

Abstract
Virulent Mycobacterium tuberculosis (Mtb) triggers necrosis in host Mφ, which is essential for successful pathogenesis. Here we demonstrate that necrosis of Mtb-infected Mφ is dependent on the action of the cytosolic kinase Receptor Interacting Protein 3 (RIPK3) and the mitochondrial Bcl-2 family member protein B-cell lymphoma - extra large (Bcl-xL). RIPK3-deficient Mφ are able to better control bacterial growth in vitro and in vivo. Cytosolic RIPK3 translocates to the mitochondria where it promotes necrosis and blocks caspase 8-activation and apoptosis via Bcl-xL. Furthermore, necrosis is associated with stabilization of hexokinase II on the mitochondria as well as cyclophilin D-dependent mitochondrial permeability transition (MPT). These events up-regulate the level of reactive oxygen species (ROS) to induce necrosis. Thus, in Mtb-infected Mφ mitochondria are an essential platform for induction of necrosis by activating RIPK3 function and preventing caspase 8 - activation.
INTRODUCTION
Cell death is triggered by regulated and highly specific intracellular and extracellular signals. Apoptosis and necrosis are two major forms of cell death that play a critical role in immunity to infection by Mycobacterium tuberculosis (Mtb) (1). These distinct cell death modalities of macrophages (Mφ) directly affect control of Mtb growth and dissemination (2–4) as well as antigen specific T cell mediated immunity via cross-presentation (5–8). Thus it is not surprising that virulent strains of Mtb have developed mechanisms that allow evasion of Mφ apoptosis by inducing necrosis (1). Furthermore, we and others have demonstrated that during infection with Mtb, mitochondrial function is critical in determining the cell death modality (2, 9–11). Infection of human Mφ with the attenuated Mtb H37Ra predominantly induces apoptosis (2, 3, 12), which is dependent on mitochondrial outer membrane permeabilization (MOMP) and release of mitochondrial cytochrome c into the cytosol (11). In contrast, infection of Mφ with the virulent Mtb strain H37Rv induces necrosis (2, 3) associated with irreversible permeabilization of the mitochondrial inner membrane (MIMP) leading to mitochondrial permeability transition (MPT) and loss of mitochondrial integrity as well as function (11). The MPT is regulated by calcium and occurs after opening of a functional pore believed to include the adenine nucleotide translocator (ANT), and a voltage-dependent anion channel (VDAC), whose functions are regulated by cyclophilin D (CypD) (13, 14). CypD, the only component of the MIMP, that is indispensable for induction of necrosis (15), is a member of the cyclophilin family of peptidyl-prolyl cis-trans isomerases and has been implicated in MPT-dependent necrotic (but not apoptotic) cell death (15, 16). We have previously shown that CypD is involved in induction of Mtb-dependent Mφ necrosis, which allows bacterial growth in virulent Mtb-infected cells (10). These studies collectively indicate that programmed necrosis is essential for evasion of immunity by Mtb and is regulated at the level of mitochondria but the molecular mechanisms involved in the necrotic pathway remain elusive.
It has recently been shown that inhibitors of apoptosis (e.g. IAP) trigger formation of a complex including RIPK3 and RIPK1, which induces a type of necrosis referred to as necroptosis (17). RIPK3 - dependent necrosis seems to be tightly regulated by caspase 8 (18) as it was shown that caspase 8 inactivates RIPK3 (19). Interestingly, an elegant study in a murine tumor model recently has demonstrated that RIPK3-mediated necroptosis is more efficient in cross-priming CD8+ T cells than apoptosis and within those dying cells RIPK1 signaling is required for initiating T cell mediated immunity (20). Death associated molecular patterns (DAMPs) released from necroptotic cells also provoke a strong inflammatory response (21). Cumulatively, little is known about the role of cell death-inducing mechanisms during host interaction with a living pathogen that initiates multiple complex signaling pathways.
NAD(P)H oxidases (NOX), electron-transporting membrane enzymes including NOX2, a phagocyte oxidase, and mitochondrial respiration are the major sources of cellular ROS (22). ROS is produced in the mitochondria predominantly by the complexes I to III of the electron transport chain and by mitochondrial dehydrogenases including glycerol-3-phosphate dehydrogenase and α-ketoglutarate dehydrogenase, a Krebs cycle enzyme (23), which produces NADH and generates ROS if the NADH/NAD+ ratio is high (24). Processing of glucose is initiated by enzymes of the hexokinase (HK) family, which has four isoforms (HK-I to HK-IV). HK-II is critical for mitochondrial energy production since its presence on the outer mitochondrial membrane couples glycolysis with oxidative phosphorylation (25). Augmented levels of HK-II increase ROS accumulation if the normal function of the mitochondrial electron chain is compromised. Interestingly, increased mitochondrial ROS production has been associated with RIPK3-dependent necrosis (26).
The mitochondrial Bcl-2 family protein B-cell lymphoma-extra large (Bcl-xL) on the mitochondria is known to block apoptosis by inhibiting the function of Bcl-2 homologous antagonist/killer (BAK) (27) via blocking Bcl-2–associated X protein (BAX) recruitment to the mitochondria (28, 29). Here we report that in Mφ infected with virulent Mtb, RIPK3 translocates to mitochondria as evidenced by its presence in mitochondria along with Bcl-xL and pro-caspase 8. Mitochondrial pro-caspase 8 in the presence of RIPK3 and Bcl-xL remains in its inactive zymogen form, suggesting that Bcl-xL is critical for preventing caspase 8 mediated apoptosis and that the presence of active RIPK3 on the mitochondria is required for induction of necrosis. Furthermore, we identify RIPK3 as an important kinase that is involved in the induction of necrosis impairing host Mφ immunity both in vitro and in vivo. Interestingly, the enhanced protection of RIPK3 deficient mice was independent of T cell mediated immunity to pulmonary Mtb infection.
Similar to our findings in human Mφ infected with virulent Mtb, Roca and Ramakrishnan have reported a comparable role of RIPK3 in a zebra fish model of infection with Mycobacterium marinum (30), which underscores the evolutionary conservation of the RIPK3-dependent necrosis pathway in Mtb pathogenesis. Collectively, our findings demonstrate that following Mtb infection RIPK3 is involved in the activation of several independent mechanisms that ultimately converge at the inhibition of apoptosis and promotion of necrosis in Mφ, which enhances the susceptibility of the host to infection.
RESULTS
RIPK3 is required for induction of necrosis in Mφ infected with virulent Mtb
A hallmark of infection of Mφ with virulent H37Rv is necrosis, which allows the bacteria to evade host defense mechanisms (1). RIPK3 is required for the induction of necrosis in a variety of cell models (18). To test whether a RIPK3-dependent pathway is directly involved in necrosis and innate control of virulent H37Rv infection, we silenced the RIPK3 gene in human Mφ using two different RIPK3 siRNAs (Figure 1A and Supplementary Figure 1A). In the absence of RIPK3, necrosis was remarkably reduced in Mtb infected Mφ (Figure 1B and Supplementary Figure 1A). This reduction of necrotic cell death also correlated with decreased bacterial growth in RIPK3 deficient Mφ (Figure 1C). Because of potential off-target effects of siRNA, we performed similar experiments using RIPK3 deficient mice. Similar to human Mφ, Mtb infected RIPK3-deficient murine Mφ revealed decreased necrosis (Figure 1D), which led to significant reduction of bacterial growth in vitro (Figure 1E). Decreased necrosis in RIPK3−/− Mφ was also associated with significant reduction of extracellular bacteria compared to Mtb-infected wild-type (WT) Mφ (Figure 1F). To determine whether reduced necrosis also occurs in the lungs of RIPK3−/− mice following Mtb infection, we infected RIPK3−/− and WT mice via the intratracheal route with H37Rv and collected bronchoalveolar lavage 2 days after infection. The levels of dead cells were significantly lower in the lungs of infected RIPK3−/− mice than WT mice (Figure 1G). We next investigated the involvement of the mixed lineage kinase domain-like protein (MLKL), which is a functional RIPK3 substrate that binds to RIPK3 through its kinase-like domain but lacks kinase activity of its own in RIPK3 mediating necrosis. Interestingly, the levels of MLKL were increased in human Mφ infected with H37Rv (Figure 1H) and silencing of MLKL significantly decreased necrosis in H37Rv-infected Mφ (Figure 1I and Supplementary Figure 1B). Collectively, these data from both in vitro human and murine Mφ as well as the in vivo experiments indicate that RIPK3 plays a critical role in the necrosis pathway during the course of Mtb infection.
Figure 1. RIPK3 impairs anti-mycobacterial defense mechanisms in Mtb infected Mφ.

