

Abstract
Computational models have earned broad acceptance for assessing chemical toxicity during early stages of drug discovery or environmental safety assessment. The majority of publicly available QSAR toxicity models have been developed for datasets including mostly drugs or drug-like compounds. We have evaluated and compared chemical spaces occupied by cosmetics, drugs, and pesticides, and explored whether current computational models of toxicity endpoints can be universally applied to all these chemicals. Our analysis of the chemical space overlap and applicability domain (AD) of models built previously for twenty different toxicity endpoints showed that most of these models afforded high coverage (>90%) for all three classes of compounds analyzed herein. Only T. pyriformis models demonstrated lower coverage for drugs and pesticides (38% and 54%, respectively). These results show that, for the most part, historical QSAR models built with data available for different toxicity endpoints can be used for toxicity assessment of novel chemicals irrespective of the intended commercial use; however, the AD restriction is necessary to assure the expected prediction accuracy. Local models may need to be developed to capture chemicals that appear as outliers with respect to global models.
Keywords: Cosmetics, Drugs, Pesticides, Chemical Space, QSAR Models, Prediction
Graphical abstract
Distribution of cosmetics, drugs, and pesticides in the chemical space.
1 Introduction
Chemical toxicity assessment is a critical point in regulatory decision making concerning the release of drugs or industrial chemicals into production, which enables their human or environmental exposure (Parasuraman, 2011). There exists also a variety of natural and synthetic substances that are exposed to humans and/or the environment that have never been evaluated in any toxicity testing protocol (Chuprina et al., 2010; Egeghy et al., 2012). Over the years, the society has tolerated the use of animals in laboratory toxicity testing. However, in recent years, there has been an increased pressure on scientists and regulatory agencies to replace potentially hazardous chemicals by safer alternatives (Collins, 2003; Schulte et al., 2013). In addition, there has been a strong push on the part of both regulatory agencies such as FDA and EPA in the United States and their counterparts around the world to avoid animal testing of every chemical as such testing has become increasingly unsustainable in terms of both cost and time needed to conduct animal trials (Burden et al., 2015).
The development of the alternative in vitro and in silico approaches has been encouraged and supported by both NIH and EPA through large-scale programs such as ToxCast project (Dix et al., 2007) and the Tox21 consortium (Tice et al., 2013). Similar programs such as Endocrine Disruptors Prioritization List (http://ec.europa.eu/environment/chemicals/endocrine/index_en.htm) and the priority substances for water safety (European Union, 2013) have been funded by the European Union. Since the acceptance of Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) legislation in 2006 by the European Union (European Union, 2007; Nicolotti et al., 2014), the use of structural alerts and statistical QSAR models (often collectively referred to as (Q)SAR) have become a major computational approach to chemical safety assessment and regulatory decision support.
The majority of publicly available models for toxicity prediction have been built for drugs or drug candidates (Benfenati et al., 2009; Melnikov et al., 2016) or environmental chemicals (Naven and Louise-May, 2015). In contrast, computational toxicity models for another large group of industrial chemicals, namely cosmetics products have been developed to a much lesser extent as the animal testing has been used as a preferred approach. However, with recent regulations banning the use of animals for testing of the cosmetics products (European Commission, 2013), there has been a resurgence of interest in employing computational models for their toxicity assessment (Bois et al., 2016; Cronin et al., 2012).
Naturally, a question can be posed as to whether toxicity prediction models built for environmental chemicals or drug molecules could be employed for the cosmetics products. The answer to this question depends on the overlap of the chemical spaces occupied by cosmetics, drugs, and environmental chemicals and the size of the applicability domain (AD) of the respective models. AD is commonly defined as the threshold of similarity between a new chemical and molecules in the training set used to develop the respective QSAR model (Netzeva et al., 2005; Tropsha, 2010; Tropsha and Golbraikh, 2007); only predictions for new molecules within the AD of QSAR models, i.e., relatively similar to the modeling set are considered reliable. Importantly, the size of the AD is fully defined by the size and diversity of the modeling set and the computational method used to develop QSAR models. For instance, it is known that the chemical space of drugs has been changing over the past few decades (Deng et al., 2013) creating a challenge for “old” models’ ability to evaluate new compounds. The applicability of current models to many new compounds was also questioned due to limited size and diversity of data available publicly for model building (Kulkarni et al., 2016).
The considerations above capture both significant advantages and challenges associated with the idea of using models developed with one group of industrial chemicals to evaluate toxicity of another group. Obvious advantages deal with significant savings in time and effort afforded by the opportunity to use previously developed models of multiple toxicity endpoints relevant to drugs and/or environmental chemicals (e.g., pesticides) to evaluate toxicity of cosmetic products. However, since chemicals used in different areas of commerce such as drug, chemical, or cosmetic industries are developed with very different applications in mind, there is no a priori reason to expect that their respective chemical spaces overlap. Taking the issue of the AD into account, investigations into studying the degree of such overlap and the applicability of models developed for one group of chemicals to predict toxicity of another group are potentially highly impactful for the respective industries, especially, cosmetics. To the best of our knowledge, such investigations have not been conducted in the public domain with large groups of industrial chemicals.
Herein, we have aimed to compare chemical spaces occupied by cosmetics, drugs, and pesticides, and analyze whether current computational models of different toxicity endpoints can be universally applied to all chemicals. To achieve these aims, we have (i) compiled, curated, and integrated chemical structures of known cosmetics, drugs, and pesticides; (ii) analyzed the distribution of these compounds in chemical space and estimated the structural similarity between the datasets; (iii) performed cluster analysis followed by toxicity annotation comparison for structurally similar compounds in the same clusters; (iv) predicted toxicities of investigated compounds with QSAR models for endpoints developed by us earlier; (v) and analyzed the coverage of these models separately for drugs, cosmetics, and pesticides. We observed that, with some exceptions, the majority of compounds in all three groups of industrial chemicals were found within the AD of QSAR models built previously for twenty different toxicity endpoints. These findings open the door for the development and employment of global toxicity models applicable to the majority of chemicals in commerce while suggesting the need to develop local models that could capture AD outliers of the global models.
