure 3). Taken together, these data demonstrate the ability to identify clonal lines in which genome editing did not interfere with the expected localization of the tagged proteins to their respective structures. Furthermore, live-cell time-lapse imaging demonstrated that proper localization occurred throughout the cell cycle and the presence of the tagged protein did not noticeably interfere with cell behavior.
Cell biological analyses to validate genome editing
As an important validation step in our gene-editing process, we sought to further address the impact of the tag on correct localization of the targeted protein by comparing the localization of the tagged protein with that of the native, unedited protein. To do so, we fixed edited clones alongside unedited cells and performed immunocytochemistry or phalloidin staining (in the case of mEGFP-tagged ACTB edited cells; Figure 4A and Supplemental Figures S5 and S6). In all 10 experiments, we observed no detectable difference in the pattern of antibody labeling between the unedited cells and the edited cell line (Figure 4A and Supplemental Figures S5 and S6). Within all edited cell lines, we were also able to compare the localization of the GFP-tagged protein with the pattern of antibody labeling (which was predicted to label both the GFP-tagged and untagged protein fractions within the same cell). In all cases, this revealed extensive colocalization (Figure 4A and Supplemental Figure S6). It was not always possible to optimize antibody staining while retaining robust GFP-tag fluorescence in fixed cells, and antibody penetration into the fixed colonies was sometimes incomplete. We conclude that any incomplete colocalization observed was due to technical reasons, including antibody background, as observed in some experiments.
FIGURE 4:

Cell biological assays to evaluate coexpression of tagged and untagged protein forms and their relative contributions to cellular proteome and structure. (A) Comparison of labeled structures in edited cells and unedited WTC parental cells. The unedited cells are shown in the left column. Representative images from edited beta actin, Tom20, and nuclear lamin B1 are shown as examples (right three columns). Labels to the left indicate tagged structure, and labels to the right indicate tagged gene and clone. Cells were stained with rhodamine phalloidin, anti-Tom20 antibody, or anti–lamin B1 antibody, as indicated (Supplemental Table S3). mEGFP fluorescence (without secondary signal amplification) in genome-edited cells and the overlay are also shown (right two columns). Scale bars: 10 µm. Additional immunofluorescence data in Supplemental Figure S6 and at the Allen Cell Explorer (Allen Institute for Cell Science, 2017). (B) Lysate from ACTB cl. 184 (left), TOMM20 cl. 27 (middle), and LMNB1 cl. 210 (right) is compared with unedited WTC cell lysate by Western blot. In all cases, blots with antibodies against the respective proteins (beta actin, Tom20, and nuclear lamin B1) are shown in the left blot, and blots with anti-GFP antibodies are shown in the right blot, as indicated. Loading controls were either alpha tubulin or alpha actinin, as indicated.
Because endogenously GFP-tagged proteins in live-imaging experiments have been shown to generate more interpretable localization data than those produced in fixed and immunostained cells (Allen Institute for Cell Science, 2017), we directly compared endogenous localization in our edited lines with cells transiently transfected with constructs expressing FP-fusion proteins (EGFP or mCherry; Supplemental Figure S7). Although transient transfection, like fixation and immunostaining, is vulnerable to artifacts, cells with low transient transgene expression exhibited similar tag localization to that observed in our gene-edited cell lines. In other cases, high transient transgene expression led to artifacts, including high diffuse cytosolic background and aggregation of the tagged protein. We used intensity level as a proxy to distinguish between low- and high-level transgene overexpression, though low-level-expressing cells were often rare. As examples, transfected cells with low EGFP-tubulin transgene expression were comparable to the gene-edited alpha tubulin cells (mEGFP-alpha tubulin), although the transfected cells contained higher cytosolic signal. Transfected cells with low desmoplakin-EGFP transgene expression revealed a similar pattern to that observed in the DSP-mEGFP gene-edited line, but the transfected cell population also contained other cells, likely expressing the transgene to a greater extent, with high cytosolic signal and increased number and size of desmosome-like puncta. Transfection and overexpression of Tom20 led to cell death and perturbed mitochondrial morphology, while the endogenously tagged cells displayed intact mitochondrial networks with both normal morphology and cell viability. These results highlight the importance of using multiple techniques to validate the localization of tagged proteins in gene-edited cell lines. They also demonstrate the advantages to using genome editing to observe cellular structures rather than conventional methods that rely on overexpression, fixation, and antibody staining.
Western blot analysis was performed on whole-cell lysates from candidate edited clones. Immunoblotting with antibodies against the endogenous protein yielded products consistent with both the anticipated molecular weight of the tagged and untagged proteins and was further confirmed in all cases using an anti-GFP antibody (Figure 4B and Supplemental Figure S8). Notably, the appropriate Tom20-GFP fusion protein product was obtained despite our inability to identify a precisely edited clone, suggesting that the additional plasmid backbone sequence did not disrupt the coding sequence of the TOMM20 gene.
The Western blot data were used to quantify the abundance of the GFP-tagged protein copy relative to the total abundance of the targeted protein (Table 2). Relative levels of the tagged/untagged protein varied between the gene-edited cell lines, but were highly reproducible. While many clones expressed the tagged protein at ∼50% of the total protein in the cell, as expected for monoallelic tagging, others did not (Table 2). In the most extreme example, although the final tagged beta actin clone expressed total levels of beta actin similar to the levels found in unedited cells, only 5% of the detected protein was tagged. This suggested that these cells adapted to any compromised function of the tagged allele while retaining normal viability and behavior.
TABLE 2:
Expression analysis of tagged proteins.
| Relative level of total protein | Relative level of tagged protein | |||||
|---|---|---|---|---|---|---|
| Protein | Clone | n | Average | 95% confidence | Average | 95% confidence |
| WTC | N/A | 100.0 | N/A | N/A | N/A | |
| Beta actin | cl. 184 | 3 | 94.38 | 15.82 | 4.50 | 0.97 |
| Fibrillarin | cl. 6 | 3 | 88.61 | 8.64 | 26.09 | 3.30 |
| Nonmuscle myosin heavy chain IIB | cl. 80 | 3 | 87.46 | 28.06 | 33.27 | 0.91 |
| Sec61 beta | cl. 55 | 1 | 147.40 | N/A | 52.90 | N/A |
| Tight junction protein ZO1 | cl. 20 | 2 | 120.75 | 27.15 | 49.80 | 1.18 |
| Alpha tubulin | cl. 105 | 4 | 114.79 | 29.75 | 54.26 | 2.20 |
| Tom20 | cl. 27 | 1 | 144.00 | N/A | 54.10 | N/A |
| Paxillin | cl. 50 | 2 | 123.80 | 27.83 | 47.25 | 2.45 |
| Nuclear lamin B1 | cl. 210 | 3 | 90.97 | 10.70 | 31.33 | 2.65 |
| Desmoplakin | cl. 65 | 0 | n.q. | n.q. | n.q. | n.q. |
Relative semiquantitative levels of the protein targeted for tagging in final candidate clones chosen for expansion and distribution compared with unedited WTC cells are as indicated. The abundances of the tagged protein relative to the untagged are as indicated. Separate quantification of untagged and tagged protein versions from the mEGFP-tagged desmoplakin clone was not possible due to the large size of the target protein. N/A, not applicable; n.q., not quantified.
The observation that the tagged allele had reduced expression in some experiments coupled with the rarity of biallelic edits in our experiments raised the possibility that the tagged protein copy has reduced function. To address this, we tested tolerance of biallelic tagging (and thus whether the tagged protein has sufficient function) by introducing a spectrally distinct red fluorescent protein tag (mTagRFP-T) into the unedited allele of two different mEGFP-tagged clonal cell lines, LMNB1 cl. 210 and TUBA1B cl. 105 (Supplemental Figure S9, top row). We reasoned that, if a GFP tag reduced protein function, then deleterious effects of the tag would be exacerbated in cells with biallelic edits. We used mTagRFP-T so that we could distinguish biallelically edited cells from those harboring only the GFP tag.
We achieved tagging with mTagRFP-T in the LMNB1 clone (Supplemental Figure S9, top left panel). Putative biallelically edited cells were FACS isolated, expanded, and imaged to confirm localization of both tags to the nuclear envelope in the enriched population (Supplemental Figure S9B). We also tested whether transfecting two unique donor plasmids (one to deliver mEGFP and another for mTagRFP-T) simultaneously could produce biallelically edited cells in a single step in unedited cells using the RNP methods described in the Genome-editing strategy section above. Both methods produced populations of mTagRFP-T+/GFP+ cells, indicating tolerance of biallelic tagging at this locus despite previously observed reduced expression of the tagged protein (Table 2 and Supplemental Figure S9A, bottom left panel).