(A) Silencing efficiency of RIPK3 siRNA. Aliquots (50 μg) of cell lysates from human Mφ treated with targeted RIPK3 siRNA or with control scrambled siRNA (Scr) were collected 12 h post6 transfection and subjected to Western blot analysis of RIPK3 levels. GAPDH was used as a loading control. (B) Human Mφ transfected with scrambled control (Scr) or RIPK3 siRNA (RIPK3 siRNA) were infected with H37Rv (MOI 5) and the cell viability was evaluated at 0 to 96 h after infection. (C) Human Mφ treated with scrambled (Scr) or RIPK3 siRNA were infected with H37Rv (MOI 5) and the replication of H37Rv was measured at 2 and 96 h post-infection. (D) BMD-Mφ from WT and RIPK3−/− mice were infected with H37Rv at an MOI of ~5. After 24 and 48h, necrosis was assessed using a commercially available Cell Death ELISA kit (Roche). (E-F) After 72 h, the growth of intracellular bacteria (E) and the number of extracellular bacteria (F) was significantly reduced in RIPK3−/− Mφ. (G) WT and RIPK3−/− mice (n=3 per group) were intratracheally infected with H37Rv (0.5 × 106 CFU) and then bronchoalveolar lavage (BAL) was performed after 48h of infection. Cell death was assessed using Live/Dead fixable dead cell stain kits (Invitrogen). Each histogram is representative of the BAL from an individual infected mouse and the numbers in histograms indicate the percentage of dead cells. (H) Increased expression of MLKL in H37Rv infected human Mϕ. (I) Human Mϕ treated with MLKL siRNA or scrambled RNA (scr) were infected with H37Rv (MOI 5 or 10) and the cell viability was evaluated at 72 h after infection. Results are expressed as mean ± SD. Data were analyzed using one-way ANOVA. *, Values of P < 0.05 were considered to be significant. Data are representative of 2–3 independent experiments.
RIPK3 and pro-caspase 8 translocate from the cytosol to the mitochondria in Mtb-infected Mφ
In cells undergoing genotoxic stress pro-caspase 8 forms a cytosolic complex with RIPK1, RIPK3 and FADD termed the “riptoptosome”, which can become a cytosolic cell death signaling platform (17, 31). We therefore investigated whether a similar complex exists in human Mφ infected with Mtb. Here we found that RIPK1, RIPK3, and pro-caspase 8 co-precipitate in the cytosol but not on mitochondria of human Mφ prior to infection (Figure 2A, Supplemental Figure. 3A) similar to the ripoptosome. At 2 h after infection of human Mφ with virulent H37Rv or avirulent H37Ra, we observed that RIPK3, RIPK1 and pro-caspase 8 translocate from the cytosol to the mitochondria (Figure 2A, right lane). In H37Rv-infected pro-necrotic Mφ, the levels of pro-caspase 8 and RIPK3 increased on the mitochondria over time (Figure 2B). In contrast, in Mφ infected with the avirulent H37Ra, the levels of pro-caspase 8 and RIPK3 on the mitochondria were markedly reduced by 24 h, which is consistent with pro-caspase 8 becoming activated at 24 h to induce apoptosis (Figure 2C). The translocation of cytosolic RIPK3 and pro-caspase 8 to the mitochondria cannot be explained by contamination of the mitochondrial fractions with cytosolic proteins, because mitochondrial preparations from uninfected Mφ do not contain significant amounts of RIPK1, RIPK3 and pro-caspase 8 (Figure 2A). Moreover, using fluorescence confocal microscopy we demonstrate that the levels of pro-caspase 8 and RIPK3 co-localizing with the mitochondria are significantly increased in Mφ infected with the virulent H37Rv strain (Figure 2D–E). Additionally, the levels of cleaved caspase 8 were significantly higher in H37Rv infected RIPK3 deficient Mφ compared to WT Mφ (Figure 2F) while there was no difference in Mφ infected with H37Ra (Supplementary Figure 2A). These data indicate that after infection of Mφ with virulent Mtb the mitochondria acquire a complex containing RIPK1, RIPK3 and pro-caspase 8 and RIPK3, which is critical in preventing caspase 8 activation.
Figure 2. translocation of a RIPK3/pro-caspase 8 containing complex to the mitochondria of H37Rv-infected Mφ.

(A) Cytosolic and mitochondrial RIPK3 and pro3 caspase-8 following H37Rv infection. Equal numbers of human Mφ were infected with H37Rv (MOI 10) and lysed for isolation of cytosolic and mitochondrial fractions after 24 h of infection. Equal amounts of protein were immunoprecipitated (IP) with anti-caspase-8 ab and the levels of RIPK3, RIPK1, and pro-caspase 8 were measured by Western blot analysis. (B-C) Time7 dependent translocation of RIPK3 and pro-caspase 8 to the mitochondria. Equal numbers of Mφ were infected with virulent H37Rv or (B) avirulent H37Ra (C) at MOI 10. The levels of pro9 caspase 8 and RIPK3 were determined by subjecting equal amounts of mitochondria isolated from infected Mφ to Western blotting. VDAC was used as a loading control. (D) Colocalization of caspase 8 and RIPK3 with the mitochondria of H37Rv-infected Mφ. Left panels: Colocalization of RIPK3 and pro-caspase 8 with mitochondria 24h after infection visualized by confocal fluorescence microscopy. Human Mφ remained either uninfected or were infected with mCherry- H37Rv (MOI 10) for 24h, fixed and stained with mitotracker (green) and anti-RIPK3 or anti caspase 8 ab (red). Scale bar, 30 μm. On the right side of every panel is the quantification of caspase 8 (top) and of RIPK3 (bottom) associated with the mitochondria. Data were analyzed using nonparametric Student t test (E) Phase-contrast images of representative infected and uninfected human primary Mφ after 24 h of infection show approximate location of the plasma membrane and the nucleus as indicated by black and white arrows, respectively. Due to incipient necrosis the plasma membrane of the H37Rv-infected Mφ is not clearly visible. (F) BMD-Mφ from WT and RIPK3−/− mice were infected with H37Rv at an MOI of ~10. After 12 h, expression of cleaved caspase 8 was assessed using flowcytometry. Results are represented as mean ± SD. Data were analyzed using one-way ANOVA. *,**,*** Values of P < 0.05, P<0.01 and P<0.001, respectively were considered to be significant. Data are representative of 2–3 independent experiments.
RIPK3 and Bcl-xL prevent activation of pro-caspase 8 on the mitochondria promoting necrosis in Mtb-infected Mφ
Bcl-xL protects mitochondria against apoptosis by inhibiting BAK activation (28). RIPK3 and pro-caspase 8 translocate to the mitochondria in H37Rv-infected Mφ as shown by their presence in mitochondrial fraction after 6h and 24h of infection (Figure 3A). Bcl-xL is required for this sequestration of pro-caspase 8 on the mitochondria in its inactive form, as silencing of the Bcl-xL-gene leads to proteolytic processing of pro-caspase 8 and disappearance of RIPK3 from the mitochondria 24 h post-H37Rv-infection (Figure 3A). Thus, by binding of pro-caspase 8 to the mitochondria Bcl-xL contributes to maintaining RIPK3 in an active form on the mitochondria (Figure 3A). In contrast, 24 h after infection with avirulent H37Ra (which promotes apoptosis), the Bcl-xL levels on the mitochondria are significantly diminished, which is also associated with disappearance of pro-caspase 8 and RIPK3 from the mitochondria (Figure 3B). These findings are in agreement with a study demonstrating that by outcompeting Bax via Bcl-xL, cells are protected against apopotosis (28). Finally, we demonstrated that silencing Bcl-xL in H37Rv-infected Mφ for 72 h significantly reduced necrosis (Figure 3C and Supplementary Figure 1C). These experiments collectively indicate that RIPK3 and Bcl-xL colocalize on the mitochondria in a mutually dependent manner and suggest that the colocalization leads to prevention of activation of pro-caspase 8 on the mitochondria and increase of necrosis in Mtb-infected Mφ.
Figure 3. Bcl-xL enables pro-caspase 8 and RIPK3 accumulation on mitochondria of Mtb2 infected Mφ.

(A) Bcl-xL is required for pro-caspase 8 and RIPK3 accumulation on the mitochondria of H37Rv infected Mφ. Human Mφ were transfected with Bcl-xL or scrambled control (Scr) siRNA and then infected with H37Rv (MOI 10). Equal amounts of purified mitochondria from non-infected and H37Rv-infected Mφ were subjected to Western blot analysis to determine the levels of pro-caspase 8 and RIPK3. (B) Caspase inhibition of H37Ra infected Mφ enables RIPK3/Bcl-xL accumulation on mitochondria. After treatment with the caspase inhibitor z-IETD pro-caspase 8, RIPK3 and Bcl-xL accumulates on the mitochondria of H37Ra infected Mφ. Mitochondria were isolated from H37Rv or H37Ra-infected Mφ treated with or without z-IETD (10 μmol) and subjected to Western blot analysis using anti caspase 8, anti-RIPK3 and anti-Bcl-xL ab. Equal amounts of the proteins were subjected to Western blot analysis for the evaluation of RIPK3 and Bcl-xL levels. (C) Mφ treated with Bcl-xL or scrambled control siRNA (Scr) were infected with H37Rv (MOI 10) and cell death was assessed after 48 h using Live/Dead fixable dead cell stain kits (Invitrogen). (D) z-IETD blocks activation of the apoptotic caspase 9 and 3 essential for apoptosis induction. Equal amounts of cell lysates of Mφ infected with H37Ra (pro-apoptotic strain) treated with or without z-IETD (10 μmol) were subjected to Western blot analysis for evaluating active caspase 3 and 9. (E) RIPK3 is required for accumulation of mitochondrial pro-caspase 8 and Bcl-xL. Mitochondria and cytosolic fraction of H37Rv-infected Mφ treated with RIPK3 or Scr siRNA were subjected to Western blot analysis and the levels of RIPK3, pro-caspase 8 and Bcl-xL were evaluated. (F) Silencing of the RIPK3 gene activates apoptosis executor caspase 3 in H37Rv infected Mφ. Mφ treated with RIPK3 or Scr siRNA were infected with H37Rv. Cell lysate was collected and equal amounts subjected to Western blot analysis for cleaved caspase 3. (G) Bid processing in H37Ra and H37Rv-infected Mφ. Mitochondria were isolated from H37Ra (right panel) or H37Rv (left panel) -infected Mφ and the kinetics of BID processing and tBID accumulation were assessed by Western blotting. (H) Silencing of the BAX gene in H37Ra-infected Mφ blocks caspase 3 activation, a marker for apoptosis. Mφ treated with BAX or Scr siRNA were infected with H37Ra or H37Rv (MOI 10). Cell lysate was collected after 0 and 24 h and equal amounts subjected to Western blot analysis for pro-caspase 3 and cleaved caspase 3. VDAC and GAPDH were used as a loading control. Results are expressed as mean ± SD. Data were analyzed using one-way ANOVA. *, Values of P < 0.05 were considered to be significant. Data are representative of four independent experiments.