2 Materials and methods
2.1 Datasets
2.1.1 Cosmetic ingredients (Dataset A)
The cosmetics ingredients were retrieved from the CosIng, European Commission database for information on cosmetic substances and ingredients (https://ec.europa.eu/growth/sectors/cosmetics/cosing_en). This dataset included 5,166 chemical records with a defined chemical structure. After curation (vide infra), 3,930 unique chemical substances were kept for this study.
2.1.2 Drugs (Dataset B)
We retrieved 7,000 chemical records from the 2014 Leadscope Marketed Drugs (http://www.leadscope.com/marketed_drugs_database/). After curation, 4,671 unique chemical substances were kept for this study.
2.1.3 Pesticides (Dataset C)
We retrieved 3,001 chemical records from the EPA’s Pesticide Product Information System Database (https://www.epa.gov/ingredients-used-pesticide-products/ppis-download-product-information-data). After curation, 2,044 unique chemical substances were kept for this study.
2.2 Data curation
The datasets were thoroughly curated using the workflows proposed by our group earlier (Fourches et al., 2016, 2015, 2010). Briefly, specific chemotypes such as aromatic and nitro groups as well as double bonds were normalized, and absolute stereo configurations removed using the ChemAxon Standardizer (v.16.10.24.0, ChemAxon, Budapest, Hungary, http://www.chemaxon.com). Polymers, substances with undefined chemical substructure, and substances with molecular weight above 1,000 DA were removed. Counterions, inorganic salts, organometallic compounds, and mixtures were removed. After structural standardization, the duplicates were identified with HiT QSAR software (Kuz’min et al., 2008) and carefully analyzed. Within the same dataset, only one record was kept and all duplicates were eliminated. The entire collection (datasets A, B, and C) comprised 9,785 unique chemical compounds. As one can see in Figure 1, 99 compounds were simultaneously labeled as cosmetics, drug, and pesticide; 220 were labeled as cosmetics and drugs; 270 were labeled as cosmetics and pesticides; 172 were labeled as drugs and pesticides; 3,341 compounds were labeled only as cosmetics; 4,180 were labeled only as drugs; and 1,503 were labeled only as pesticides.
Figure 1.
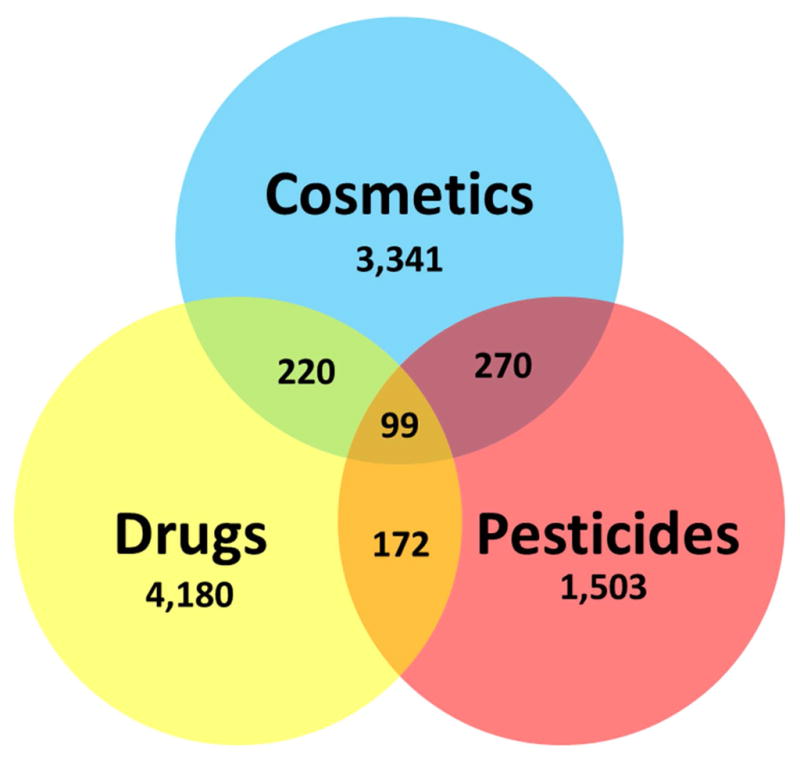
Distribution of investigated compounds on cosmetics, drugs, and pesticides.
2.3 Molecular descriptors
We have calculated the same molecular descriptors as in our previously built QSAR models of toxicity endpoints used in this study (see Table 1 for more detailed information about descriptors, models, and respective references). Majority of the models were built using DRAGON descriptors (Talete SRL, 2007). hERG models were built using Morgan fingerprints and human skin sensitization models were developed with whole-molecule descriptors and QNA (quantitative neighborhoods of atoms) descriptors calculated in GUSAR software (Filimonov et al., 2009). Daphnia magna and fathead minnow models were built using whole-molecule, QNA, and “biological” descriptors, which represent multiple bioactivity predictions by the PASS (prediction of activity spectra of substances) software (Lagunin et al., 2009). Occasionally, several models based on different types of descriptors were built in the same study. We decided to use limited number of descriptor’s types per dataset for simplicity.
Table 1.