In contrast to LMNB1, we were unable to recover mTagRFP-T+/GFP+ cells after attempted editing of the TUBA1B-mEGFP clonal cell line with the TUBA1B-mTagRFP-T donor plasmid, nor were we able to isolate mTagRFP-T+/GFP+ cells when both donors were codelivered to unedited cells, despite the prevalence of both mTagRFP-T+ and GFP+ cells as separate edited populations (Supplemental Figure S9A, right panels). From these data, we conclude that cells cannot tolerate biallelic editing of TUBA1B, in contrast to LMNB1. More generally, we surmise that genomic loci likely vary widely in their tolerance for biallelic tagging and that cells may compensate for monoallelic tags by reducing expression of the tagged protein, as we observed (Table 2). However, though the ratio of the expression of tagged protein to untagged protein varies by the edited line, the total amount of a (tagged plus untagged) protein in an edited line remains similar to the (untagged) amount in unedited cells (Table 2 and Supplemental Figure S8).
These observations raised the possibility of allele-specific loss of expression in clonally derived cultures due to perturbed function of the tagged protein copy. To assess this, we maintained cultures of four cell lines displaying unequal tagged/untagged protein copy abundance (and mEGFP-alpha tubulin as a control) that were otherwise identical but had been in culture for differing lengths of time. We imaged these two sets of cultures and discovered no difference in the signal intensity or tag localization in cultures separated by four passages (14-d culture time) (Supplemental Figure S10). We likewise found no significant difference in the relative abundance of the tagged and untagged protein in immunoblotting experiments performed on cultures that differed with respect to length of passage time (Supplemental Figure S11). Additionally, the ratio of tagged to untagged protein abundance in four to five independently edited clonal lines was consistent between the final clone chosen for expansion and alternative, independently generated clones (Supplemental Figure S11). Flow cytometry confirmed that GFP-negative cells were indistinguishably scarce in cultures at both passage numbers in each of five experiments and that the overall fluorescence intensity of the GFP-tagged protein was unaltered (Supplemental Figure S12A). The consistency in expression across clones and passaging time provides further confidence in the stability of expression (Supplemental Figure S11).
To assess whether cell growth might be altered due to editing, we used flow cytometry and microscopy to assess cell cycle profiles of clones (Supplemental Figure S12, B and C; Chen et al., 2013; Yang et al., 2016). Among clones with noticeably diminished tagged protein abundance, we detected no significant differences in cell cycle profile in either assay in any clone. Additionally, in growth-curve assays, we found no noteworthy differences in culture kinetics between unedited cells and the final expanded clones from all 10 experiments (Supplemental Figure S12D).
Stem cell quality-control analyses
Upon validating the expression and localization of the GFP-tagged protein in each of the genome-edited lines, we focused on ensuring that each expanded candidate clonal line retained stem cell properties comparable to the unedited WTC cells. Assays included morphology, growth rate, expression of pluripotency markers, and differentiation potential (Figure 5 and Supplemental Figure S12D). Undifferentiated stem cell morphology was defined as colonies retaining a smooth, defined edge and growing in an even, homogeneous monolayer (Figure 5A). Clones with morphology consistent with spontaneous differentiation were rejected (Thomson et al., 1998; Smith, 2001; Brons et al., 2007; Tesar et al., 2007). Such cultures typically displayed colonies that were loosely packed with irregular edges and larger, more elongated cells compared with undifferentiated cells, as observed with 1 PXN clone (a confirmed biallelic edit; Figure 5A, rightmost image). We also assayed for the expression of established pluripotency stem cell markers, including the transcription factors Oct3/4, Sox2, and Nanog, and cell-surface markers SSEA-3 and TRA-1-60 (Figure 5B and Table 3). We found high levels of penetrance in the expression of each marker (>86% of cells) in all final clonal lines from the 10 different genome edits, similar to that of the unedited cells (Figure 5B and Table 3). Consistent with these results, we also found low penetrance (≤9% of cells) of the early differentiation marker SSEA-1 by flow cytometry in both the edited and control WTC cells (Figure 5B and Table 3). All 39 clones satisfied commonly used guidelines of >85% pluripotency-marker expression and <15% cells expressing the differentiation marker SSEA-1 used by various stem cell banks (Baghbaderani et al., 2015).
Table 3:
Quantitative assessment of pluripotency and cardiomyocyte differentiation for final 10 clonal lines.
| Pluripotency markers, % positive (n) | Germ-layer markers, % positive (n) | Cardiomyocyte differentiation metrics (n) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell line | Oct3/4 | Nanog | Sox2 | SSEA-3 | TRA-1-60 | SSEA-1 | Brachyury (mesoderm) | Sox17 (endoderm) | Pax6 (ectoderm) | % cTnT + | % of experiments with beating cells | Range in day of beating initiation |
| Unedited (WTC) | >95 (7) | >97 (7) | >97 (7) | >97 (7) | >97 (7) | <2 (7) | 84–97 (3) | 47–70 (3) | 95–99 (3) | 81–98 (12) | 93 (41) | d6–d15 |
| PXN cl. 50 | >97 (4) | >99 (4) | >98 (4) | 100 (4) | >86 (4) | <4 (4) | 96 | 58 | 65 | 66–98 (4) | 75 (16) | d6–d17 |
| TUBA1B cl. 105 | >98 (2) | 100 (2) | >99 (2) | 100 (2) | >88 (2) | <6 (2) | 92 | 80 | 89 | 87–98 (5) | 75 (12) | d6–d20 |
| LMNB1 cl. 210 | >95 (2) | >99 (2) | 100 (2) | >98 (2) | >86 (2) | <4 (2) | 93 | 81 | 72 | 83–98 (4) | 80 (10) | d7–d12 |
| TOMM20 cl. 27 | >98 (3) | 100 (3) | >99 (3) | >98 (3) | >91 (3) | <9 (3) | 94 | 82 | 93 | 80–95 (2) | 75 (12) | d7–d13 |
| DSP cl. 65 | >98 (3) | >99 (3) | >99 (3) | >97 (3) | >86 (3) | <5 (3) | 91 | 57 | 78 | 92–95 (2) | 50 (10) | d7–d15 |
| ACTB1 cl. 184 | 95 | 100 | 100 | 100 | 94 | 8 | >94 (2) | 48 (2) | >98 (2) | 80–96 (4) | 100 (4) | d7–d8 |
| SEC61B cl. 55 | 97 | 100 | 100 | 95 | 97 | 1 | >97 (2) | 76–79 (2) | >97 (2) | 91–97 (5) | 80 (5) | d7–d8 |
| FBL cl. 6 | 97 | 100 | 100 | 96 | 91 | 2 | >96 (2) | 57–75 (2) | >98 (2) | 85–96 (3) | 75 (4) | d7–d9 |
| MYH10 cl. 80 | 96 | 98 | 98 | 100 | 99 | 4 | >94 (2) | 52–64 (2) | 98 (2) | 75–99 (4) | 100 (4) | d7–16 |
| TJP1 cl. 20 | 100 | 99 | 100 | 100 | 94 | 4 | >94 (2) | 53–66 (2) | >99 (2) | 85–98 (4) | 100 (6) | d7–d17 |
Final clones chosen for distribution were assessed for pluripotency, embryonic germ-layer differentiation, and cardiomyocyte differentiation using immunostaining analyzed by flow cytometry. The percentage of stained cells above the positive staining threshold for the indicated marker is listed. The number of trials is shown as (n) where more than one trial was performed. Values reflect either the lowest measured percentage (when indicated by >), the highest measured percentage (when indicated by <), or the range observed across multiple experiments. Unedited WTC cells accompanied edited cell lines in all trials. The fraction of cTnT-positive cells reported is from trials with confirmed contractile cells.
We performed in vitro differentiation assays to confirm the pluripotency of these cell lines, because the more conventional teratoma assays are less stringent, more expensive, and less scalable (Buta et al., 2013). We compared directed germ-layer differentiation between unedited cells and the final selected edited clonal line representing each of the 10 targeted structures. Each cell line was differentiated for 5–7 d under defined conditions to mesoderm, endoderm, and ectoderm using differentiation media specific to each lineage. The cells were stained for early markers of germ-layer differentiation (Brachyury, Sox17, and Pax6) and analyzed by flow cytometry (Figure 5C and Table 3; Showell et al., 2004; Murry and Keller, 2008; Zhang et al., 2010; Viotti et al., 2014). While the differentiation into each germ layer was variable, all three germ-layer markers in the edited clones showed increased expression relative to undifferentiated cells (Figure 5C). In all edited clones tested, ≥91% of cells expressed Brachyury after mesodermal differentiation, ≥47% expressed Sox17 after differentiation to endoderm, and ≥65% expressed Pax6 upon ectoderm differentiation (Figure 5C and Table 3). Directed differentiation of edited clones into each germ-layer lineage was generally comparable to unedited cells.
We additionally investigated whether each clone could robustly differentiate into cardiomyocytes, a more developmentally advanced tissue type that will be the biological context of future experiments within the Allen Institute for Cell Science. We followed established differentiation protocols using a combination of growth factors and small molecules (Lian et al., 2015; Palpant et al., 2015) and evaluated cultures for spontaneous beating (days 6–20) and cardiac troponin T (cTnT) expression (days 20–25) to evaluate the robustness of cardiomyocyte differentiation. Clonal lines generally displayed successful cardiomyocyte differentiation, with cTnT expression and qualitative spontaneous contractility comparable to the parental WTC line (Figure 5, D and E, and Table 3).