The dual function of caspase 8 in initiation of apoptosis and suppression of RIPK3-dependent necrosis (32–34), which has been shown to be required for normal murine embryonic development and survival (35) is well documented. Active caspase 8 blocks necrosis by proteolytically inactivating RIPK1 and RIPK3 (19, 36, 37). Thus we next inhibited caspase 8 activation in H37Ra-infected Mφ using the selective caspase 8 inhibitor z-IETD-FMK and then measured the levels of mitochondria-associated RIPK3. As expected, in Mφ infected with avirulent H37Ra pro-caspase 8 (Fig. 3B) and both apoptosis-executor caspases 9 and 3 were not activated in the presence of z-IETD-FMK (Figure 3D) confirming an essential role for the caspases 8, 9 and 3 in apoptosis of Mtb-infected Mφ. Importantly, in H37Ra-infected Mφ treated with z-IETD-FMK the levels of mitochondria-associated RIPK3, Bcl-xL, and pro-caspase 8 were all increased to the similar levels found in Mφ infected with the virulent strain H37Rv (Figure 3B) indicating that inhibition of caspase 8, 9 and 3 - activation is required for the RIPK3-dependent necrotic pathway to be initiated.
Finally, to test whether RIPK3 is also required for regulation of Bcl-xL-mediated pro-caspase 8-persistence on the mitochondria we silenced the RIPK3 gene in H37Rv-infected Mφ. At 24 h after infection, reduction of mitochondrial Bcl-xL-accumulation correlated with reduced pro-caspase 8-accumulation on the mitochondria (Figure 3E) suggesting that proteolytic processing of pro-caspase 8 initiates apoptosis. Additionally, silencing of the RIPK3 gene leads to caspase 3-activation, which is a key indicator of apoptosis (Figure 3F). In contrast to H37Rv infection, apoptosis was significantly increased in H37Ra-infected Mφ as measured by caspase 8 and 3 activation (Supplementary Figure 2A–C) as well as increased apoptotic surface marker (Supplementary Figure 2D). However, presence or absence of RIPK3 had no significant effect in Mφ infected with H37Ra. Collectively these data indicate that induction of necrosis in Mtb-infected Mφ is critically dependent on blocking activation of pro-caspases 8-, 9- and 3- mediated apoptosis via RIPK3.
To determine whether in H37Ra-infected Mφ caspase 8-activation is part of the intrinsic mitochondria-dependent apoptotic pathway, which includes activation of BID to tBID and depends on BAX (29) we evaluated activation of BID and BAX in Mφ infected with H37Ra or H37Rv. After 10 min of infection with H37Ra pro-caspase 8-activation led to activation of BID on the mitochondria to tBID (Figure 3G, right panel). However, tBID was not detectable on the mitochondria of H37Rv-infected Mφ (Figure 3G, left panel). Most importantly, silencing of the BAX gene in H37Ra-infected Mφ inhibits apoptosis-executor caspase 3 (Figure 3H) indicating that activation of pro-caspase 8 on the mitochondria is initiated by the intrinsic apoptotic pathway via BAX translocation, which is part of the mitochondrial amplification loop of apoptosis and triggers degradation of RIPK3 from the mitochondria.
RIPK3 mediates hexokinase II translocation to the mitochondria via Bcl-xL in H37Rv-infected Mφ
As RIPK3 regulates ROS production following TNF-α treatment and because ROS play an important role in regulation of cell death programs (26), we next studied whether necrosis in Mtb infected Mφ is ROS-dependent. We found that ROS accumulation in Mφ infected with virulent H37Rv is significantly increased in comparison to Mφ infected with the avirulent strain H37Ra (Figure 4A) and that RIPK3 is required to increase ROS accumulation in H37Rv-infected Mφ because silencing of the RIPK3 gene significantly reduced ROS production (Figure 4B). This increased production of ROS was independent of mitochondrial mass as there was no difference in fluorescence emission between the groups using MitoTracker Red FM, which is ROS-insensitive (data not shown). Increased ROS accumulation triggers necrosis in H37Rv-infected Mφ because scavenging of ROS with Tiron significantly reduces necrosis (Figure 4C). As hexokinase II (HKII) is the limiting enzyme of glucose metabolism on the mitochondria and is essential for mitochondrial energy production (25), we next focused on HKII to determine whether it contributes to ROS accumulation. HKII binds to the voltage dependent anion channel (VDAC) in the mitochondrial outer membrane. We first investigated whether HKII is required for ROS accumulation. Kinetic studies of HKII-translocation to the mitochondria indicate that the levels of HKII were significantly increased and maintained on the mitochondria of H37Rv-infected Mφ compared to H37Ra-infected Mφ (Figure 4D). Increased binding of HKII to mitochondria in Mφ infected with virulent H37Rv correlated with enhanced ROS production, which was reduced after HKII gene silencing (Figure 4E). As NADH accumulation is required for ROS production in a HKII-dependent manner (25), NADH levels were diminished after silencing of the HKII gene and the RIPK3 gene (Supplementary Figure 3C and 3D). Further, NADH oxidase inhibitor Diphenyleneiodonium (DPI) abolished ROS production that reduces necrosis in H37Rv infected Mφ (Figure 4F and 4G). Interestingly, in H37Rv-infected Mφ, silencing of the Bcl-xL gene diminished HKII recruitment to the mitochondria at 24h post infection (Figure 4H), while silencing of the RIPK3 gene prevented HKII recruitment to the mitochondria as early as 6h post infection that persisted up to 24h post infection (Figure 4I), which is an indication of a potentially differential role for RIPK3 and Bcl-xL in HKII recruitment. Collectively, these data indicate that in Mφ infected with virulent Mtb, RIPK3 mediates HKII translocation to the mitochondria via Bcl-xL and RIPK3 that leads to increased ROS production.
Figure 4. RIPK3 is required for ROS-dependent necrosis in Mφ infected with virulent Mtb via upregulation of mitochondrial HKII-levels.

(A) Mφ were infected with H37Ra or H37Rv (MOI 5 and 10) and after 48 h ROS accumulation was determined by FACS analysis using the fluorescent dye CM-H2XRos. (B) ROS production was significantly reduced in RIPK3 deficient Mφ infected with H37Rv. Human Mφ were transfected with RIPK3 or scrambled control siRNA infected with H37Rv (MOI 2.5, 5 and 10) and ROS accumulation was determined after 48 h of infection. (C) Scavenging ROS increases Mφ viability following H37Rv infection. Mφ were treated with or without 0.05 mM Tiron and infected with H37Rv (MOI 10). Mφ viability was measured at 0, 48 and 72 h post-infection. Data are represented as mean ± standard error. All experiments were repeated at least three times with similar results. (D) HKII recruitment to the mitochondria in H37Ra and H37Rv infected Mφ. Equal amounts of mitochondrial extracts from H37Ra and H37Rv-infected Mφ were subjected to Western blot analysis of HKII at 0, 2, 6 and 24h post-infection (top). (E) Mφ were silenced with HKII siRNA or treated with scrambled control (Scr) siRNA and then infected with H37Rv (MOI 10:1). Mφ deficient in HKII produced significantly less ROS after H37Rv infection. After 48 h ROS accumulation was determined by FACS analysis using the fluorescent dye CM-H2XRos. (F–G) Mφ were treated with NADH oxidase inhibitor (DPI) for 2 h prior to infection with H37Rv (MOI 10) and ROS accumulation and viability was determined after 48 h and 72h. (H) Silencing of the Bcl-xL gene in H37Rv-infected Mφ prevents HKII recruitment to the mitochondria at 24 h after infection; (I) silencing of the RIPK3 gene prevents HKII recruitment to the mitochondria at as early as 2 h post-infection and at 24 h after infection. Mitochondria were isolated from H37Rv-infected Mφ treated with RIPK3, or scrambled control siRNA and were then subjected to Western blot analysis for HKII. MOI’s for all experiments were 10:1. VDAC was used as a loading control. Results are expressed as mean ± SE. using the nonparametric Student t test. *, Values of P < 0.05 were considered to be significant. Data are representative of three independent experiments.