List of datasets and respective molecular descriptors used in this study to compare the chemical space of cosmetics, drugs, and pesticides.
| Endpoint | Molecular descriptor | QSAR modeling reference |
|---|---|---|
| Ames mutagenicity | DRAGON | (Sushko et al., 2010) |
| Aquatic toxicity | ||
| Daphnia magna | Whole-molecule, QNA, and PASS | (Zakharov et al., 2014) |
| Fathead minnow | Whole-molecule, QNA, and PASS | (Zakharov et al., 2014) |
| Tetrahymena pyriformis | DRAGON | (Zhu et al., 2008) |
| Hepatotoxicity | DRAGON | (Low et al., 2011) |
| hERG | Morgan | (Braga et al., 2015) |
| Skin sensitization (human data) | Whole-molecule and QNA | (Alves et al., 2016) |
| Skin sensitization (murine data) | DRAGON | (Alves et al., 2015a) |
| AhR, AR, ARE, AR_LBD, aromatase, ATAD5, ER, ER_LBD, HSE, MMP, p53, PPAR-gamma | DRAGON | (Capuzzi et al., 2016) |
2.4 Chemical space of cosmetics, drugs, and pesticides
Chemical space formed by Datasets A, B, and C was analyzed by plotting the barycentric coordinates of all the 9,785 structures, which were defined by the DRAGON descriptors. Barycentric coordinates correspond to the location of the points of a simplex (a triangle, tetrahedron, etc.) in the space, defined by the vertices (Vityuk et al., 1999). In this case, a simplex is defined by all the DRAGON descriptors of a particular chemical substance. Barycentric coordinates were determined using Methods of Data Analysis module of HiT QSAR software (Kuz’min et al., 2008). In addition, a similarity map was generated using OSIRIS DataWarrior software (Sander et al., 2015).
2.5 How well does the applicability domain of toxicity QSAR models cover cosmetics, drugs, and pesticides?
We have assessed if Datasets A, B, and C were inside the AD of QSAR models built previously. The ADs were calculated as Dcutoff=<D>+Zs, where Z is a similarity threshold parameter defined by a user (0.5 in this study), and <D> and s are the average and standard deviation, respectively, of all Euclidian distances in the multidimensional descriptor space between each compound and its nearest neighbors for all compounds in the training set (Golbraikh et al., 2003; Tropsha and Golbraikh, 2007). This analysis involved datasets used to build models for Ames mutagenicity (Sushko et al., 2010), aquatic toxicity (Daphnia magna, fathead minnow (Zakharov et al., 2014), and Tetrahymena pyriformis (Zhu et al., 2008)), hepatotoxicity (Low et al., 2011), hERG (Braga et al., 2015), and human (Alves et al., 2016) and murine (Alves et al., 2015a) skin sensitization. In addition, twelve stress response and nuclear receptor signaling pathways toxicity datasets used to generate QSAR models published by our group as part of the 2014 Tox21 Challenge (Capuzzi et al., 2016) were used as well. These respective endpoints included androgen receptor (AR), androgen receptor-ligand binding domain (AR_LBD), aromatase, aryl hydrocarbon receptor (AhR), ATPase family AAA Domain-containing 5 (ATAD5); estrogen receptor alpha-full (ER), estrogen receptor alpha-ligand binding domain (ER_LBD), peroxisome proliferator-activated receptor gamma (PPAR-gamma), nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (ARE), heat shock factor response element (HSE), mitochondrial membrane potential (MMP), and tumor suppressor p53.
2.6 Cluster analysis
Chemical clusters were generated by the Sequential Agglomerative Hierarchical Non-overlapping method implemented in the ISIDA/Cluster software (Varnek et al., 2008). Briefly, the software generates a dendrogram of the parent-child relationships between clusters and a heat map of the proximity matrix colored according to the pairwise chemical similarity between compounds. To better visualize the clusters, the distance matrix of the 9,785 compounds from datasets A, B, and C was calculated and the compounds were clustered into 100 clusters. A stratified sample containing 500 compounds representative of all clusters was taken, with similar proportion of cosmetics, drugs, and pesticides. Then, 500 compounds from the training sets of QSAR models analyzed in this study were randomly selected. The total clustering set was composed of 1000 compounds. This method was applied to check the structural diversity of compounds in cosmetics, drugs and pesticides and whether compounds from training sets of QSAR models, not overlapping with the datasets A, B, and C, would cluster with them.
3 Results
3.1 Analysis of chemical space of cosmetics, drugs, and pesticides
A plot of calculated logP (ClogP) vs. molecular weight (MW) is shown in Figure 2. As one can see, there is a big overlap between all the industrial classes of compounds, as well as with compounds from datasets used to develop historical QSAR models. At higher MW, drugs and cosmetics separate from pesticides. Drugs present the same range of ClogP, even at higher MW, while cosmetics tend to have higher ClogP, i.e., include compounds with low solubility. In Figure 2B, the difference between drugs and pesticides almost disappear albeit pesticides are spread more. Most of drugs, cosmetics, and pesticides occupy the space from the bottom left to the center. In the top right part of the plot, there is a region of the chemical space that is covered mostly by drugs and cosmetics; the number of pesticides is very limited there. Apparently, QSAR models based on drugs and cosmetics could also be used to predict pesticides, but the opposite may not work for a fraction of drugs and pesticides dissimilar from pesticides.
Figure 2.

A) Chemical space of investigated compounds defined by ClogP and MW. B) Chemical space of investigated compounds in barycentric coordinates obtained from 2D DRAGON descriptors. Shadowed area represent the chemical space occupied by compounds from datasets used to generate current toxicity QSAR models. Two outliers (coordinates 1631, 160 and 960, −794) from the training sets of QSAR models are not shown.