During our quality-control process, we tested a total of 39 edited lines, of which 38 displayed successful cardiomyocyte differentiation. The single exception was one of three tested DSP clones that did not meet our criterion for spontaneous beating. While the other two clones from this experiment satisfied our criteria, these clones nevertheless showed reduced propensity for beating (50% experiments with beating for clone 65; Table 3) compared with the other edited clones. Because desmoplakin is known to be an important structural protein in cardiomyocytes, this may indicate a sensitivity to tagging that does not emerge until differentiation into a cardiomyocyte fate (Al-Jassar et al., 2013; Samuelov and Sprecher, 2015). These cardiomyocyte differentiation data combined with pluripotency-marker expression and germ-layer differentiation data, support the conclusion that fusing GFP with these endogenously expressed proteins via monoallelic tagging does not appear to disrupt pluripotency or differentiation potential of these edited hiPSC cells.
In summary, of the 39 clones analyzed for genomic stability, expression of the tagged protein, proper subcellular localization, stem cell morphology, and pluripotency, three clones were rejected due to changes in either stem cell morphology (biallelically edited PXN clone), abnormal karyotype (one of five TUBA1B clones), and impaired differentiation into cardiomyocytes (one of three DSP clones) (Table 4). Therefore these data underscore the ability of hiPSCs to tolerate monoallelic GFP tags in key proteins while retaining properties of pluripotent stem cells. Of the 36 fully validated clones, 20 (two per gene/structure) were expanded for banking and reanalyzed for genomic stability by karyotype analysis and sterility. Final clonal lines encompassing all 10 structures (one of the two final clones corresponding to each structure) described in this report are now openly available to the research community (Coriell Medical Institute Biorepository, 2017).
DISCUSSION
Endogenous gene tagging is a revolutionary approach for understanding complex processes in living cells (Doyon et al., 2011; Dambournet et al., 2014; Dean and Palmer, 2014; Ratz et al., 2015; Cho et al., 2016; Gan et al., 2016; White et al., 2017). However, its broad feasibility, particularly for introducing large tags, is unclear, because it is described in only a limited number of reports, which encompass a small number of genes using different cell types and protocols. Our editing of 10 genes with full-length GFP tags using a scalable editing protocol demonstrates the feasibility of large-tag gene editing in hiPSCs as well and reveals many useful observations about the process, which we enumerate below.
With respect to experimental design, 1) expressed loci are generally amenable to HDR with RNP Cas9/crRNA delivery using a homology donor plasmid and an efficient FACS enrichment strategy. We typically observed a 0.1% to 4% rate of HDR, with the exception of 24% HDR at one locus (SEC61B), suggesting that editing rate can be locus dependent. A robust selection approach can result in successful recovery of edited clones despite inefficient HDR (e.g., MYH10, 0.1% HDR). Because this approach is limited to tagging genes with sufficient expression, we are developing an editing strategy to accommodate genes expressed only upon differentiation. 2) The likelihood of editing success can be increased by testing multiple crRNAs in parallel. This approach was essential for obtaining eight of the gene edits discussed in this study. We did not observe clear trends in editing success with regard to the proximity between the Cas9 cleavage site and the designed point of insertion. Our use of multiple crRNAs with low-promiscuity crRNA sequences led to successful editing for these 10 genes.
Our genetic analysis of edited clones also revealed useful trends: 3) Donor plasmid backbone integration was common across all loci, necessitating assays for its detection. This problem was particularly severe for some targets (e.g., TOMM20). We also noticed that incorrect integration of the donor plasmid backbone occurred much more commonly at the targeted locus, as opposed to randomly. 4) ddPCR in conjunction with junctional PCR efficiently identified precisely edited clones. While Southern blotting, which is lower throughput, provides definitive confirmation of precise editing, we elected to use ddPCR, because the assay requires no target-specific optimization, allows higher throughput screening, and provides adequate confidence in monoallelic-editing precision. Additionally, the rationale behind this approach can be adapted to conventional PCR/qPCR. 5) Expression and localization of the anticipated fusion protein is not a faithful indicator of precise editing, because we often observed clones with correct localization that harbored exogenous sequences at the targeted locus. 6) Our low frequency of NHEJ damage (23%) in untagged alleles among tagged clones suggests that the frequency of double-strand breaks induced with our RNP protocol is below saturation. This conclusion is supported by the preponderance of monoallelic edits observed among clones. It may be possible to alter this protocol to accomplish greater biallelic editing by optimizing Cas9 activity, although with greater risk for both off-target and on-target mutations.
Our quality-control analyses in clones chosen for expansion, imaging and distribution also revealed useful observations: 7) No off-target mutations at select sequenced sites predicted by Cas-OFFinder were detected, likely due to the mild levels of double-strand break induction and our selection of low-promiscuity crRNA target sites. 8) Tagged protein function may be perturbed in some cases, as suggested by reduced expression of the tagged allele in four of the 10 cell lines. While reduced, expression of the tagged allele appeared stable during continuous passaging in all four of the lines, and displayed no changes in growth dynamics. Our interpretation that biallelic editing of TUBA1B is lethal to cells, while LMNB1 (a cell line with reduced tagged allele expression) tolerated biallelic editing, suggests that equivalently expressed tagged protein copies (e.g., mEGFP-alpha tubulin) may nevertheless have perturbed function. 9) Clones satisfied stem cell quality-control criteria, including pluripotency-marker expression, karyotypic integrity, differentiation capacity, and normal morphology and growth, showing that hiPSCs tolerate monoallelic edits in diverse genes. Despite all of our quality control, undetected global effects of the tag and perturbed function of the tagged protein remain possible. These will emerge from detailed interrogation by investigators using these cell lines. We also plan to identify potential changes to each cell line resulting from editing using transcriptome and whole-exome sequencing.
In conclusion, we established a robust gene-tagging protocol and produced clonal GFP-tagged hiPSC lines spanning 10 target genes that were comparable to unedited cells despite harboring endogenous gene tags. This general finding is consistent with experiments performed in other systems, including a study of a human cancer cell line using a similar approach (Ratz et al., 2015). The endogenously tagged fusion proteins faithfully localize to the appropriate subcellular structure and are providing unprecedented imaging clarity for studying stem cell organization and dynamics. Studies using these lines will likely benefit from the absence of overexpression and staining artifacts and the use of healthy diploid cells to reveal novel aspects of cell biology (Gibson et al., 2013).
One of our goals is to create a shared resource for open science. To that end, we have made the cell lines, the plasmids used to derive them, and the associated methodologies available through the Allen Cell Collection (Coriell Medical Institute Biorepository, 2017), the Allen Cell Collection Plasmids (Addgene, 2017), and the Allen Cell Explorer (Allen Institute for Cell Science, 2017), respectively.