RIPK3 mediates MPT in Mφ infected with H37Rv
Increased mitochondrial permeability transition (MPT), a sign of mitochondrial damage, induces pyridine nucleotide release from the mitochondria (38), which causes decreases in mitochondrial respiration and in cellular ATP-levels and leads to necrotic cell death (39). MPT increases electron leakage predominantly from mitochondrial complex I and III and enhances ROS production in the presence of sufficient NADH (39). We have previously shown that virulent Mtb induce MPT-dependent necrosis and that cyclosporin A (CsA), a selective inhibitor of the peptidyl-prolyl cis-trans isomerase activity of cyclophilin D (CypD), blocks MPT mediated necrosis (10, 11, 40). The critical role of CypD in necrosis was demonstrated in Ppif null mice, which lack the gene encoding CypD, as these mice were protected against necrosis (15,16). Thus we were wondering whether there is a link between RIPK3, CypD, and MPT in Mtb-infected Mφ. We first showed that silencing of the RIPK3 gene blocks H37Rv-induced MPT in Mφ after 48 and 72h post-infection (Figure 5A). As we showed that induction of MPT by RIPK3 is required for initiation of necrosis, we next sought to determine how virulent Mtb initiates MPT. In H37Rv-infected Mφ treatment with CsA inhibits ROS accumulation (Figure 5B) and necrosis (Figure 5C). CsA inhibits the interaction of CypD with ANT in the inner mitochondrial membrane, which correlates with a block of MPT by closing the permeability transition pore (PTP) (41). We also found that in CsA-treated Mφ infected with H37Rv, the levels of mitochondrial RIPK3, pro-caspase 8, Bcl-xL, and HKII were diminished (Figure 5D). These data correlate with a lack of CypD-binding to ANT in the mitochondria of H37Ra-infected Mφ in comparison to H37Rv-infected Mφ (Figure 5E). Moreover, immunoprecipitation of ANT from the mitochondria of H37Rv infected Mφ treated with CsA showed that CypD binding to ANT was significantly decreased in comparison to mitochondria of H37Rv infected Mφ in absence of CsA (Figure 5F). These data suggest that RIPK3 acts as a regulator of CypD function to induce MPT leading to inhibition of ATP generation and contributing to necrosis. To test whether RIPK3 causes MPT by initiating CypD - binding to ANT and MPT- pore opening, we silenced RIPK3 in H37Rv-infected Mφ and assessed the interaction of CypD with ANT. In the absence of RIPK3 in H37Rv-infected Mφ the CypD/ANT interaction was diminished (Figure 5G), which correlated with the reduction of MPT (Figure 5A).
Figure 5. RIPK3 induces CypD-dependent MPT in H37Rv-infected Mφ.

(A) RIPK3 is required for MPT in H37Rv infected Mφ. Mφ were transfected with RIPK3 or scrambled (Scr) control siRNA and were then infected with H37Rv (MOI 10). Cationic dye (DiOC6(3)) release from mitochondria (a measure for MPT) was measured at 48 and 72 h after infection. (B and C) Inhibition of CypD reduces ROS-dependent necrosis. (B) Equal numbers of CsA-treated (5 μM) and untreated Mφ were infected with H37Rv (MOI 5 or 10). After 48 h of infection ROS accumulation (B) was measured by FACS analysis using the fluorescent dye CM-H2XRos and cell viability (C) was determined using the Live-Dead Assay. (D) CypD inactivation reduces translocation of RIPK3, pro-caspase 8, Bcl-xL and HKII to the mitochondria in H37Rv-infected Mφ at 24 h. Equal amounts of mitochondria from H37Rv-infected Mφ treated with or without CsA (5 μM) were subjected to Western blot analysis for RIPK3, pro-caspase 8, Bcl-xL and HKII after 24 h of infection. (E) Mitochondria from H37Ra or H37Rv infected Mφ were subjected to IP with anti-ANT ab and were then subjected to Western blot analysis for CypD. (F) CypD - ANT interaction is augmented in H37Rv-infected Mφ and is inhibited by inactivation of CypD with CsA (5 μM). (G) Top panel: RIPK3 is required for CypD - ANT interaction on the mitochondria. After 24h of infection, mitochondria from H37Rv-infected Mφ transfected with RIPK3 or scrambled (Scr) control siRNA were subjected to IP with anti-ANT ab and were then analyzed by Western blot for CypD. ANT was used as a loading control. Bottom panel: Silencing efficiency of RIPK3 siRNA. VDAC was used as a loading control. Results are expressed as mean ± SE, using nonparametric Student t test. *, Values of P < 0.05 were considered to be significant. Data are representative of three independent experiments.
Thus RIPK3 induces necrosis in H37Rv-infected Mφ, by at least two independent, but interrelated mechanisms: 1) RIPK3 increases Bcl-xL-dependent HKII recruitment to the mitochondria possibly leading to enhanced glucose consumption and increased NADH accumulation and increase of ROS generation; and 2) RIPK3 induces MPT in a CypD dependent manner, which in presence of abundant NADH boosts accumulation of ROS by uncoupling of the mitochondrial electron chain.
RIPK3 deficient Mφ mediate resistance to pulmonary Mtb infection
To translate our in vitro observation to in vivo, we have utilized two in vivo models of Mtb infection. In the first model, mice were intravascularly infected with the high dose of H37Rv (1×106 CFU) and after 4 weeks of infection, there was a significant reduction in the pulmonary bacterial burden of RIPK3−/− compared to WT mice (Figure 6A). Interestingly, the number of neutrophils (cellular marker of necrotic lung tissues) was significantly reduced in Mtb infected RIPK3−/− mice (Figure 6B and Supplementary Figure 4). However, with the exception of TNFα, which was significantly reduced in RIPK3−/− mice, the expression levels of other cytokines, including IL-1β, IL-6, and IL12 were not markedly changed (Figure 6C). We next infected WT and RIPK3−/− mice with a low dose (~50 CFU) of the virulent strain H37Rv. After 5 weeks of infection, RIPK3−/− mice had significantly lower lung bacterial burden (~half log) in comparison to WT mice (Figure 7A). Considering the important role of T cells in protection against Mtb infection, we next examined the potential contribution of T cell mediated immunity in protection of RIPK3−/− mice. Similar to other studies (20, 42), we also found that the presence or absence of RIPK3 had no effect on T cell proliferation in vitro (Supplementary Figure 5A). At 5 weeks post-Mtb infection there was no significant difference in frequency of pulmonary CD4+/CD8+ T cells (Figure 7B) as well as TB10.4-specific CD8+ T cells (Figure 7C) or ESAT6-specific CD4+ T cells between WT and RIPK3−/− mice (Figure 7D). Moreover, there was no difference in total cell numbers of T cells or antigen-specific T cells (Supplementary Figure 5B–C). Furthermore, ELISPOT experiments revealed that the function of T cells was intact as they produced similar levels of IFN-γ following stimulation with Mtb-antigens recognized by either CD4+ or CD8+ T cells (Supplementary Figure 5D). Thus these data indicate that the function of T cells was not affected by RIPK3 and the increased protection of RIPK3−/− mice after Mtb infection was independent of T cell mediated immunity.
Figure 6. Reduced bacterial burden in the lungs of RIPK3−/− mice infected with high dose of Mtb.
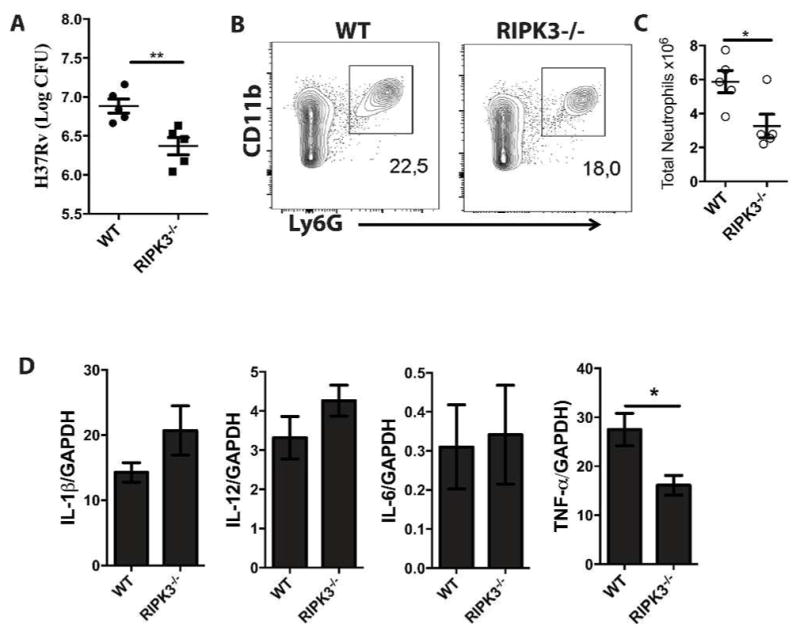
(A) RIPK3−/− mice were infected with Mtb (1×106 CFU) through intravenous injection. After 4 weeks of infection, mice were sacrificed to quantify (A) bacterial load in the lungs. (B) Lung cells were immunophenotyped to assess recruitment of neutrophils by gating CD11b+Ly6G+ cells. (C) Total number of neutrophils in the lungs of infected mice. (D) After 4 weeks of infection, the level of expression of cytokines in the lungs of infected animals (IL-6, TNF-α, IL-12, IL-1β was determined at mRNA level using qPCR. Results are expressed as mean ± SE, using nonparametric Student t test. *,** Values of P < 0.05 and P<0.01 were considered to be significant, respectively.
Figure 7. RIPK3-deficient Mφ mediate host resistance to pulmonary Mtb infection.