The overlap between QSAR datasets and cosmetics, drug, and pesticides (See Table 2) has shown that drugs are well represented in almost all cases, except for aquatic toxicity (D. magna, fathead minnow, and T. pyriformis) and skin sensitization (both human and murine) datasets. D. magna and fathead minnow predominantly include pesticides, while T. pyriformis and skin sensitization predominantly include cosmetics. Hepatotoxicity and hERG datasets mainly include drugs and drug-like compounds. Nine out of twenty datasets (D. magna, fathead minnow, hepatotoxicity, hERG, AhR, AR_LBD, Aromatase, HSE, and MMP) included higher number of pesticides than cosmetics. The other eleven datasets had higher number of cosmetics than pesticides.
Table 2.
Overlap between QSAR datasets and cosmetics, drug, and pesticides.
| Models | Industrial class | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Endpoint | Dataset size | Cosmetic | Drug | Pesticide | Cosmetic and drug | Cosmetic and pesticide | Drug and pesticide | Cosmetic, drug, and pesticide | Total |
| Ames mutagenicity | 4361 | 265 | 382 | 217 | 50 | 93 | 68 | 56 | 1131 |
| Aquatic toxicity | |||||||||
| Daphnia magna | 283 | 16 | 11 | 80 | 1 | 21 | 31 | 6 | 166 |
| Fathead minnow | 659 | 59 | 17 | 121 | 7 | 52 | 38 | 16 | 310 |
| Tetrahymena pyriformis | 644 | 161 | 9 | 25 | 5 | 50 | 5 | 14 | 269 |
| Hepatotoxicity | 102 | 0 | 71 | 2 | 4 | 1 | 9 | 3 | 90 |
| hERG | 5984 | 0 | 141 | 1 | 4 | 1 | 6 | 2 | 155 |
| Skin sensitization (human data) | 109 | 33 | 4 | 5 | 0 | 20 | 11 | 14 | 87 |
| Skin sensitization (murine data) | 254 | 33 | 10 | 10 | 0 | 22 | 3 | 12 | 90 |
| AhR | 1194 | 78 | 323 | 132 | 30 | 8 | 35 | 8 | 614 |
| AR | 422 | 28 | 211 | 8 | 10 | 3 | 8 | 1 | 269 |
| ARE | 342 | 17 | 163 | 6 | 8 | 3 | 9 | 2 | 208 |
| AR_LBD | 1386 | 80 | 382 | 148 | 34 | 11 | 36 | 14 | 705 |
| Aromatase | 268 | 18 | 82 | 40 | 11 | 0 | 12 | 2 | 165 |
| ATAD5 | 404 | 27 | 123 | 21 | 12 | 5 | 10 | 2 | 200 |
| ER | 1070 | 94 | 337 | 67 | 28 | 14 | 17 | 5 | 562 |
| ER_LBD | 476 | 39 | 168 | 23 | 16 | 2 | 8 | 4 | 260 |
| HSE | 486 | 34 | 134 | 43 | 16 | 8 | 11 | 8 | 254 |
| MMP | 1436 | 97 | 401 | 158 | 33 | 19 | 36 | 19 | 763 |
| p53 | 620 | 40 | 207 | 37 | 18 | 7 | 22 | 8 | 339 |
| PPAR-gamma | 266 | 16 | 79 | 15 | 7 | 1 | 8 | 3 | 129 |
The estimation of AD showed that most of the models used in this study provided high coverage (>90%) for all three classes of compounds analyzed in this study (see Table 3). Only the model for T. pyriformis (Zhu et al., 2008) has shown a big difference between cosmetics and drugs and pesticides. In this case, the coverage for cosmetics was 90%, while the coverage of drugs and pesticides was significantly smaller (38% and 54%, respectively). The fathead minnow model (Zakharov et al., 2014) presented a slightly lower coverage (by ~9%) for drugs (86.7%), when compared to cosmetics (95%) and pesticides (95.9%). Chemical structures of all compounds and toxicity predictions made by all twenty models (considering AD restriction) are available in the Supplementary Materials.
Table 3.
Percentage of compounds inside the AD of studied QSAR models.
| Model | Cosmetic | Drug | Pesticide | Cosmetic and drug | Cosmetic and pesticide | Drug and pesticide | Cosmetic, drug, and pesticide | Total coverage |
|---|---|---|---|---|---|---|---|---|
| Ames mutagenicity | 99.9% | 98.2% | 96.5% | 100.0% | 99.6% | 99.4% | 100.0% | 98.6% |
| Aquatic toxicity | ||||||||
| Daphnia magna | 99.9% | 99.9% | 100.0% | 100.0% | 99.3% | 100.0% | 100.0% | 99.9% |
| Fathead minnow | 95.0% | 86.7% | 95.9% | 94.1% | 94.4% | 95.9% | 96.0% | 91.6% |
| Tetrahymena pyriformis | 89.6% | 37.8% | 54.4% | 78.2% | 91.5% | 60.5% | 92.9% | 61.4% |
| Hepatotoxicity | 96.2% | 96.0% | 96.1% | 93.6% | 97.8% | 95.3% | 96.0% | 96.1% |
| hERG | 99.6% | 100.0% | 99.9% | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% |
| Skin sensitization (human data) | 99.6% | 99.8% | 98.9% | 99.5% | 98.9% | 98.8% | 98.0% | 99.5% |
| Skin sensitization (murine data) | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% |
| AhR | 97.3% | 95.8% | 91.0% | 95.9% | 93.0% | 94.8% | 89.9% | 95.4% |
| AR | 97.1% | 93.9% | 85.0% | 95.0% | 95.2% | 88.4% | 93.9% | 93.6% |
| ARE | 97.4% | 95.7% | 92.8% | 96.8% | 96.7% | 95.9% | 97.0% | 95.9% |
| AR_LBD | 96.7% | 92.2% | 83.7% | 95.0% | 95.9% | 90.1% | 93.9% | 92.6% |
| Aromatase | 96.6% | 93.5% | 89.6% | 92.3% | 90.7% | 92.4% | 86.9% | 93.8% |
| ATAD5 | 96.6% | 93.8% | 89.3% | 95.9% | 94.4% | 94.2% | 92.9% | 94.1% |
| ER | 98.7% | 96.2% | 93.9% | 97.3% | 98.1% | 97.7% | 97.0% | 96.8% |
| ER_LBD | 97.6% | 96.3% | 94.1% | 96.4% | 95.2% | 97.1% | 92.9% | 96.4% |
| HSE | 97.6% | 95.2% | 94.7% | 95.0% | 97.8% | 94.8% | 94.9% | 96.0% |
| MMP | 97.5% | 95.7% | 92.3% | 96.8% | 96.7% | 94.2% | 94.9% | 95.8% |
| p53 | 98.4% | 96.0% | 86.6% | 98.2% | 96.7% | 90.1% | 93.9% | 95.3% |
| PPAR-gamma | 96.9% | 94.3% | 89.6% | 94.5% | 95.2% | 90.7% | 92.9% | 94.4% |
The cluster analysis made with 500 representatives of cosmetics, drugs, and pesticides and 500 compounds from training sets of QSAR models showed high structural diversity (Figure 3). This analysis was made using a sample containing 500 compounds from the 100 clusters produced from the initial dataset containing 9,785 compounds and 500 compounds from training sets of QSAR models not overlapping with dataset A, B, and C. The 500 compounds from training sets spread across all the 27 smaller clusters, revealing an overlap of the chemical space (see Supplementary Materials).