MATERIALS AND METHODS
Cell culture
All work with hiPSC lines was approved by internal oversight committees and performed in accordance with applicable National Institutes of Health, National Academy of Sciences, and International Society for Stem Cell Research guidelines. The parental WTC hiPSC cell line was generated by the Bruce R. Conklin Laboratory at the Gladstone Institutes and University of California–San Francisco (UCSF) and maintained using described methods (Kreitzer et al., 2013). Upon receipt, we authenticated the cell line with the donor fibroblasts using short tandem repeat analysis (WiCell). The original cells received were at passage 33, and all passage numbers indicated in this study reflect additional subsequent passages. Edited cell lines described in this report can be obtained by visiting the Allen Cell Explorer (Allen Institute for Cell Science, 2017). WTC hiPSCs were cultured in a feeder-free system on tissue culture dishes or plates coated with GFR Matrigel (Corning) diluted 1:30 in cold DMEM/F12 (Life Technologies). Undifferentiated cells were maintained with mTeSR1 medium (STEMCELL Technologies) supplemented with 1% (vol/vol) penicillin–streptomycin (P/S) (Life Technologies). Cells were not allowed to reach confluency greater than 85% and were passaged every 3–4 d by dissociation into single-cell suspension using StemPro Accutase (Life Technologies). When in single-cell suspension, cells were counted using a Vi-CELL-XR Series Cell Viability Analyzer (Beckman Coulter). After passaging, cells were replated in mTeSR1 supplemented with 1% P/S and 10 µM ROCK inhibitor (Stemolecule Y-27632, ReproCELL) for 24 h. Medium was replenished with fresh mTeSR1 medium supplemented with 1% P/S daily. Cells were maintained at 37°C and 5% CO2. A detailed protocol can be found at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
Donor plasmids, crRNAs, and Cas9 protein
Donor plasmids were designed uniquely for each target locus, with each following a similar design strategy. HAs 5′ and 3′ of the desired insertion site were each 1 kb in length and designed using the GRCh38 reference genome. WTC-specific variants (SNPs and insertions and deletions [INDELs]) were identified from publicly available exome data (UCSC Genome Browser, n.d.). In cases in which the WTC-specific variant was heterozygous, the reference genome variant was used in the donor plasmid; when the WTC-specific variant was homozygous, the WTC-specific variant was used in the donor plasmid. Linkers for each protein were unique and were used to join the terminus of the protein with the GFP-encoding sequence (inserted 5′ of GFP for C-terminal tags and 3′ of GFP for N-terminal tags). To prevent crRNAs from targeting donor plasmid sequence, we introduced mutations to disrupt Cas9 recognition or crRNA binding; when possible, these changes did not affect the amino acid sequence. Because NHEJ is a common editing outcome and null alleles of the genes we targeted might affect our interpretation of editing data if highly frequent, we examined where double-strand breaks were anticipated between the third and fourth nucleotides of the crRNA targeting sequence proximal to the protospacer-adjacent motif (PAM-3 location) with each crRNA used. In most cases, null alleles, which could arise from NHEJ, were unlikely, because the break site resided in the 5′ untranslated region (UTR) (N-terminal tagging) or in the 3′ UTR sequences or just upstream of the stop codon (C-terminal tagging). SEC61B and MYH10 were exceptions, because double-strand breaks were anticipated 3′ of the start codon. All plasmids and design criteria used to construct them are available at Addgene (Addgene, 2017). The plasmids were initially created either by In-Fusion (Clontech) assembly of gBlock pieces (IDT) into a pUC19 backbone (New England Biolabs) or were synthesized and cloned into a pUC57 backbone by Genewiz. Plasmid DNA for transfection was prepared using endotoxin-free purification kits (NucleoBond Xtra Maxi EF; Clontech). Custom synthetic crRNAs and their corresponding tracrRNAs were ordered from either IDT or Dharmacon. Recombinant wild-type Streptococcus pyogenes Cas9 protein was purchased from the University of California–Berkeley QB3 Macrolab. All tagging experiments discussed in the current report used the mEGFP (K206A) sequence, except PXN, which was performed with EGFP instead. Pilot LMNB1 and TUBA1B biallelic tagging experiments also used mTag-RFP-T with the same linkers as those used with mEGFP tag. Detailed information on editing design can be found at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
Transfection and enrichment by FACS
Cells were dissociated into single-cell suspension using Accutase as described in Cell culture above. Transfections were performed using the Neon transfection system (ThermoFisher Scientific). We evaluated various delivery methods, including CRISPRMax (ThermoFisher Scientific), GeneJuice Transfection Reagent (EMD Millipore), Amaxa (Lonza), and Neon (ThermoFisher Scientific) and concluded that Neon electroporation resulted in favorable co-introduction of protein, RNA, and plasmid into hiPSCs as measured by transfection of a control reporter plasmid and T7 assays as a readout for Cas9 activity (unpublished data). Cas9:crRNA:tracrRNA precomplexed 1:1:1 and cotransfected with 2 µg of donor plasmid optimally balanced editing efficiency with cell survival after transfection (unpublished data), and we chose this platform for all editing experiments. A detailed protocol for the RNP transfection can be also be found at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
A cell pellet of 8 × 105 cells was resuspended in 100 µl Neon Buffer R with 2 µg donor plasmid (1:1 in biallelic-editing experiments), 2 µg Cas9 protein, and duplexed crRNA:tracrRNA in a 1:1 M ratio to Cas9. Before addition to the cell suspension, the Cas9/crRNA:tracrRNA RNP was precomplexed for a minimum of 10 min at room temperature. Electroporation was with one pulse at 1300 V for 30 ms. Cells were then immediately plated onto GFR Matrigel–coated six-well dishes with mTeSR1 medium supplemented with 1% P/S and 10 µM ROCK inhibitor. Transfected cells were cultured as described under Cell culture above for 3–4 d until the transfected culture had recovered to ∼70% confluence.
Cells were harvested for FACS using Accutase as described under Cell culture above. The cell suspension (0.5–1.0 × 106 cells/ml in mTeSR1 with ROCK inhibitor) was filtered through a 35-µm mesh filter into polystyrene round-bottomed tubes. Cells were sorted using a FACSAriaIII Fusion (BD Biosciences) with a 130-µm nozzle and FACSDiva software (BD Biosciences). Forward scatter and side scatter (height vs. width) were used to exclude doublets, and the GFP+ gate was set using live, untransfected WTC cells such that <0.1% of untransfected cells fell within the gate. Sorted populations were plated into GFR Matrigel–coated 96-well plates (<2 × 103 cells recovered) or 24-well plates (<1 × 104 cells recovered) for expansion of the whole enriched population before clone isolation. In some cases (e.g., PXN) coisolation of presumptively unedited cells was tolerated due to the weak GFP fluorescence intensity of tagged protein. To determine % HDR, we analyzed data using FlowJo version 10.2 (TreeStar).
Clonal cell line generation
FACS-enriched populations of edited cells were seeded at a density of 1 × 104 cells in a 10-cm GFR Matrigel–coated tissue culture plate. After 5–7 d, clones were manually picked with a pipette and transferred into individual wells of 96-well GFR Matrigel–coated tissue culture plates with mTeSR1 supplemented with 1% P/S and 10 µM ROCK inhibitor for 1 d. After 3–4 d of normal maintenance with mTeSR1 supplemented with 1% P/S, colonies were dispersed with Accutase and transferred into a fresh GFR Matrigel–coated 96-well plate. After recovery, the plate was divided into daughter plates for ongoing culture, freezing, and gDNA isolation.
For cryopreservation of clones in a 96-well format, when cells were 60–85% confluent, they were dissociated and pelleted in 96-well V-bottom plates. Cells were then resuspended in 60 µl mTeSR1 supplemented with 1% P/S and 10 µM ROCK inhibitor. Two sister plates were frozen using 30 µl cell suspension per plate, added to 170 µl CryoStor CS10 (Sigma) in non–GFR Matrigel coated 96-well tissue culture plates. Plates were sealed with Parafilm and stored at −80°C.
Genetic screening with ddPCR
During clone expansion, more than 1500 cells were pelleted from a 96-well plate for total gDNA extraction using the PureLink Pro 96 Genomic DNA Purification Kit (ThermoFisher Scientific). ddPCR was performed using the Bio-Rad QX200 Droplet Reader, Droplet Generator, and QuantaSoft software. The reference assay for the two-copy, autosomal gene RPP30 was purchased from Bio-Rad (assay ID dHsaCP1000485, cat. no. 10031243). For primary ddPCR screening the assay consisted of three hydrolysis probe–based PCR amplifications targeted to 3 different genes: GFP (insert), AMP or KAN (backbone), and the genomic reference RPP30. The following primers were used for the detection of GFP (5′-GCCGACAAGCAGAAGAACG-3′, 5′-GGGTGTTCTGCTGGTAGTGG-3′) and hydrolysis probe (/56-FAM/AGATCCGCC/ZEN/ACAACATCGAGG/3IABkFQ/). This assay was run in duplex with the genomic reference RPP30-HEX. The PCR for detection of the AMP gene used the primers (5′-TTTCCGTGTCGCCCTTATTCC-3′, 5′-ATGTAACCCACTCGTGCACCC-3′) and hydrolysis probe (/5HEX/TGGGTGAGC/ZEN/AAAAACAGGAAGGC/3IABkFQ/). The PCR for detection of the KAN gene used the primers (5′-AACAGGAATCGAATGCAACCG-3′, 5′-TTACTCACCACTGCGATCCC-3′) and hydrolysis probe (/5HEX/GTGAAAATA/ZEN/TTGTTGATGCGCTGG/3IABkFQ/).
PCRs were prepared using the required 2× Supermix for probes with no UTP (Bio-Rad) with a final concentration of 400 nM for primers and 200 nM for probes, together with 10 units of HindIII and 3 µl of sample (30–90 ng DNA) to a final volume of 25 µl. Each reaction before cycling was loaded into a sample well of an eight-well disposable droplet generation cartridge followed by 70 µl of droplet generator oil into the oil well (Bio-Rad). Droplets were then generated using the QX200 droplet generator. The resulting emulsions were then transferred to a 96-well plate, sealed with a pierceable foil seal (Bio-Rad), and run to completion on a Bio-Rad C1000 Touch thermocycler with a Deep Well cycling block. The cycling conditions were: 98°C for 10 min, followed by 40 cycles (98°C for 30 s, 60°C for 20 s, 72°C for 15 s) with a final inactivation at 98°C for 10 min. After PCR, droplets were analyzed on the QX200, and data analysis was preformed using QuantaSoft software.