(A) Bacterial burden in the lung after 5 weeks of aerosolized Mtb infection (H37Rv, 50–100 CFU) was significantly lower in RIPK3−/− mice compared to WT mice. The data were pooled from two independent experiments. (B) After 5 weeks of Mtb infection the frequency of CD3+CD4+ T cells (Left panel) and CD3+CD8+ T cells (Right panel) was determined in the lungs. (C-D) Representative flow cytometry plots of (C) TB10.44–11 MHC class I tetramer staining of CD8+ T cells (D) IAbESAT61–20 MHC class II tetramer staining of CD4+ T cells and in the lungs 5 weeks post-Mtb infection. (E) The anti-necrotic properties of Mtb-infected RIPK3−/− Mφ reflect the innate control of infection in vivo. Bacterial burden in the lung 4 weeks after intratracheal (i.t.) transfer of H37Rv-infected RIPK3−/− or WT Mφ into Rag1−/− mice. Results are expressed as mean ± SE. Data were analyzed using nonparametric Student t test. *, Values of P < 0.05 were considered to be significant.
To directly evaluate the protective role of RIPK3 in Mφ innate immunity in the absence of any contribution from adaptive immune responses, we utilized our established model of the adoptive transfer of Mtb-infected Mφ (3, 5, 43). We infected WT and RIPK3−/− murine alveolar Mφ in vitro with a low dose of the virulent strain H37Rv (MOI~1), then transferred the cells by the intratracheal route into recipient mice deficient in recombinase-activating gene 1 (Rag1−/−) mice. The number of bacteria in WT and RIPK3−/− alveolar Mφ was identical prior to transfer (Supplementary Figure 5E). However, four weeks after adoptive transfer, the pulmonary bacterial burden was significantly lower in Rag1−/− mice that received Mtb-infected RIPK3−/− Mφ than in the recipients of Mtb-infected WT Mφ (Figure 7E). This experiment demonstrates that transfer of infected RIPK3−/− Mφ, which are resistant to necrosis restrict bacterial replication independent of adaptive immunity in vivo. Thus RIPK3 plays an important role in early Mφ immunity and protection against Mtb infection.
DISCUSSION
Mtb hijacks Mφ cell death pathways to maintain an environment for propagation within the host. We and others have previously demonstrated that Mtb actively inhibits apoptosis, which plays a protective role in both innate and adaptive immunity to Mtb infection (44). By inducing necrosis, virulent Mtb is able to exit the Mφ and therefore escape the Mφ’s anti-mycobacterial defense mechanisms (1). Although we have shown that Mtb induces necrosis by targeting the mitochondrial inner membrane (10, 11, 40) and the plasma membrane repair mechanisms (3), the molecular mechanisms involved in necrosis remain largely unknown. Here we have extended these studies and show that Bcl-xL and RIPK3 are involved in triggering Mφ programmed necrosis during infection with virulent Mtb.
Recently a novel form of necrosis, necroptosis, was described, which depends on the action of a cytosolic complex containing RIPK1 and RIPK3. Formation of this complex is inhibited by the RIPK1 inhibitor necrostatin-1 (17). However, similar to another infection model (45), we found no effect of RIPK1 on necrosis either by using necrostatin-1 or silencing RIPK1 (data not shown). As necroptosis seems to be independent of the mitochondria and is a rapid early event (46) the mechanisms involved in the Mtb-induced necrosis, which develops slowly and reaches its maximum between 72 – 96 h after infection, might be different from classical necroptosis. Interestingly, a recent study suggests that induction of necroptosis by RIPK3 requires inactivation of caspase-8 (47). Similarly, our findings indicate that caspase 8 determines the fate of Mφ infected with virulent Mtb. Following infection with Mtb RIPK3 translocates from the cytosol to the mitochondria of Mφ. Bcl-xL, an inhibitor of BAK (27, 28) leading to a block of caspase 8 activation and to apoptosis (48) protects RIPK3 from caspase 8-dependent proteolysis (19). We also found that mitochondrial Bcl-xL protects RIPK3 and pro-caspase 8 on the mitochondria and prevents caspase 8 activation inducing necrosis in Mtb-infected Mφ. On the other hand in human lymphoblastoid cell lines caspase 8 was shown to be associated with cardiolipin rafts on the mitochondria located at contact points of the inner and outer mitochondrial membranes leading to its activation. Cardiolipin was found to be responsible for the translocation of caspase 8 to the mitochondria and for caspase 8 activation following death receptor stimulation (49). Targeting of tBid to the mitochondria and mobilization of cytochrome c are also cardiolipin dependent indicating their importance in the transduction of the apoptotic signaling cascades (50). Although binding of Bcl-xL to caspase 8 was reported earlier (48), we failed to find any binding of Bcl-xL to pro-caspase 8 (data not shown).
Consistent with these studies we found that in Mφ infected with an avirulent strain of Mtb, H37Ra, pro-caspase 8 is proteolytically processed, which not only correlates with activation of the effector caspases 9 and 3 leading to apoptosis, but also with preventing necrosis by inactivation of RIPK3. We currently do not know the exact mechanism of mitochondrial caspase 8 activation in Mφ infected with an avirulent strain of Mtb. Interestingly, Niture and colleagues (51) have reported that the levels of Bcl-xL on the mitochondria are regulated by a member of the phosphoglycerate mutase family 5 (PGAM5), which forms a bridge between Bcl-xL and Kelch-like ECH-associated protein-1 (a sensor of chemical and radiation-induced stress) and targets Bcl-xL degradation by activating an E3 ubiquitin ligase. The involvement of this or additional mechanisms in Bcl-xL-degradation or the role of signaling platforms such as cardiolipin that promotes mitochondria-dependent caspase 8 activation remains to be determined. Additionally, it is currently unclear whether Bcl-xL is the only anti-apoptotic component on the mitochondria involved in this mechanism. It is likely that other Bcl-2 family members including Mcl-1 and Bcl-2 (52, 53) may also contribute to RIPK3 mediated caspase 8 - inactivation in mitochondria leading to the induction of necrosis.
The mitochondria are a major source of energy and ROS production, and disruption of mitochondrial function significantly contributes to ROS generation as well as to cell death. For example, blocking ROS accumulation in L929 cells prevents necrosis (26). Our results indicate that in Mφ infected with virulent Mtb, increased ROS accumulation contributing to induction of necrosis. Virulent Mtb induces necrosis by several mechanisms including inhibition of ATP production, e.g. due to blocking of mitochondrial ADP transport by ANT (54), sustained JNK activation (55) and release of toxic proteins from the permeable mitochondria (56).
In cells with increased glucose consumption, NADH levels are drastically increased, which can result in increased accumulation of ROS (39). Our results indicate that in H37Rv-infected Mφ increased ROS accumulation requires activation of two independent mechanisms associated with mitochondria. First, the generation of sufficient amounts of electron donors and increased HKII levels on the mitochondria lead to enhanced production of glucose-6-phosphate, the rate-limiting step of glycolysis (25). HKII is predominantly associated with VDAC on the mitochondria and couples oxidative phosphorylation to glycolysis essential for ROS production. Thus reduction of mitochondrial HKII down-regulates glycolysis as well as ROS generation. Interestingly, similar to H37Rv-infected Mφ, HKII is highly up-regulated on the mitochondria of many tumors, which leads to increased aerobic glycolysis known as the Warburg effect (25). Second, RIPK3 induces MPT, which interrupts the electron chain in mitochondrial complex I leading to electron-leakage and allows molecules with a size up to ~1.5 kDa to exit the mitochondrial matrix. MPT is part of a cellular suicide mechanism and its contribution to pathophysiology of several diseases including heart injury (14) is well documented. Although it was proposed that induction of MPT is a mechanism, by which aging mitochondria are earmarked for removal, there is no evidence of this mechanism in studies using CypD knockout mice (15). Thus the role of MPT under physiological conditions is still unclear. In contrast, under certain pathological conditions including Mφ infection with virulent Mtb, MPT triggers induction of necrosis, which is essential for the release of the bacilli from the host cell ensuring the vicious cycle of infection. Here we have identified two different complementary mechanisms, MPT and HKII accumulation, which act in concert to increase ROS accumulation triggering a necrotic cell death program in Mtb-infected Mφ (Figure 8).
Figure 8. Model of RIPK3-dependent programmed necrosis and caspase 8-dependent apoptosis in Mφ infected with Mtb.

In Mφ infected with virulent Mtb RIPK3 and pro-caspase 8 present in the cytosol translocate to the mitochondria in presence of Bcl-xL and RIPK3 is activated RIPK3 enhances binding of HKII to VDAC on the outer mitochondrial membrane controlling mitochondrial glycolysis. At the same time activated RIPK3 triggers CypD-dependent formation of the mitochondrial permeability transition (MPT) pore via interaction between ANT and VDAC leading to leakage of the electron chain. Both mechanisms seem to be required for increasing ROS-dependent necrosis (right). In Mφ infected with avirulent Mtb the RIPK3 and caspase 8 also translocate to the mitochondria but this step is quickly followed by activation of caspase 8 and degradation of RIPK3. Oligomerization of BAX and BAK, which in turn allows the release of pro-apoptotic molecules (e.g. cytochrome c) leads to apoptosis (left). The exact action mechanism of Bcl-xL function is unknown.