Figure 3.

Results of cluster analysis of 1,000 compounds including cosmetics, drugs, and pesticides. Heatmap and dendrogram of the distance matrix are both colored according to structural similarity (blue/violet = similar; yellow/red = dissimilar).
4 Discussion
4.1 Compounds simultaneously used as cosmetics, drugs, or pesticides
We shall note that compounds labeled as cosmetics, drugs, and pesticides may not be the active ingredients, but rather excipients used in the formulations of final products, e.g., mannitol or stearic acid. This explains the big overlap between these three categories. In addition, we fully realize that defining these labels as “categories” is an oversimplification, since these terms do not reflect chemical classes, but rather their final use. For instance, several compounds such as methane, trichloromethane, benzene, urea, formaldehyde, formic acid, etc., are related to multiple industrial chemical processes, which may be the reason of their multi-labelling. On other hand, there were a few drugs related to pesticides as well. For instance, diazepam is used in the treatment of intoxication of organophosphorus ester pesticide poisoning (Marrs, 2003). Phenobarbital is indicated to cause insecticide resistance in house flies (Hayaoka and Dauterman, 1982) and Aedes aegypti (Sousa-Polezzi and Bicudo, 2004). Conversely, difenacoum is labelled as both drug and pesticide. This compound is a warfarin analog used as a rodenticide (Feinstein et al., 2016). Apparently, the EPA’s Pesticide Product Information System Database includes any chemical associated with pesticides. Leadscope Marketed Drugs database comprises the historically marketed drug records from FDA and parent compounds of the active ingredients, which explains why a few compounds overlapped.
4.2 Overlap between QSAR datasets and cosmetics, drug, and pesticides
Despite the low number of QSAR studies totally focused on cosmetics, our results presented on Figure 1 and Table 2 demonstrate that existing QSAR models, in general, could predict cosmetic products well. Drugs and drug-like are well-represented in the majority of studied models, except for aquatic toxicity and skin sensitization. D. magna and fathead minnow predominantly contain pesticides, while T. pyriformis and both skin sensitization datasets mostly contain cosmetics.
It is understandable that D. magna and fathead minnow contain mostly pesticides, since both endpoints are related to aquatic toxicity and represent important ecotoxicity assays (EPA, 2002). On the other hand, it is surprising that most of the compounds for T. pyriformis are composed of cosmetics and not of pesticides (see the next section for additional discussion). Although our findings show that current QSAR models contains a significant number of cosmetics, there is still a lack of QSAR studies focused on this industrial chemical class. Recently, our group has extensively studied skin sensitization (Alves et al., 2016, 2015a, 2015b). Skin sensitization is an autoimmune inflammatory reaction, which is caused by topical exposure to chemical allergens (Hennino et al., 2005), therefore, this endpoint has high importance to cosmetic industry (Vandebriel and van Loveren, 2010). In addition, the animal testing has been completely banned for cosmetics in Europe (European Commission, 2013), which explains the high number of cosmetics in this dataset.
Surprisingly, despite the fact that drugs are the most common industrial class present in the hERG dataset, this analysis showed that the number of drugs with public hERG data was very low. The hERG channels have a key hole in the mediating the repolarization of cardiac action potential. Its blockage is related to heart arrhythmia and death (Picard et al., 2011). This is one of the most important anti-targets to be considered in the early stages of the drug development process due to its high ligand promiscuity, mainly due to its large hydrophobic intracellular binding pocket and its multiple states (open, inactive, and closed) (Mitcheson et al., 2000). As hERG safety testing is a mandatory FDA-required procedure (FDA, 2005a, 2005b), scientific community would benefit from models developed based on marketed drugs.
4.3 Analysis of chemical space of cosmetics, drugs, and pesticides
The analysis of chemical space characterized by all descriptors revealed a huge overlap between cosmetics, drugs, and pesticides (Figure 2A–C). The overlap shown in both figures indicates that distinguishing these compounds by simple analysis, such as using ClogP and MW is impossible, reinforcing the importance of using QSAR models instead. Although some regions of chemical space are not covered by pesticides, current global QSAR models for toxicity could be used to predict cosmetics, drugs, and pesticides, since most of these models were build using drugs or compounds designed to be drugs. Cluster analysis revealed high structural diversity and the distribution of the 500 query compounds from training sets through all the 27 clusters (see Supplementary Materials).