The AMP or KAN signal was determined to be from residual nonintegrated/background plasmid when the ratio of AMP/RPP30 or KAN/RPP30 fell below 0.2 copies/genome, because this was the maximum value of nonintegrated plasmid observed at the time point used for screening in control experiments (unpublished data). To ensure that no significant amplification bias existed between the GFP and AMP amplicons, a dilution series was performed using a known plasmid containing both the GFP and AMP sequence. Copies of plasmid (78–5000) were loaded per well, and both GFP and AMP primers and probes were multiplexed together to ensure that the value returned corresponded to the copies of plasmid loaded. For primary screening, the ratios of (copies/µlGFP)/(copies/µlRPP30) were plotted against (copies/µlAMP)/(copies/µlRPP30) to identify cohorts of clones for ongoing analysis. A detailed protocol for these methods is available at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
Genetic screening with tiled junctional PCR
PCR was used to amplify the tagged allele in two tiled reactions spanning the left and right HAs, the GFP and linker sequence, and portions of the distal genomic region 5′ of the left HA and 3′ of the right HA (Figure 2) using gene-specific primers (Supplemental Table S2). Both tiled junctional PCR products were Sanger sequenced (Genewiz) bidirectionally with PCR primers when their size was validated by gel electrophoresis and/or fragment analysis (Fragment Analyzer; Advanced Analytics Technologies). In final clones, a single, nontiled junctional PCR using the gene-specific external 5′ and 3′ junctional primers (Supplemental Table S2) was used to amplify both the edited and wild-type allele in a single reaction. All PCRs described in this section were prepared using PrimeStar (Takara) 2× GC buffer, 200 µM DNTPs, 1 unit PrimeStar HS polymerase, 800 nM primers, and 10 ng gDNA in a final volume of 25 µl. Cycling conditions were as follows (98°C for 10 s, 70°C for 5 s, 72°C for 60 s) × 6 cycles at −2°C/cycle annealing temperature (98°C for 10 s, 54°C for 5 s, 72°C for 60 s) × 32 cycles, 12°C hold.
Screening for clones with wild-type untagged allele sequences
PCR was also used to amplify the untagged allele using gene-specific primers (Supplemental Table S2). These primers did not selectively amplify the unmodified locus, as was the case for tiled junctional PCR amplification of the tagged allele, but rather amplified both untagged and tagged alleles. PCR was performed with the same PrimeStar reagents and cycling conditions as described in the Genetic screening with tiled junctional PCR section above. Tracking of INDELs by decomposition (TIDE) analysis was performed manually on the amplification reaction after bidirectional Sanger sequencing in order to determine the sequence of the untagged allele. For all final clones with wild-type untagged alleles, the PCR product corresponding to the untagged allele was gel isolated and sequenced to confirm the initial result from TIDE analysis.
Off-target PCR screening
Cas-OFFinder was used to identify potential off-targets (NRG PAMs with up to 3 mismatches and 1 DNA or RNA bulge) in GRCh38 genome build (GCA_000001405.15). Cas-OFFinder output was further filtered to identify the most problematic off-targets with the fewest number of flaws (flaw = mismatch or bulge) relative to the on-target crRNA. Problematic off-targets were defined as off-targets with up to 1 flaw in the seed region (10 nucleotides at 3′ end) and up to 2 flaws in the nonseed region (10 nucleotides at 5′ end) with an NGG or NAG PAM (Supplemental Figure S2A) (Graham and Root, 2015). We selected 8–10 of these off-targets for sequencing with the goal of checking ∼4 off-targets that fell close to exons (within 50 base pairs) or within exons (exon feature in GRCh38 NCBI annotation 107) and ∼4 off-targets that were closest in sequence to on-target crRNA. Approximately 300 base pairs of sequence flanking each off-target was amplified by PCR and Sanger sequenced. A list of sequenced off-targets and primers used to amplify them is available at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
Off-target categorization
We categorized off-targets based on their PAM-3 location (expected double-strand break site): exonic = PAM-3 site inside exon or within 50 base pairs of exon; genic = not in exonic category but either inside gene (intron) or within 200 base pairs of gene; nongenic = outside of exons or genes). The locations of exons and genes were identified from GRCh38 assembly (GCA_000001405.15) NCBI annotations version 107. We also categorized off-targets based on their sequence profile: number of flaws (mismatches or DNA/RNA bulges), location of flaws (seed vs. nonseed), and PAM sequence (NGG vs. NAG; Supplemental Figure S2A). The same off-target site (same = same strand and same double-strand break site) can have a slightly different sequence depending on the position of the bulge and the ability to accommodate either a bulge or a mismatch flaw, and Cas-OFFinder will report this same site multiple times. We collapsed these multiple outputs into a single off-target site and used the sequence profile ranking scheme in Supplemental Figure S2A to categorize the resulting off-target.
Cell plating for imaging
Cells were plated on glass-bottom multiwell plates (1.5H glass; Cellvis) coated with phenol red–free GFR Matrigel (Corning) diluted 1:30 in phenol red–free DMEM/F12 (Life Technologies). Cells were seeded at a density of 2.5 × 103 in 96-well plates and (12.5–18) × 103 in 24-well plates and fixed or imaged 3–4 d later. A detailed protocol can be found at the Allen Cell Explorer (Allen Institute for Cell Science, 2017).
Live-cell imaging
Cells were maintained with phenol red–free mTeSR1 medium (STEMCELL Technologies) 1 d before live-cell imaging. Cells were imaged on a Zeiss spinning-disk microscope with a Zeiss 10×/0.3 EC Plan-NEOFLUAR, 20×/0.8 NA Plan-Apochromat, or 100×/1.25 W C-Apochromat Korr UV Vis IR objective, a CSU-X1 Yokogawa spinning-disk head, and Hamamatsu Orca Flash 4.0 camera. Microscopes were outfitted with a humidified environmental chamber to maintain cells at 37°C with 5% CO2 during imaging.
Mitotic index
Unedited and edited clonal cells were cultured in triplicate in 96-well plates for 4 d as described in the section Cell plating for imaging. Cells were fixed with 4% paraformaldehyde for 10 min (Electron Microscopy Sciences) followed by a 4′,6-diamidino-2-phenylindole (DAPI) stain (1× NucBlue Fixed Cell Stain; ThermoFisher Scientific). All colonies within a well were imaged using a Zeiss laser-scanning (LSM) 800 confocal microscope with a 20×/0.8 NA Plan-Apochromat objective. Three-dimensional maximum-intensity projections were analyzed with CellProfiler software to determine the total numbers of nuclei per colony (Carpenter et al., 2006). Total mitotic cells were manually identified and counted in 7–20 colonies per cell line within a certain size range (300–1000 cells) across the replicate wells to calculate the mitotic index (percentage of dividing cells) of each cell line. One-way analysis of variance (ANOVA) was performed to test whether mitotic index was significantly different between cell lines.
Immunocytochemistry and fixed-cell imaging
All cell lines except for TUBA1B were fixed and permeabilized in 24- or 96-well plates with a solution of 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and 0.5% Triton X-100 (EMD Millipore) for 10–15 min. TUBA1B cells were fixed in −20°C methanol for 5 min. Following fixation and permeabilization, all cells were blocked with 1% or 5% bovine serum albumin (BSA; ThermoFisher Scientific) in 1× PermWash Buffer (BD Biosciences) and incubated in primary antibody overnight at 4°C, followed by incubation in Alexa Fluor–conjugated secondary antibodies (Supplemental Table S3) and DAPI (1× NucBlue Fixed Cell Stain; ThermoFisher Scientific) for 2 h at room temperature. In the case of ACTB, cells were stained with rhodamine phalloidin (ThermoFisher Scientific) at 1:1000. SlowFade Gold (ThermoFisher Scientific) mounting medium was added to the cells after a final washing step, and cells were stored at −20°C until imaged, as described in the Live-cell imaging section above. All fixed cells, except for DSP and SEC61B, were imaged on a 3i system with a Zeiss 100×/1.46 NA alpha Plan-Apochromat oil objective, CSU-W1 Yokogawa spinning-disk head, and a Hamamatsu Orca Flash 4.0 camera. DSP cells (and corresponding controls) were imaged on a Zeiss laser-scanning (LSM) 880 confocal microscope with a Zeiss C-Apochromat 40×/1.2 W Korr FCS M27 objective. SEC61B cells were imaged with a Zeiss 100×/1.25 W C-Apochromat Korr UV Vis IR objective, a CSU-W1 Yokogawa spinning-disk head, and a Hamamatsu Orca Flash 4.0 camera. Images were processed in Fiji. Brightness and contrast values were not recorded and were not equivalent when comparing edited and unedited cells.