How does RIPK3 induce MPT? Increased HKII binding to VDAC on the outer mitochondrial membrane induced by virulent Mtb not only increases generation of glucose-6-phosphate but also enhances CypD binding to the ANT (57) in the inner mitochondrial membrane, which opens the permeability transition pore causing MPT. The proline isomerase CypD, a target for CsA, is essential for induction of MPT (15). CypD keeps the MPT pore in the mitochondrial inner membrane in the “open” position by binding to ANT (58). We confirm here CypD-binding to ANT in virulent Mtb-infected-Mφ undergoing MPT and demonstrate that RIPK3 is required for binding of CypD to ANT to initiate ROS mediated necrosis. Considering that several components of MPT are kinase substrates (59) it is tempting to speculate that RIPK3 may phosphorylate some components involved in MPT to disrupt their functions generating increased levels of ROS.
RIPK3−/− mice were more resistant than WT mice in both the low-dose aerosol and high-dose intravascular models of Mtb infections. In addition, we observed significant decrease in the total number of neutrophils in the lungs of Mtb infected RIPK3−/− mice. Reduced number of neutrophils further support the notion of less necrosis in RIPK3−/− Mtb infected animals. However, this protection was mainly dependent on Mφ immunity, considering that there were no qualitative or quantitative differences in T cell mediated immunity between Mtb-infected RIPK3−/− or WT mice. Additionally, Mφ transfer experiments directly demonstrate that the fate of Mtb-infected Mφ was the key determinant of the relative resistance of these mice. Interestingly, a recent study by Albert’s group has elegantly shown that RIPK3-mediated necroptosis is more effective in cross-priming T cells than apoptosis (20). However, we speculate that during Mtb infection the host potentially may compensate for the lack of necrosis-dependent T cell mediated immunity via apoptosis to maintain an adequate T cell response to Mtb infection (5–7). The links between apoptotic/necrotic Mφ, DC, and T cell immunity in the control of Mtb infection certainly requires further investigation.
The dramatic differences in behavior of avirulent versus virulent strains of Mtb in induction of apoptosis and necrosis, respectively, suggest the involvement of a specific pathogen-encoded inducer of RIPK3-dependent necrosis, which needs to be elucidated. Several virulent strains of Mtb encode specific genes, such as the nuoG gene, which actively inhibits the apoptotic death pathway in Mφ (60). The nuoG gene of Mtb encodes the type I NADH dehydrogenase of Mtb, which neutralizes NOX-2 dependent ROS production in the Mφ required for induction of apoptosis (61). It will be interesting to know whether the nuoG gene or other genes are involved in RIPK3-dependent necrosis.
There are several differences between our study and other previous published data using various cell lines including HeLa, 293A, L929, and 3T3-cells (26, 46, 62, 63). We envision that these differences are mainly due to using cell lines and soluble ligands versus primary Mφ and live-replicating bacteria. For instance, addition of caspase inhibitors to L929 cells induces necrosis due to autocrine production of TNF in the absence of active pro-apoptotic caspases (35). In contrast, addition of caspase inhibitors to primary human Mφ in the absence of Mtb infection does not induce necrosis as autocrine TNF-production is minimal indicating that the pathways leading to necrosis in certain cell lines and in primary Mφ are significantly different.
Despite the world - wide application of BCG vaccination and other anti-Mtb interventions, Mtb remains one of the most successful human pathogens. More than 2 billion persons carry latent tuberculosis infection, and approximately 20 million persons have currently active tuberculosis, of which 2 million people die every year. (64). The success of this pathogen is closely linked to its ability to alter the intracellular environment of Mφ including induction of cell death. Thus dissecting the molecular mechanisms involving mitochondria as entities, which determine whether Mtb-infected Mφ undergo apoptosis or necrosis is of significant importance in identifying the outcome of host immunity to Mtb infection and might pave the road to inhibiting the necrotic death program as a novel therapy.
MATERIALS AND METHODS
Materials
Anti-caspase 8 (551242), and anti-RIP1 (551042) antibodies were from BD Biosciences. Anti-caspase 8 (52183, for immunofluorescence), anti-RIPK3 (56164), and anti-adenine nucleotide translocase (ANT, 109864) antibodies were from Abcam. Anti-CypD (AP1035) antibody was from Calbiochem. Anti-hexokinase II (2106), anti-BAX (2772), anti-Bcl-xL (2762), anti-caspase 3 (9662), anti-caspase 9 (9502), anti-BID (2002), anti-GAPDH (2118), and anti-caspase 8 (Asp387, 14071, for flowcytometry) were from Cell Signaling Technology. Anti-VDAC (SP5361P) antibody was from Acris Antibodies. Active caspase 3 for flowcytometry was from BD. Anti-human mitochondria antibody (HMS-0100) was from Immunovision. Dylight 650-conjugated Donkey anti-rabbit IgG antibody and Alexa fluro 488-conjugated Donkey anti-human IgG antibody were from Jackson Immuno Research Laboratory. The selective caspase 8 inhibitor z-IETD-FMK (FMK007) was from R&D Systems. Gammabind protein G sepharose (17-0885-01) was from GE Health Care. Cyclosporin A (30024), Tiron (89460) and Protease Inhibitor Cocktail (P2714) from Calbiochem and Phosphatase Inhibitor Cocktail 1 (P2850) was from SIGMA-ALDRICH. Mitochondria/Cytosol Fractionation Kit (ThermoFisher Scientific Mitochondrial Isolation kit #89874) and MitoTracker Red CMXRos (M7512), MitoTracker Red CM-H2XRos (M7513), and LIVE/DEAD Fixable Dead Cell Stain Kits (MP34955) from Invitrogen.
In vivo M. tuberculosis infection
Six to ten week old C57BL/6 or Rag1−/− mice were obtained from Jackson Labs (Bar Harbor, ME); RIPK3−/− mice were obtained from Genentech. Mice were infected with virulent M. tuberculosis (H37Rv) via the aerosol route using a nose-only exposure unit and received approximately 50–100 CFU/mouse (65) or intratracheally (0.5 × 106 CFU) or intravenously (1×106 CFU)3. Mice were euthanized by CO2 inhalation and the lung, spleen, and lymph node were aseptically removed, minced, and pressed through 70-μl cell strainer followed by red blood cell lysis to obtain single cell suspension. Bronchoalveolar lavage was performed with PBS in mice infected intratracheally (3) (8).
Adoptive transfer models of infection
Alveolar Mφ were collected from the BAL of naive WT and RIPK3−/− mice and were immediately suspended at a concentration 0.5 × 106 cells/500 μl RPMI medium with 10% FCS and infected in suspension using virulent H37Rv (MOI ~ 1) for 30 minutes. Free bacteria were then removed by 6 washes with cold PBS, each time followed by centrifugation for 10 min at 1000 RPMI at 4°C. Cells were resuspended in PBS at 0.5 × 106/40 μl and transferred by the intratracheal route into naive C57BL/6 mice (3) (5).
CFU determination
Following infection, mice were euthanized and their lungs removed and individually homogenized in 0.9% NaCl-0.02% Tween 80 with a Mini-Bead-Beater-8 (BioSpec Products). Viable bacteria were enumerated by plating 10-fold serial dilutions of organ homogenates onto 7H11 agar plates (Remel). Colonies were counted after 3 weeks of incubation at 37°C.
Bacteria
The virulent Mtb strain H37Rv and attenuated stain H37Ra (American Type Culture) were grown in Middlebrook 7h9 broth (BD Biosiences) with BBL Middlebrook OADC Enrichment (Becton Dickson) and 0.05% (vol/vol) Tween 80 (Difco). Aliquots were distributed into small tubes and stored at −80 °C. 100μl of the aliquot was serially diluted 10-fold with 0.02% Tween 80 in PBS and plated on 7H11 agar plates (Thermo Fisher Scientific Remel Products, Lenexa, KS, USA). The bacterial concentration of each batch was determined by colony counting after 4-weeks.
Cells and cell culture
Human mononuclear leukocytes from buffy coat preparations (Research Blood Component, Boston) were cultured for 7 days in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% (vol/vol) human AB serum (Gemini bio-Products) at a density of 2.0×106 cells per ml in six-well plates and 2.0×107 cells per ml in 10 cm plate, and then were changed to IMDM with 2% (vol/vol) human AB serum 1 day before challenged with varying MOI of Mtb. Bone marrow from 8- to 10-week-old mice was harvested from femurs and differentiated into Mφ for 7 days in RPMI-1640 (Invitrogen) supplemented with 10% L929 cell (ATCC)-conditioned medium, 10% FBS, 2 mM L-glutamine and 1 mM sodium pyruvate, 1% essential and non-essential amino acids (Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin.
In vitro infection
Adherent human Mφ were infected with H37Rv or H37Ra at different MOIs. After 4h the Mφ were washed 3 times with HBSS to remove non-adherent bacilli and replaced with fresh IMDM with 2% AB. Adherent murine Mφ were infected with H37Rv at different MOIs. After 4h the Mφ were washed 3 times with warm PBS to remove non-adherent bacilli and replaced with fresh media containing FBS.
FACS analysis
Cells were stained as previously described using antibodies specific for mouse CD3, CD4, CD8, and CD19 (BD Biosciences) Ly6G, Ly6c, CD11b, F4/80, CD11c (ebioscience) (5). Antigen-specific CD4+ and CD8+ T cells were identified using IAbESAT61–20 and H2-KbTB10.44–11 tetramers, respectively (National Institutes of Health Tetramer Core Facility, Emory University Vaccine Centre, Atlanta, GA). Cleaved Caspase 8 Staining was performed in WT and RIPK3−/− Mϕ. Cells were washed, collected and suspended in formaldehyde to obtain a final concentration of 4% for 30 min at RT. Cells were permeabilized by adding ice-cold 100% methanol slowly to pre-chilled cells, while gently vortexing, to a final concentration of 90% methanol by incubating 30 min on ice. Cells were washed by with buffer and PE-conjugated cleaved caspase 8 Ab was added for 1h at RT. Cell were then washed and resuspended in buffer to be analyzed by flow cytometer. Flow cytometry was performed by using a BD LSR-II (BD Biosciences) and FlowJo analysis software (Tree Star).