The low coverage of drugs for T. pyriformis is probably due to the high similarity of compounds used to generate this model. Our analysis reveals that T. pyriformis dataset of 644 compounds contains 161 cosmetics, 9 drugs, 25 pesticides, 5 cosmetics and drugs, 50 cosmetics and pesticides, 5 drugs and pesticides, and 14 cosmetics, drugs, and pesticides. As shown on Figure 4A, compounds in T. pyriformis dataset could be easily clustered. The most representative clusters are shown on Figure 4A and C. Cluster 1 contains primary alcohols and other mid and long chain compounds; cluster 2 – fatty acids and aldehydes; cluster 3 – nitro aromatics; and cluster 4 – halogenated aromatics. Compounds in clusters 1 and 2 are mostly cosmetic products, which is confirmed by the higher similarity with cosmetics that is shown on Figure 4B. Cluster 3 and 4 presents high dissimilarity with most compounds outside its cluster. The representatives of Clusters 1–4: hexanol (cosmetic), octanoic acid (cosmetic), 1,3-dinitrobenzene (explosive), and 2,4-dichloroaniline (pesticide) are shown on Figure 4C. As could be seen from there, in general cosmetics, unlike drugs and pesticides, are structurally similar to compounds from T. pyriformis dataset, which explains the high coverage for cosmetics and low coverage for compounds from two other industrial classes.
Figure 4.

A) Distribution of cosmetics, drugs, pesticides, and T. pyriformis dataset (644 compounds) in chemical space. Four clusters of highly similar compounds are highlighted by black circles and numbered. B) Distribution of Tanimoto coefficients between industrial compounds and their nearest neighbor in the T. pyriformis dataset. C) Representative compounds for clusters 1–4.
Earlier, our group (Golbraikh et al., 2003; Muratov et al., 2010; Tropsha and Golbraikh, 2007) and others (Gadaleta et al., 2016; Mathea et al., 2016) have demonstrated the importance of AD in QSAR modeling. In most of QSAR models used in this study, the use of AD resulted in increase of prediction accuracy at the expense of coverage, except skin sensitization and hERG models (Alves et al., 2015b; Braga et al., 2015, 2014), where significant reduction in coverage was not accompanied by an improvement in the predictivity. Thus, we decided to use the model’s coverage of our collection as a measure of model’s capability to predict a specific class of industrial chemicals. Our results demonstrate (see Table 3) that majority of models provided high (>90%) coverage for all three classes of industrial compounds, except for the model for fathead minnow (Zakharov et al., 2014) and T. pyriformis (Zhu et al., 2008). Fathead minnow showed a slightly lower coverage for drugs (86.7%) than for cosmetics and pesticides (95% and 95.6%, respectively). This dataset includes smaller number of drugs (cf. Table 2), which explains the slight reduction in coverage. The T. pyriformis model had better coverage for cosmetics (90%) than for drugs and pesticides (38% and 54%, respectively). These results allowed us to draw a conclusion that most of existing publicly available QSAR models could predict chemical toxicity of drugs, pesticides, and cosmetics with similar success.
5 Conclusions
Vast majority of current QSAR models of various toxicity endpoints have been developed to predict toxicity of drugs, drug-like compounds, and, less frequently, pesticides, or other environmental chemicals. The ability of these models to predict toxicity for another big class of industrial chemicals – cosmetics, was not examined previously. The analysis of chemical space revealed a huge overlap between cosmetics, drugs, and pesticides. Our results also show that drugs and cosmetics are more structurally dissimilar than pesticides. In addition, we found that the datasets used for building existing toxicity models contain many drugs, while cosmetics and pesticides are less represented. However, the similarity of cosmetics, drugs, and pesticides to compounds in QSAR datasets is reasonably high. This is reflected in high (>90%) coverage of all three classes of chemicals by all the studied models except T. pyriformis. These results allow us to conclude that, because of high structural similarity between cosmetics; drugs; and pesticides, publicly available QSAR models of various toxicity endpoints (typically built either for drugs or for pesticides) could be successfully used to predict respective toxicities for all three classes of industrial chemicals. We posit that this conclusion is especially valuable for the cosmetic industry where toxicity modeling has been limited in the past but where the demand for alternative, non-animal toxicity testing is very high. Thus, our findings provide critical support for reusing existing toxicity models, irrespective of the classes of compounds they have been developed for, to predict cosmetics’ toxicity.
Supplementary Material
The majority of publicly available QSAR models for toxicity endpoints were built on datasets consisted mostly of drugs, drug-like compounds, and pesticides;
There is a lack of data for cosmetics and the question we would like to address whether current models could be applied for predicting toxicity of cosmetics;
The analysis of chemical space revealed a huge overlap between cosmetics, drugs, and pesticides;
Our results indicate that current QSAR models could be used to predict chemical toxicity for cosmetics, drugs, and pesticides.
Acknowledgments
This study was supported in part by NIH (grant 1U01CA207160). VA thanks FAPEG (grant 201310267001095), CNPq (grant 400760/2014-2), and CAPES. A.Z. acknowledge Intramural Research Program, National Center for Advancing Translational Sciences, National Institutes of Health (1ZIATR000058-02). The authors express sincere gratitude to Drs. Glenn Myatt and Nora Aptula for providing datasets used in this study. The authors are also grateful for Drs. Vladimir Poroikov and Dmitri Filimonov providing the GUSAR Software and appreciate ChemAxon for providing free academic licenses for their products.