Western blotting
Whole-cell lysate was extracted from cell lines using M-PER buffer (ThermoFisher Scientific) supplemented with 1X Halt protease and phosphatase inhibitors (ThermoFisher Scientific), 0.16 M MgCl2, and 100 U Pierce universal nuclease (ThermoFisher Scientific) on ice for 30 min or with urea sample buffer (USB: 8 M urea, 1% SDS, 10% glycerol, 0.06 M Tris, pH 6.8, 0.001% pyronin Y). M-PER–based cell lysates were boiled for 5 min, then diluted with 1:1 Bolt LDS running buffer (ThermoFisher Scientific), heated at 70°C for 10 min, and stored at −20°C. Before gel electrophoresis, M-PER–based samples were diluted to a final concentration of 1X Bolt LDS running buffer, supplemented with 1X Bolt sample-reducing agent (ThermoFisher Scientific), and heated to 70°C for 10 min. USB-based cell lysates were triturated with a syringe and a 27-g needle, and stored at −20°C. For electrophoresis, USB-based samples were supplemented with 5% beta mercaptoethanol. First, lysates were separated on 4–12% Bolt Bis-Tris Plus gels (ThermoFisher Scientific), using 1X MOPS SDS running buffer (ThermoFisher Scientific) with Bolt antioxidant (ThermoFisher Scientific) or NuPAGE Novex 3-8% Tris-acetate gels (ThermoFisher Scientific), using 1× Tris acetate SDS running buffer (ThermoFisher Scientific), with Bolt antioxidant. Second, gels were transferred onto 0.45-µm nitrocellulose membranes (ThermoFisher Scientific). A XCell SureLock Mini-Cell System (ThermoFisher Scientific) was used for both steps. Untagged and GFP-tagged proteins were detected using protein-specific and GFP-specific antibodies in PBS-T with 5% milk overnight at 4°C (see Supplemental Table S3 for details). Beta actin was used as the loading control for PXN, SEC61B, TUBA1B, TJP1, and MYH10, while alpha tubulin was used for LMNB1, FBL, ACTB, and DSP, and alpha actinin was used for TOMM20 (Supplemental Table S3). Goat polyclonal anti-mouse Alexa Fluor 647–conjugated secondary antibody or goat polyclonal anti-rabbit Alexa Fluor 647–conjugated antibody were used as secondary antibodies as described in Supplemental Table S3. Blots were imaged at different exposure times using the ChemiDoc MP system (Bio-Rad Laboratories), and appropriate exposure times were used for semiquantitative analysis of protein levels.
Transient transfection for live-cell imaging
The constructs pmEGFP-a-tubulin-C1 (gift from Daniel Gerlich, IMBA Austrian Academy of Sciences; Addgene plasmid #21039), 1136-Desmoplakin-GFP (gift from Kathleen Green, Feinberg School of Medicine, Northwestern University; Addgene plasmid #32227), and mCherry-TOMM20-N-10 (gift from Michael Davidson, Florida State University; Addgene plasmid #55146) were used for transient transfections (Godsel et al., 2005; Steigemann et al., 2009). Bacterial strains containing the constructs were grown in 200 ml Terrific Broth (ThermoFisher Scientific) supplemented with 100 µg/ml carbenicillin (ThermoFisher Scientific) or 100 µg/ml kanamycin (ThermoFisher Scientific) overnight in a shaking incubator at 37°C. DNA plasmids were extracted from the bacterial cultures using an EndoFree Plasmid Maxi Kit (Qiagen) according to the manufacturer’s instructions and resuspended in the provided endotoxin-free water. DNA concentrations were quantified using a NanoDrop8000 (ThermoFisher Scientific) and stored at −20°C. A working DNA stock for transfections was made by diluting the DNA to a final concentration of 0.25 µg/µl in Opti-MEM (ThermoFisher Scientific) and stored at −20°C. Three days after cells were plated as described in Cell Plating for Imaging, they were transfected using GeneJuice Transfection Reagent (EMD Millipore). Medium was replaced with phenol red–free mTeSR1 30–60 min before transfection. GeneJuice transfection reagent (1.5 μl ) was diluted in 25 μl Opti-MEM (ThermoFisher Scientific) and incubated at room temperature for 5 min. DNA (1 μg) was added to the GeneJuice-Opti-MEM solution and incubated for 10 min. Of this final transfection solution, 6 μl was added per 100 μl medium in the well of a 96-well plate. Live cells were imaged as specified in the Live-cell imaging section above, 1 d after transfection.
Pluripotency testing by flow cytometry
Cells were dissociated with Accutase as described in Cell culture above, fixed with CytoFix Fixation Buffer (BD Biosciences) for 30 min at 4°C, and frozen at −80°C in KnockOut Serum Replacement (Life Technologies) with 10% dimethyl sulfoxide (DMSO). After being thawed, cells were washed with 2% BSA in Dulbecco’s phosphate-buffered saline (DPBS), and half of the cells were stained with anti–TRA-1-60 Brilliant Violet 510, anti–SSEA-3 Alexa Fluor 647, and anti–SSEA-1 Brilliant Violet 421 (all BD Biosciences) for 30 min at room temperature, then washed with 2% BSA in DPBS. The other half of the cells were permeabilized with 0.1% Triton X-100 and 2% BSA in DPBS for 30 min at room temperature followed by staining with anti–Nanog Alexa Fluor 647, anti–Sox2 V450, and anti–Oct3/4 Brilliant Violet 510 (all BD Biosciences) for 30 min at room temperature. Intracellularly stained cells were washed with BD Perm/Wash buffer (BD Biosciences). All cells received a final wash of 2% BSA in DPBS before being resuspended in 2% BSA in DPBS for acquisition. Cells were acquired on a FACSAriaIII Fusion (BD Biosciences) equipped with 405-, 488-, 561-, and 637-nm lasers and analyzed using FlowJo version 10.2 (Treestar). Approximately one-fourth of samples were stained with a panel designed for a different FACSAriaIII configuration: anti–TRA-1-60 PerCP-Cy5.5, anti–SSEA-3 PE, anti–SSEA-1 Brilliant Violet 650, anti–Nanog PE, anti–Sox2 Alexa Fluor 647, and anti–Oct3/4 PerCP-Cy5.5 (all BD Biosciences). Cells were distinguished from debris using forward-scatter area versus side-scatter area. Doublets were excluded using forward scatter and side scatter (height vs. width). Positive staining thresholds were established using fluorescence minus one (FMO) controls in which all staining reagents are included except the reagent of interest. For each reagent of interest, the positive gate was set to include 1% of FMO control cells. The cells stained with the reagent of interest that fell within this gate were used to calculate the number of positive cells. Antibody details are listed in Supplemental Table S3.
Trilineage differentiation of parental and edited WTC human iPSCs
WTC edited and unedited parental hiPSCs were assessed for pluripotency using the STEMdiff Trilineage Differentiation Kit (STEMCELL Technologies) as a means of abbreviated directed differentiation to endoderm (EN), mesoderm (M), and ectoderm (EC), with an undifferentiated (U) control. Cells were cultured according to the STEMdiff Trilineage Differentiation Kit protocol in 1:30 diluted GFR Matrigel–coated 24-well plates (Corning). Before flow analysis, cells were collected by Accutase detachment, washed in DPBS, and fixed with BD Cytofix Fixation Buffer (BD Biosciences) for 30 min at 4°C. Cells were washed once with DPBS before being pelleted and frozen at −80°C in 50 µl KSR with 10% DMSO. After being thawed, cells were washed with 2% BSA in DPBS. EC cells were permeabilized with 0.1% Triton X-100 and 2% BSA in DPBS for 30 min at room temperature. EN and M cells were permeabilized with BD Perm/Wash buffer (BD Biosciences) for 30 min at room temperature. Cells were stained with a lineage-specific antibody (EC: anti–Pax6 Alexa Fluor 647 [BD Biosciences]; EN: anti–Sox17 APC [R&D Systems]; M: anti–Brachyury APC [R&D Systems]) or equal mass of isotype control for 30 min at room temperature and then washed with their corresponding permeabilization buffer. All cells received a final wash of 2% BSA in DPBS before being resuspended in 2% BSA in DPBS for acquisition. Cells were acquired on a FACSAriaIII Fusion (BD Biosciences) and analyzed using FlowJo version 10.2 (Treestar). Samples were gated to exclude debris and cell doublets as described in Transfection and enrichment by FACS above. A gate containing 1% of isotype control-positive cells defined the positive staining threshold. Antibody details are given in Supplemental Table S3.
In vitro directed differentiation of hiPSCs to cardiomyocytes
We followed previously reported methods for cardiomyocyte differentiation (Palpant et al., 2015). Briefly, cells were seeded onto GFR Matrigel–coated six-well tissue culture plates at a density of (0.75–2) × 106 cells per well in mTeSR1 supplemented with 1% P/S, 10 µM ROCK inhibitor, and 1 µM CHIR99021 (Cayman Chemical). The following day (designated day 0), directed cardiac differentiation was initiated by treating the cultures with 100 ng/ml ActivinA (R&D Systems) in RPMI medium (Invitrogen) containing 1:60 diluted GFR Matrigel (Corning), and insulin-free B27 supplement (Invitrogen). After 17 h (day 1), cultures were treated with 5 ng/ml BMP4 (R&D systems) in RPMI medium containing 1 µM CHIR99021 and insulin-free B27 supplement. At day 3, cultures were treated with 1 µM XAV 939 (Tocris Biosciences) in RPMI medium supplemented with insulin-free B27 supplement. On day 5, the medium was replaced with RPMI medium supplemented with insulin-free B27. From day 7 onward, medium was replaced with RPMI medium supplemented with B27 with insulin (Invitrogen) every 2–3 d.