In vitro T Cell Proliferation
CD3+ T cells were purified from spleens of naive WT and Annex1−/− mice using MACS column purification procedure by negative selection (Pan T cell kit - MACS Miltylene). The purity of CD3+ T cells was usually between 92–97%. Purified CD8+ T cells were stained with 5 M CFSE in PBS containing 0.1% BSA at room temperature. Staining was stopped after 8 min of incubation by addition of RPMI medium containing 10% FBS. T cells were then cultured with different concentration of anti-CD3 and anti-CD28. After 3 days, T cell proliferation was measured by reduction in CFSE expression (CFSElow) using flow cytometry.
ELISPOT assay
The IFN-γ Elispot assay was conducted according to the manufacturer’s instructions (R&D Sytems). Briefly, isolated cells were seeded into a 96-well plate pre-coated with mouse IFN-γ capturing Abs. Cells were then incubated for 24 h with or without antigenic stimulation. The plate was developed and the spots were enumerated using a series A immunospot plate reader, Image Acquisition version 4.0, and Immunospot version 3.2 analysis software (Cellular Technology).
Isolation of mitochondria from Mφ
Mitochondria were isolated using the Mitochondria/Cytosol Fractionation Kit (ThermoFisher Scientific Mitochondrial Isolation kit #89874) according to the manufacturer’s recommendations. Briefly, cells were washed twice with ice-cold PBS and then 1ml of Isotonic Mitochondrial Buffer (containing protease inhibitors) was added. Cells were homogenized on ice by passing through a 25-gauge needle (Becton Dickinson) after detachment from the culture dish by using a cell scraper. The lysate was centrifuged at 600 × g for 10 min at 4 °C to remove the nuclei, debris and intact cells. The supernatants were centrifuged at 10,000 × g for 30 min at 4 °C. The enriched mitochondrial fraction was washed and lysed in 100 μl of a buffer containing Tris-HCl (50mM, pH7.4), NaCl (150 mM), EDTA (5 mM), Triton X-100 (1%), sodium ortho-vanadate (0.2 mM) and protease inhibitor cocktail (Sigma) at 4°C and stored at − 80 °C together with the cytosolic fraction. The purity of the mitochondria was determined by taking 10 μg of protein from the mitochondrial and cytosolic fractions and measuring Bcl-xL and GAPDH levels by Western blotting.
Silencing of the genes encoding human RIPK3, Bcl-xL, hexokinase II, MLKL and BAX
The target siRNA sequences for human RIPK3 specific siRNA (5′-GGAAUGCCUACCAAAAACU-3′), the Bcl-xL siRNA (5′-GGAGAUGCAGGUAUUGGUG-3′)(66), and for hexokinase II (5′-CACGAUGAAAUUGAACCUGGU-3′) (67), MLKL siRNA is a pool of 3 different duplexes (5′-GGAAUACCGUUUCAGAUGUtt-3′, 3′-ACAUCUGAAACGGUAUUCCtt-5′; 5′-GGAAUAGUGAGGUUCACUUtt-3′, 3′-AAGUGAACCUCACUAUUCCtt-5′; 5′-GAAGGCUGUGAUUCUAAGAtt-3′, 3′-UCUUAGAAUCACAGCCUUCtt-5′) and RIPK3 second siRNA pool (5′-CUGAGUGGCUAAACAAACUtt-3′, 3′-AGUUUGUUUAGCCACUCAtt-5′; 5′-GGAGACAACAACUACUUGtt-3′, 3′-UCAAGUAGUUGUUGUCUCCtt-5′; 5′-CUCCAAGAGUUACGAGUUtt-3′, 3′-UAACUCGUAACUCUUGGAGtt-5′ were generated by IDT. Human BAX-specific siRNA (#6321) was from Cell Signaling Technology. Mφ were cultured in IMDM with 10% (vol/vol) human AB serum and in IMDM with 2% human AB serum 1 d before transfection. All siRNA was transfected at 37 °C into the cells at a final concentration of 50 nM with Lipofectamine RNAiMAX (13778-150, Invitrogen). Mφ were infected with Mtb 48–72 h after transfection.
Immunoblot analysis
Protein was measured with the Bradford assay. Samples (5 μg) were fractionated by SDS-PAGE and protein was transferred to a PVDF membrane at 4 °C. The membrane was blocked with 5 % dried milk in Tris HCl buffer (10 mM), pH 8.0 containing NaCl (150 mM) and Tween 20 (0.5%) and was incubated with the appropriate primary antibodies overnight at 4 °C. After three washes blots were incubated with the corresponding HRP-conjugated secondary antibodies for 1 h at room temperature. Enhanced chemiluminescence reagent was added and the blots were exposed to Denville Hyblot CL film. Western input of Mφ infected with H37Rv for RIPK3, RIPK1, procaspase8, and loading control (actin and VDAC) is incorporated in supplementary (Supplementary Figure 3A).
Co-immunoprecipitation
After centrifugation at 18 000 × g for 30 min the Mφ lysates were immunoprecipitated overnight with immobilized monoclonal antibody under gentle rotation at 4 °C. 50 μl of protein G beads - slurry (50%) was added to the sample and the mixture incubated for another 1 h at 4 °C. To remove non-specific bound material the beads were washed with lysis buffer 3 times. The beads were then gently mixed with lysis buffer, centrifuged at 4 °C and the supernatant removed. Twenty μl of SDS-sample buffer was added to the samples and the samples were then loaded on SDS-PAGE gels after boiling.
Assessment of MPT
MPT was measured as described (11). Briefly, human Mφ were cultured at a density of 1.5 × 106 mononuclear cells per well with 2 ml medium containing IMDM with 2% AB serum in 6-well cluster plates (Corning Glass). After 7 days cells were pre-loaded with 1.5 nM DiOC6(3) in IMDM for 20 min at 37 °C, washed, and incubated at 37°C for 10 min in medium containing 15 μg/ml digitonin, washed, and fixed with 1% paraformaldehyde for 1 h at r t. Mφ were then dislodged with a rubber policeman, washed with PBS and resuspended in PBS with 1% (wt/vol) BSA. A FACSort flow cytometer (BD Biosciences) was used for flow cytometry.
Assay for apoptosis
Apoptosis was measured by determining caspase 3-activation using Western blotting and flow cytometry because the caspases 3/10 are the direct executioners of pro-apoptotic pathways. Apoptosis was also determined by flow cytometry with Annexin V staining. Briefly, RIPK3−/− Mϕ were infected with H37Ra (MOI 5) for 4h followed by washing and incubation for 96h in BMDM media. Cells were harvested and incubated with Annexin V dye in buffer recommended by manufacturer for 30 min. Mϕ were resuspended with 7AAD for dead cell staining and acquired using flowcytometer within 1h.
Assay for necrosis
Mφ necrosis was evaluated in vitro and in vivo by flow cytometry with the LIVE/DEAD fixable Dead Cell Stain kit (Invitrogen, L34955) according to the instructions of the manufacturer. Briefly, adherent infected and uninfected human Mφ (1.5 × 105 per well) were washed, 0.5 ml of 1% of reconstituted fixable violet fluorescent reactive dye was added and the mixture incubated at room temperature for 30 min in dark. Mφ were washed and fixed with 3.7% paraformaldehyde for 1 h at room temperature. Mφ were dislodged with a rubber policeman, washed and resuspended in PBS with 1% BSA. A FACSort flow cytometer was used for flow cytometry. In some experiments necrosis was measured with cell death detection ELISAPLUS photometric enzyme immunoassay (11 920685 001; Roche Applied Science).
ROS assay
ROS accumulation was measured by using reduced rosamine-Mitotracker probes (Invitrogen, M7513). Adherent Mφ were washed with IMDM medium, stained with 200 nM Mitotracker Red CM-H2XRos in IMDM without serum at 37° C for 30 min. Then the medium was removed, and the cells were washed and fixed with 3.7% paraformaldehyde for 1 hour at room temperature. After fixation, Mφ were washed and re-suspended in 0.5 ml PBS with 1% BSA. Fluorescence intensity of ROS was measured using a flow cytometer. To control for possible changes in the mitochondrial mass under the different conditions the cells were tested with the ROS-insensitive mitotracker dye red FM (Invitrogen). No differences in in fluorescence emission were seen indicating that the mitochondrial mass was not different in the Mφ subjected to the different experimental conditions.
NADH determination
NADH in Mφ extracts was measured with the NAD+/NADH Quantification Colorimetric Kit from BioVision according to the recommendation of the manufacturer.