Abbreviations
- AD
applicability domain
- AhR
aryl hydrocarbon receptor
- AR
androgen receptor
- ARE
nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element
- AR_LBD
androgen receptor—ligand binding domain
- ATAD5
ATPase family AAA Domain containing 5
- ClogP
calculated logP
- ER
estrogen receptor alpha—full
- ER_LBD
estrogen receptor alpha—ligand binding domain
- HSE
heat shock factor response element
- MMP
mitochondrial membrane potential
- MW
molecular weight
- PASS
prediction of activity spectra of substances
- p53
tumor suppressor p53
- PPAR-gamma
peroxisome proliferator-activated receptor gamma
- QNA
quantitative neighborhoods of atoms
- QSAR
quantitative structure-activity relationship
Footnotes
Supplementary materials are available online; these include curated chemical datasets for cosmetics, drugs, and pesticides, predictions along with AD estimation, and chemical clusters.
Conflict of interests
The authors declare no actual or potential conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alves VM, Capuzzi SJ, Muratov EN, Braga RC, Thornton TE, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. QSAR models of human data can enrich or replace LLNA testing for human skin sensitization. Green Chem. 2016;18:6501–6515. doi: 10.1039/C6GC01836J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves VM, Muratov EN, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds. Toxicol Appl Pharmacol. 2015a;284:262–272. doi: 10.1016/j.taap.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves VM, Muratov EN, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Predicting chemically-induced skin reactions. Part II: QSAR models of skin permeability and the relationships between skin permeability and skin sensitization. Toxicol Appl Pharmacol. 2015b;284:273–280. doi: 10.1016/j.taap.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati E, Benigni R, Demarini DM, Helma C, Kirkland D, Martin TM, Mazzatorta P, Ouédraogo-Arras G, Richard AM, Schilter B, Schoonen WGEJ, Snyder RD, Yang C. Predictive models for carcinogenicity and mutagenicity: frameworks, state-of-the-art, and perspectives. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:57–90. doi: 10.1080/10590500902885593. [DOI] [PubMed] [Google Scholar]
- Bois FY, Ochoa JGD, Gajewska M, Kovarich S, Mauch K, Paini A, Péry A, Benito JVS, Teng S, Worth A. Multiscale modelling approaches for assessing cosmetic ingredients safety. Toxicology. 2016 doi: 10.1016/j.tox.2016.05.026. [DOI] [PubMed] [Google Scholar]
- Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Liao LM, Tropsha A, Andrade CH. Pred-hERG: A Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol Inform. 2015;34:698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Tropsha A, Andrade CH. Tuning HERG out: antitarget QSAR models for drug development. Curr Top Med Chem. 2014;14:1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden N, Sewell F, Chapman K. Testing Chemical Safety: What Is Needed to Ensure the Widespread Application of Non-animal Approaches? PLoS Biol. 2015;13:e1002156. doi: 10.1371/journal.pbio.1002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuzzi SJ, Politi R, Isayev O, Farag S, Tropsha A. QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front Environ Sci. 2016;4 doi: 10.3389/fenvs.2016.00003. [DOI] [Google Scholar]
- Chuprina A, Lukin O, Demoiseaux R, Buzko A, Shivanyuk A. Drug- and lead-likeness, target class, and molecular diversity analysis of 7.9 million commercially available organic compounds provided by 29 suppliers. J Chem Inf Model. 2010;50:470–479. doi: 10.1021/ci900464s. [DOI] [PubMed] [Google Scholar]
- Collins T. The importance of sustainability ethics, toxicity and ecotoxicity in chemical education and research. Green Chem. 2003;5:G51–G52. doi: 10.1039/b307694f. [DOI] [Google Scholar]
- Cronin MTD, Madden JC, Richarz A-N. The COSMOS Project: A Foundation For The Future Of Computational Modelling Of Repeat Dose Toxicity [WWW Document] [accessed 12.21.16];AltTox.org. 2012 http://alttox.org/the-cosmos-project-a-foundation-for-the-future-of-computational-modelling-of-repeat-dose-toxicity/
- Deng ZL, Du CX, Li X, Hu B, Kuang ZK, Wang R, Feng SY, Zhang HY, Kong DX. Exploring the Biologically Relevant Chemical Space for Drug Discovery. J Chem Inf Model. 2013;53:2820–2828. doi: 10.1021/ci400432a. [DOI] [PubMed] [Google Scholar]
- Dix DJ, Houck Ka, Martin MT, Richard AM, Setzer RW, Kavlock RJ. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci. 2007;95:5–12. doi: 10.1093/toxsci/kfl103. [DOI] [PubMed] [Google Scholar]
- Egeghy PP, Judson R, Gangwal S, Mosher S, Smith D, Vail J, Hubal EAC. The exposure data landscape for manufactured chemicals. Sci Total Environ. 2012;414:159–166. doi: 10.1016/j.scitotenv.2011.10.046. [DOI] [PubMed] [Google Scholar]
- EPA. [accessed 3.1.17];Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms [WWW Document] 2002 https://www.epa.gov/sites/production/files/2015-08/documents/acute-freshwater-and-marine-wet-manual_2002.pdf.
- European Commission. On the animal testing and marketing ban and on the state of play in relation to alternative methods in the field of cosmetics [WWW Document] [accessed 2.9.16];Commun from commision to Eur Parliam Counc. 2013 http://ec.europa.eu/consumers/sectors/cosmetics/files/pdf/animal_testing/com_at_2013_en.pdf.