For measuring cardiac troponin T (cTnT) expression, cells were harvested using 2.5% trypsin (Invitrogen), diluted to 0.25% with Versene (Invitrogen). Cells were washed with PBS and incubated with 0.25% trypsin/Versene for 8–20 min at room temperature. Cells were gently titurated up to 15× and pelleted at 90 × g for 3 min at room temperature. Cells were then fixed with 4% paraformaldehyde in DPBS for 10 min at room temperature, washed twice with 5% FBS in DPBS, and incubated in BD Perm/Wash buffer containing anti-cardiac troponin T Alexa Fluor 647 or equal mass of mouse Immunoglobulin G1, k AF647 isotype control (all BD Biosciences) for 30 min at room temperature (Supplemental Table S3). After staining, cells were washed with BD Perm/Wash buffer, then 5% FBS in DPBS, and resuspended in 5% FBS in DPBS with DAPI (2 µg/ml). Cells were acquired on a FACSAriaIII Fusion (BD Biosciences) and analyzed using FlowJo software version 10.2. (Treestar). Nucleated particles were identified as a sharp, condensed peak on a DAPI histogram and were then gated to exclude doublets as described in Transfection and enrichment by FACS above. The cTnT-positive gate was set to include 1% of cells in the isotype control sample.
G-banding karyotype analysis
Karyotype analysis was performed by Diagnostic Cytogenetics. A minimum of 20 metaphase cells were analyzed per clone. In addition to confirming stable karyotype with the original unedited parental cells, karyotyping was performed twice for each edited clonal line at passages indicated within parentheses: PXN cl.50 (p22, p29); TUBAIB cl.105 (p29, p32); TOMM20 cl.27 (p28, p35); DSP cl.65 (p29, p32); ACTB cl.184 (p31, p34); FBL cl.6 (two independent instances of p26); LMNB1 cl210 (p26, p32); MYH10 cl.80 (p27, p29); Sec61B cl.55 (p21, p24); and TJP1 cl.20 (p25, p28). The later passage reflects the karyotype of cells from the final bank we created for distribution.
RNA-Seq analysis
Two clonal populations (one at passage 8 and one at passage 14) were sequenced from the WTC unedited parental line. After dissociation of cell cultures with Accutase, (2–3) × 106 cells were pelleted, washed once with DPBS, resuspended in 350 µl of Qiagen RLT plus lysis buffer, and then flash-frozen in liquid nitrogen before storage at −80°C. Sequencing libraries were prepared using an Illumina TruSeq Stranded mRNA Library Prep kit, and 101 base pairs paired end reads were generated on an Illumina HiSeq 2500 at a depth of 30 million read pairs (Covance). Adaptors were trimmed using Cutadapt (Martin, 2017). Reads were mapped to human genome build GRCh38 (GCA 000001405.15) and NCBI annotations 107 using STAR aligner (Dobin et al., 2013). Gene- and isoform-level transcript abundances were estimated using Cufflinks (Trapnell et al., 2010).
Cell cycle analysis
WTC edited and unedited parental hiPSCs were cultured as described in Cell culture above, until they reached 50–60% confluency. Cells were dissociated into single-cell suspension using Accutase, collected into 15-ml conical vials, pelleted, and resuspended in 50 µl DPBS. The cells were vortexed while ice-cold 70% ethanol was added dropwise to obtain a final concentration of 106 cells/ml. Cells were fixed overnight at 4°C and then transferred to −20°C until staining. A total of 2 × 105 cells per sample were aliquoted into wells of a round-bottom 96-well plate and centrifuged at 850 × g at 4°C for 10 min. Cells were washed twice with cold DPBS, resuspended in 50 µl Accutase, and incubated at 37°C for 5 min. Propidium iodide/RNase/DPBS working solution (150 µl) was added to each well for a final concentration of 50 µg/ml propidium iodide (Molecular Probes) and 250 µg/ml RNase (ThermoFisher Scientific). Cells were incubated at 37°C for 30 min, filtered through 35-µm mesh caps (Corning) and acquired on a FACSAriaIII Fusion (BD Biosciences), and analyzed using FlowJo version 10.2 (Treestar). Samples were gated to exclude debris and cell doublets at described in Transfection and enrichment by FACS above, and then singlets were gated for G1 and G2/M based on negative controls. Percent S phase was calculated by subtracting percent G1 and G2/M from 100%.
Growth-curve measurements
Edited and unedited WTC hiPSCs were grown to ∼75–80% confluence in a 10-cm plate and passaged via Accutase detachment on day 0 of growth. A total of 4 × 103 cells were then plated in triplicate on four GFR Matrigel–coated 96-well plates (one for each of the terminal time points: 0, 48, 72, and 96 h). A standard curve was also plated in triplicate as a twofold serial dilution from 2 × 105 cells to 98 cells. The ATP-based CellTiter-Glo (Promega) kit was used as an indirect measure of cell growth. Briefly, the CellTiter-Glo reagent was added to the live cells at a 1:4 dilution at each of the time points, and luminescence counts were read with a Perkin-Elmer Enspire plate reader. The standard curve plate and 0-h plates were read within 2 h of plating. Cell numbers were extrapolated from the linear portion of the standard curve for each experiment, and the following equation was used to calculate cell doubling time where Tf is the final time in hours, Xf is the final cell count, and Xi is the initial cell count:
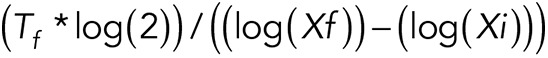 |
Reported doubling time was calculated using counts at time of seeding (0 h) and at 96 h after seeding. Two independent experiments were performed for each edited cell line. Triplicate counts from each independent experiment were averaged (leaving two data points per edited cell line and three for unedited WTC), and a one-way ANOVA was performed to test whether doubling times between cell lines were significantly different.
Supplementary Material
Acknowledgments
We thank Michelle Baird, Bruce Conklin, Jacob Corn, Gaudenz Danuser, Daphne Dambournet, Mirek Dundr, Lisa Godsel, Lawrence Goldstein, Kathleen Green, Dirk Hockemeyer, Tony Hyman, Boaz Levi, Miguel Vicente-Manzanares, Wallace Marshall, Tom Misteli, Charles Murry, Lil Pabon, Sean Palecek, Ina Poser, Jennifer Lippincott-Schwartz, William Skarnes, Tim Stearns, Annie Truong, Fyodor Urnov, Xiulan Yang, Jessica Young, and Becky Zaunbrecher for many insightful discussions, advice, and reagents. We also thank Susan Bort, Colette DeLizo, Chris Jeans, Melissa Hendershott, and Nadiya Shapovalova for technical support and Thao Do for illustration. We thank Nikki Bialy and Nathalie Gaudreault for critical reading of the manuscript. The WTC line that we used to create our gene-edited cell lines was provided by the Bruce R. Conklin Laboratory at the Gladstone Institute and UCSF. We thank the Allen Institute for Cell Science founder, Paul G. Allen, for his vision, encouragement, and support.
Abbreviations used:
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- crRNA
CRISPR RNA
- cTnT
cardiac troponin T
- DAPI
4′,6-diamidino-2-phenylindole
- ddPCR
droplet digital PCR
- DMSO
dimethyl sulfoxide
- DPBS
Dulbecco’s phsophate-buffered saline
- EC
ectoderm
- En
endoderm
- ER
endoplasmic reticulum
- FACS
fluorescence-activated cell sorting
- FMO
fluorescence minus one
- GFP
green fluorescent protein
- gRNA
guide RNA
- HA
homology arm
- HDR
homology-directed repair
- hiPSC
human induced pluripotent stem cell
- INDELs
insertions and deletions
- M
mesoderm
- mEGFP
monomeric enhanced green fluorescent protein
- NHEJ
nonhomologous end-joining
- PAM
protospacer-adjacent motif
- P/S
penicillin–streptomycin
- RFP
red fluorescent protein
- RNP
ribonuclear protein
- SNP
single-nucleotide polymorphism
- TIDE
tracking of INDELs by decomposition
- tracrRNA
trans-activating crRNA
- U
undifferentiated
- UTR
untranslated region
- WTC
Wild Type C.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-03-0209) on August 16, 2017.