Immunostaining and confocal microscopy
Mφ were cultured on poly-D lysine-coated cover slips in 24-well plates at a density of 1.5 × 105 cells/well and were infected for 24 h with mCherry labeled H37Rv. Mφ were then fixed for 1 h with 4% paraformaldehyde and blocked over night at 4 °C with PBS containing 10% horse serum. Cover slips were incubated for 2h at room temperature with anti-mitochondria, anti-caspase 8 or anti-RIPK3 (dilution, 1:100) ab. Mφ were then washed and stained for 1h at room temperature with fluorescent secondary ab. Mφ were mounted and then subjected to confocal and transmission light microscopy. Microscope images were acquired at the Brigham and Women’s Confocal Core Facility with a Nikon TE2000-U inverted microscope, Nikon C1 Plus confocal system, 60 × Nikon Plan Apochromat objective, 10-mW Spectra Physics 488-nm argon laser, Melles Griot Red HeNe 543-nm laser, Chroma 515-nm/30-nm and 543-nm emission filters and a 30-μm pinhole. Images were acquired under identical exposure conditions and micrographs were compiled and analyzed with Nikon EZ-C1 v3.8 and Adobe Photoshop v10.0.1. Colocalization of RIPK3 and pro-caspase 8 on the mitochondria was quantified using Metamorph. Each channel (R, G, B) was considered separately with each pixel assigned an intensity value (0–255) with care taken to assure that no pixel was oversaturated in any image used for quantification. The lower threshold is set whereby areas of the image without staining are excluded. The software then calculates that area of caspase 8 or RIPK3 with mitotracker and reports it in terms of percent positive cells. Quantification was done on 100–200 cells for every condition, and representative images are shown from at least three independent experiments.
Quantitative real time PCR
Total RNA was isolated with Trizol reagent from lungs of Mtb infected animals. RNA was quantified with the help of a NanoDrop spectrophotometer. The A260/A280 ratio of all samples was in the range of 1.90 to 2.00. DNA contamination from RNA samples was removed by amplification grade DNase (Abm according to manufacturer’s instructions). Briefly, RNA samples (1μg) were incubated with DNase (1U) for 15 min in the reaction buffer. After the incubation, DNase activity was terminated by stop solution. Further, the samples were heated to 70°C for 10 min to inactivate DNase activity. Real time PCR was performed using SYBR Green Master Mix Kit. Melting curve analysis was performed to eliminate the possibility of non-specific amplification. Results are represented in the arbitrary units. Analysis was done by comparative Ct method, where Ct values were normalized against the house-keeping control GAPDH.
Statistics
Results are expressed as mean ± SD or SEM. The data were analyzed by using Microsoft Excel Statistical Software (Jandel, San Rafael, CA) using the t test for normally distributed data with equal variances. In some experiments, one-way analysis of variance (ANOVA) with Dunnett’s posttest or with Bonferroni’s posttest was performed using Prism version 5 for Windows (Graph-Pad Software).
Ethics Statement
All animal experiments were conducted in accordance with Canadian Council on Animal Care guidelines. The animal used protocol was approved by the McGill Animal Care Committee (protocol number: 2010–5860).
Supplementary Material
AUTHOR SUMMARY.
Despite the world-wide application of BCG vaccination and other anti–Mycobacterium tuberculosis (Mtb) interventions, Mtb remains one of the most successful human pathogens. Approximately two million people die of tuberculosis annually and eight to ten million new cases of active tuberculosis occur each year due to the large reservoir of asymptomatic people chronically infected with Mtb. The success of this pathogen is closely linked to its ability to alter the intracellular environment of the alveolar macrophage (Mφ) including cell death programs. Virulent Mtb escapes the hostile milieu of the host Mφ by inducing necrosis, a type of cell death that favors Mtb survival and facilitates bacillary spread into the surrounding tissue for a new cycle of infection and dissemination. Additionally, virulent Mtb actively inhibits apoptosis, an alternate form of Mφ death beneficial to the host, by killing intracellular bacilli and enhancing adaptive immunity. In the current study we show that virulent Mtb induces necrosis by involving receptor interacting protein kinase 3 (RIPK3) and the Bcl-family member Bcl-xL, which prevents caspase 8 mediated apoptosis. We furthermore demonstrate that inhibition of RIPK3 activity significantly enhances immunity to Mtb infection. Thus dissecting the molecular mechanisms involving Mtb-infected Mφ death modality is of significant importance in identifying the outcome of host immunity to Mtb infection and may lead to the development of novel therapies for this devastating disease.
Acknowledgments
We are grateful to V. W. Hsu, E. Remold-O’Donnell and C. Geadas for helpful discussions and Nancy Kedersha for supplying anti-mitochondrial antibody. This study was supported by the US National Institute of Health Grant AI50216 to H.G.R. and the Canadian Institute of Health Research (CIHR) Foundation Grant (FDN-333189) to M.D.
References
- 1.Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8(9):668–74. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205(12):2791–801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10(8):899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. 2012;12(3):289–300. doi: 10.1016/j.chom.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 2010;11(8):751–8. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest. 2007;117(8):2279–88. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winau F, Kaufmann SH, Schaible UE. Apoptosis paves the detour path for CD8 T cell activation against intracellular bacteria. Cell Microbiol. 2004;6(7):599–607. doi: 10.1111/j.1462-5822.2004.00408.x. [DOI] [PubMed] [Google Scholar]
- 8.Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest. 2015;125(2):752–68. doi: 10.1172/JCI77014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8(7):267–71. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 10.Gan H, He X, Duan L, Mirabile-Levens E, Kornfeld H, Remold HG. Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J Infect Dis. 2005;191(8):1292–300. doi: 10.1086/428906. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol. 2006;176(6):3707–16. doi: 10.4049/jimmunol.176.6.3707. [DOI] [PubMed] [Google Scholar]
- 12.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164(4):2016–20. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–12. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 15.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 17.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Vandenabeele P, Declercq W, Van HF, Vanden BT. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3(115):re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 19.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278(51):51613–21. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 20.Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350(6258):328–34. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage13 associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20(24):8972–9. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2335–45. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robey RB, Hay N. Mitochondrial hexokinases: guardians of the mitochondria. Cell Cycle. 2005;4(5):654–8. doi: 10.4161/cc.4.5.1678. [DOI] [PubMed] [Google Scholar]
- 26.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation24 driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145(1):104–16. doi: 10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7(12):1166–73. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 30.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521–34. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salvesen GS, Walsh CM. Functions of caspase 8: the identified and the mysterious. Semin Immunol. 2014;26(3):246–52. doi: 10.1016/j.smim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kikuchi J, Yamada S, Koyama D, Wada T, Nobuyoshi M, Izumi T, et al. The novel orally active proteasome inhibitor K-7174 exerts anti-myeloma activity in vitro and in vivo by down-regulating the expression of class I histone deacetylases. J Biol Chem. 2013;288(35):25593–602. doi: 10.1074/jbc.M113.480574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13(19):2514–26. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di LF, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276(4):2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 39.Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem. 2004;279(17):17197–204. doi: 10.1074/jbc.M310329200. [DOI] [PubMed] [Google Scholar]
- 40.Duan L, Gan H, Golan DE, Remold HG. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J Immunol. 2002;169(9):5181–7. doi: 10.4049/jimmunol.169.9.5181. [DOI] [PubMed] [Google Scholar]
- 41.Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268(1):153–60. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci U S A. 2010;107(29):13034–9. doi: 10.1073/pnas.1005997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, et al. Targeted Prostaglandin E2 Inhibition Enhances Antiviral Immunity through Induction of Type I Interferon and Apoptosis in Macrophages. Immunity. 2014;40(4):554–68. doi: 10.1016/j.immuni.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 44.Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol. 2013;783:103–20. doi: 10.1007/978-1-4614-6111-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, 3rd, Ingram JP, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20(1):13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5(4):878–85. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 48.Stegh AH, Barnhart BC, Volkland J, geciras-Schimnich A, Ke N, Reed JC, et al. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J Biol Chem. 2002;277(6):4351–60. doi: 10.1074/jbc.M108947200. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183(4):681–96. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788(10):2022–31. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Niture SK, Jaiswal AK. Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglycerate mutase 5 and controls cellular apoptosis. J Biol Chem. 2011;286(52):44542–56. doi: 10.1074/jbc.M111.275073. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17(12):1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gotow T, Shibata M, Kanamori S, Tokuno O, Ohsawa Y, Sato N, et al. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 2000;7(7):666–74. doi: 10.1038/sj.cdd.4400694. [DOI] [PubMed] [Google Scholar]
- 54.Temkin V, Huang Q, Liu H, Osada H, Pope RM. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol. 2006;26(6):2215–25. doi: 10.1128/MCB.26.6.2215-2225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 56.Ricci JE, Waterhouse N, Green DR. Mitochondrial functions during cell death, a complex (I-V) dilemma. Cell Death Differ. 2003;10(5):488–92. doi: 10.1038/sj.cdd.4401225. [DOI] [PubMed] [Google Scholar]
- 57.Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006;281(20):14314–20. doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- 58.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336( Pt 2):287–90. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65(22):10545–54. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 60.Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007;3(7):e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller JL, Velmurugan K, Cowan MJ, Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 2010;6(4):e1000864. doi: 10.1371/journal.ppat.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 63.Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF36 induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7(12):845–55. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-Deficient Mice Have Impaired Resistance to Mycobacterium tuberculosis Infection through Defective Innate and Adaptive Immunity. Journal of Immunology. 2008;181(10):7157–65. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 66.Zhu H, Guo W, Zhang L, Davis JJ, Wu S, Teraishi F, et al. Enhancing TRAIL-induced apoptosis by Bcl-X(L) siRNA. Cancer Biol Ther. 2005;4(4):393–7. doi: 10.4161/cbt.4.4.1616. [DOI] [PubMed] [Google Scholar]
- 67.Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–26. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.