REFERENCES
- Addgene. The Allen Cell Collection Plasmids. 2017. www.addgene.org/allen-institute-cell-science (accessed 27 September 2017)
- Al-Jassar C, Bikker H, Overduin M, Chidgey M. Mechanistic basis of desmosome-targeted diseases. J Mol Biol. 2013;425:4006–4022. doi: 10.1016/j.jmb.2013.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Institute for Cell Science. Allen Cell Explorer. 2017. www.allencell.org (accessed 27 September 2017)
- Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghbaderani BA, Tian X, Neo BH, Burkall A, Dimezzo T, Sierra G, Zeng X, Warren K, Kovarcik DP, Fellner T, et al. cGMP-manufactured human induced pluripotent stem cells are available for pre-clinical and clinical applications. Stem Cell Rep. 2015;5:647–659. doi: 10.1016/j.stemcr.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Buta C, David R, Dressel R, Emgard M, Fuchs C, Gross U, Healy L, Hescheler J, Kolar R, Martin U, et al. Reconsidering pluripotency tests: do we still need teratoma assays. Stem Cell Res. 2013;11:552–562. doi: 10.1016/j.scr.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Qiu C, Huang Y, Jiang L, Huang Q, Guo L, Liu T. Human amniotic epithelial cell feeder layers maintain iPS cell pluripotency by inhibiting endogenous DNA methyltransferase 1. Exp Ther Med. 2013;6:1145–1154. doi: 10.3892/etm.2013.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WK, Jayanth N, Mullen S, Tan TH, Jung YJ, Cisse II. Super-resolution imaging of fluorescently labeled, endogenous RNA polymerase II in living cells with CRISPR/Cas9-mediated gene editing. Sci Rep. 2016;6:35949. doi: 10.1038/srep35949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coriell Medical Institute Biorepository. Allen Cell Collection. 2017. https://catalog.coriell.org/1/AllenCellCollection (accessed 27 September 2017)
- Dambournet D, Hong SH, Grassart A, Drubin DG. Tagging endogenous loci for live-cell fluorescence imaging and molecule counting using ZFNs, TALENs, and Cas9. Methods Enzymol. 2014;546:139–160. doi: 10.1016/B978-0-12-801185-0.00007-6. [DOI] [PubMed] [Google Scholar]
- Dean KM, Palmer AE. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat Chem Biol. 2014;10:512–523. doi: 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, Misteli T, Olson MO. The dynamics of postmitotic reassembly of the nucleolus. J Cell Biol. 2000;150:433–446. doi: 10.1083/jcb.150.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay SD, Vincent KM, Berman JR, Postovit LM. A digital PCR-based method for efficient and highly specific screening of genome edited cells. PloS ONE. 2016;11:e0153901. doi: 10.1371/journal.pone.0153901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu BX, St. Onge RP, Fire AZ, Smith JD. Distinct patterns of Cas9 mismatch tolerance in vitro and in vivo. Nucleic Acids Res. 2016;44:5365–5377. doi: 10.1093/nar/gkw417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z, Ding L, Burckhardt CJ, Lowery J, Zaritsky A, Sitterley K, Mota A, Costigliola N, Starker CG, Voytas DF, et al. Vimentin intermediate filaments template microtubule networks to enhance persistence in cell polarity and directed migration. Cell Systems. 2016;3:500–501. doi: 10.1016/j.cels.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gibson TJ, Seiler M, Veitia RA. The transience of transient overexpression. Nat Methods. 2013;10:715–721. doi: 10.1038/nmeth.2534. [DOI] [PubMed] [Google Scholar]
- Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, Huen AC, Thorne ME, Gaudry CA, Park JK, Myung K, et al. Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol. 2005;171:1045–1059. doi: 10.1083/jcb.200510038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DB, Root DE. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015;16:260. doi: 10.1186/s13059-015-0823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassart A, Cheng AT, Hong SH, Zhang F, Zenzer N, Feng Y, Briner DM, Davis GD, Malkov D, Drubin DG. Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. J Cell Biol. 2014;205:721–735. doi: 10.1083/jcb.201403041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane RN, Lizarraga SB, Wiese C, Wilde A, Zheng Y. γ-Tubulin complexes and their role in microtubule nucleation. Curr Top Dev Biol. 2000;49:55–73. doi: 10.1016/s0070-2153(99)49004-0. [DOI] [PubMed] [Google Scholar]
- Hendriks WT, Warren CR, Cowan CA. Genome editing in human pluripotent stem cells: approaches, pitfalls, and solutions. Cell Stem Cell. 2016;18:53–65. doi: 10.1016/j.stem.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell. 2016;18:573–586. doi: 10.1016/j.stem.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz R, Johnson GT. Whole cell maps chart a course for 21st-century cell biology. Science. 2017;356:806–807. doi: 10.1126/science.aan5955. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, et al. Versatile protein tagging in cells with split fluorescent protein. Nat Commun. 2016;7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Merdes A, Mourey L, Agard DA. Microtubule nucleation by gamma-tubulin complexes. Nat Rev Mol Cell Biol. 2011;12:709–721. doi: 10.1038/nrm3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer FR, Salomonis N, Sheehan A, Huang M, Park JS, Spindler MJ, Lizarraga P, Weiss WA, So PL, Conklin BR. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am J Stem Cells. 2013;2:119–131. [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci USA. 2016;113:E3501–E3508. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Bao X, Zilberter M, Westman M, Fisahn A, Hsiao C, Hazeltine LB, Dunn KK, Kamp TJ, Palecek SP. Chemically defined, albumin-free human cardiomyocyte generation. Nat Methods. 2015;12:595–596. doi: 10.1038/nmeth.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2017;17:10–12. [Google Scholar]
- Merkle FT, Neuhausser WM, Santos D, Valen E, Gagnon JA, Maas K, Sandoe J, Schier AF, Eggan K. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015;11:875–883. doi: 10.1016/j.celrep.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, Berman JR, Cooper SB, Mayerl SJ, Chan AH, Zhang B, Karlin-Neumann GA, Conklin BR. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci Rep. 2016;6:23549. doi: 10.1038/srep23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, Conklin BR. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Musch A. Microtubule organization and function in epithelial cells. Traffic. 2004;5:1–9. doi: 10.1111/j.1600-0854.2003.00149.x. [DOI] [PubMed] [Google Scholar]
- Oceguera-Yanez F, Kim SI, Matsumoto T, Tan GW, Xiang L, Hatani T, Kondo T, Ikeya M, Yoshida Y, Inoue H, et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods. 2016;101:43–55. doi: 10.1016/j.ymeth.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Palpant NJ, Pabon L, Roberts M, Hadland B, Jones D, Jones C, Moon RT, Ruzzo WL, Bernstein I, Zheng Y, et al. Inhibition of beta-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development. 2015;142:3198–3209. doi: 10.1242/dev.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep. 2015;5:9592. doi: 10.1038/srep09592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesen FK, Rothen-Rutishauser B, Wunderli-Allenspach H. A ZO1-GFP fusion protein to study the dynamics of tight junctions in living cells. Histochem Cell Biol. 2002;117:307–315. doi: 10.1007/s00418-002-0398-y. [DOI] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov L, Sprecher E. Inherited desmosomal disorders. Cell Tissue Res. 2015;360:457–475. doi: 10.1007/s00441-014-2062-y. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showell C, Binder O, Conlon FL. T-box genes in early embryogenesis. Dev Dyn. 2004;229:201–218. doi: 10.1002/dvdy.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
- Soares FA, Sheldon M, Rao M, Mummery C, Vallier L. International coordination of large-scale human induced pluripotent stem cell initiatives: Wellcome Trust and ISSCR workshops white paper. Stem Cell Rep. 2014;3:931–939. doi: 10.1016/j.stemcr.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RD. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Toya M, Takeichi M. Organization of non-centrosomal microtubules in epithelial cells. Cell Struct Funct. 2016;41:127–135. doi: 10.1247/csf.16015. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCSC Genome Browser. WTC Public Exome and Genome. http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=alexgraehl&hgS_otherUserSessionName=po%2Dlin%2DWTC%2Dhg19%2Dpublic%2Djan%2D16 (accessed 27 September 2017)
- Viotti M, Nowotschin S, Hadjantonakis AK. SOX17 links gut endoderm morphogenesis and germ layer segregation. Nat Cell Biol. 2014;16:1146–1156. doi: 10.1038/ncb3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Adelstein RS. Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol Biol Cell. 2000;11:3617–3627. doi: 10.1091/mbc.11.10.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbein U, Benvenisty N, Ben-David U. Quality control: genome maintenance in pluripotent stem cells. J Cell Biol. 2014;204:153–163. doi: 10.1083/jcb.201310135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CW, Vanyai HK, See HB, Johnstone EKM, Pfleger KDG. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci Rep. 2017;7:3187. doi: 10.1038/s41598-017-03486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng X, et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333:307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LS. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5:974–985. doi: 10.1016/j.celrep.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Scavuzzo MA, Chmielowiec J, Sharp R, Bajic A, Borowiak M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci Rep. 2016;6:21264. doi: 10.1038/srep21264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Goldstein LS. Alzheimer’s disease in a dish: promises and challenges of human stem cell models. Hum Mol Genet. 2012;21:R82–R89. doi: 10.1093/hmg/dds319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang CT, Chen J, Pankratz MT, Xi J, Li J, Yang Y, Lavaute TM, Li XJ, Ayala M, et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell. 2010;7:90–100. doi: 10.1016/j.stem.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.