- European Union. Directive 2013/39/EU. Off J Eur Union. 2013:1–17. [Google Scholar]
- European Union. Regulation (EC) No 1907/2006. Off J Eur Union. 2007:3–280. [Google Scholar]
- FDA. Guidance for industry. S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. Rockville, MD: 2005a. [PubMed] [Google Scholar]
- FDA. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Rockville, MD: 2005b. [PubMed] [Google Scholar]
- Feinstein DL, Akpa BS, Ayee MA, Boullerne AI, Braun D, Brodsky SV, Gidalevitz D, Hauck Z, Kalinin S, Kowal K, Kuzmenko I, Lis K, Marangoni N, Martynowycz MW, Rubinstein I, van Breemen R, Ware K, Weinberg G. The emerging threat of superwarfarins: history, detection, mechanisms, and countermeasures. Ann N Y Acad Sci. 2016;1374:111–122. doi: 10.1111/nyas.13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filimonov DA, Zakharov AV, Lagunin AA, Poroikov VV. QNA-based “Star Track” QSAR approach. SAR QSAR Environ Res. 2009;20:679–709. doi: 10.1080/10629360903438370. [DOI] [PubMed] [Google Scholar]
- Fourches D, Muratov E, Tropsha A. Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J Chem Inf Model. 2016;56:1243–1252. doi: 10.1021/acs.jcim.6b00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourches D, Muratov E, Tropsha A. Curation of chemogenomics data. Nat Chem Biol. 2015;11:535–535. doi: 10.1038/nchembio.1881. [DOI] [PubMed] [Google Scholar]
- Fourches D, Muratov E, Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model. 2010;50:1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O. Applicability Domain for QSAR Models: Where Theory Meets Reality. Int J Quant Struct Relationships. 2016;1:45–63. doi: 10.4018/IJQSPR.2016010102. [DOI] [Google Scholar]
- Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J Comput Aided Mol Des. 2003;17:241–253. doi: 10.1023/A:1025386326946. [DOI] [PubMed] [Google Scholar]
- Hayaoka T, Dauterman WC. Induction of glutathione S-transferase by phenobarbital and pesticides in various house fly strains and its effect on toxicity. Pestic Biochem Physiol. 1982;17:113–119. doi: 10.1016/0048-3575(82)90015-3. [DOI] [Google Scholar]
- Hennino A, Vocanson M, Chavagnac C, Saint-Mezard P, Dubois B, Kaiserlian D, Nicolas J. Update on the pathophysiology with special emphasis on CD8 effector T cells and CD4 regulatory T cells. An Bras Dermatol. 2005;80:335–347. doi: 10.1590/S0365-05962005000400003. [DOI] [Google Scholar]
- Kulkarni SA, Benfenati E, Barton-Maclaren TS. Improving confidence in (Q)SAR predictions under Canada’s Chemicals Management Plan – a chemical space approach $ SAR QSAR Environ Res. 2016;27:851–863. doi: 10.1080/1062936X.2016.1243152. [DOI] [PubMed] [Google Scholar]
- Kuz’min VE, Artemenko AG, Muratov EN. Hierarchical QSAR technology based on the Simplex representation of molecular structure. J Comput Aided Mol Des. 2008;22:403–421. doi: 10.1007/s10822-008-9179-6. [DOI] [PubMed] [Google Scholar]
- Lagunin A, Filimonov D, Zakharov A, Xie W, Huang Y, Zhu F, Shen T, Yao J, Poroikov V. Computer-Aided Prediction of Rodent Carcinogenicity by PASS and CISOC-PSCT. QSAR Comb Sci. 2009;28:806–810. doi: 10.1002/qsar.200860192. [DOI] [Google Scholar]
- Low Y, Uehara T, Minowa Y, Yamada H, Ohno Y, Urushidani T, Sedykh A, Muratov E, Kuz’min V, Fourches D, Zhu H, Rusyn I, Tropsha A. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem Res Toxicol. 2011;24:1251–1262. doi: 10.1021/tx200148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs TC. Diazepam in the treatment of organophosphorus ester pesticide poisoning. Toxicol Rev. 2003;22:75–81. doi: 10.2165/00139709-200322020-00002. [DOI] [PubMed] [Google Scholar]
- Mathea M, Klingspohn W, Baumann K. Chemoinformatic Classification Methods and their Applicability Domain. Mol Inform. 2016;35:160–180. doi: 10.1002/minf.201501019. [DOI] [PubMed] [Google Scholar]
- Melnikov F, Kostal J, Voutchkova-Kostal A, Zimmerman JB, Anastas TP. Assessment of predictive models for estimating the acute aquatic toxicity of organic chemicals. Green Chem. 2016;18:4432–4445. doi: 10.1039/C6GC00720A. [DOI] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratov EN, Artemenko AG, Varlamova EV, Polischuk PG, Lozitsky VP, Fedchuk AS, Lozitska RL, Gridina TL, Koroleva LS, Sil’nikov VN, Galabov AS, Makarov VA, Riabova OB, Wutzler P, Schmidtke M, Kuz’min VE. Per aspera ad astra: application of Simplex QSAR approach in antiviral research. Future Med Chem. 2010;2:1205–1226. doi: 10.4155/fmc.10.194. [DOI] [PubMed] [Google Scholar]
- Naven R, Louise-May S. Computational toxicology: Its essential role in reducing drug attrition. Hum Exp Toxicol. 2015;34:1304–1309. doi: 10.1177/0960327115605440. [DOI] [PubMed] [Google Scholar]
- Netzeva TI, Worth A, Aldenberg T, Benigni R, Cronin MTD, Gramatica P, Jaworska JS, Kahn S, Klopman G, Marchant CA, Myatt G, Nikolova-Jeliazkova N, Patlewicz GY, Perkins R, Roberts D, Schultz T, Stanton DW, van de Sandt JJM, Tong W, Veith G, Yang C. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern Lab Anim. 2005;33:155–173. doi: 10.1177/026119290503300209. [DOI] [PubMed] [Google Scholar]
- Nicolotti O, Benfenati E, Carotti A, Gadaleta D, Gissi A, Mangiatordi GF, Novellino E. REACH and in silico methods: an attractive opportunity for medicinal chemists. Drug Discov Today. 2014;19:1757–1768. doi: 10.1016/j.drudis.2014.06.027. [DOI] [PubMed] [Google Scholar]
- Parasuraman S. Toxicological screening. J Pharmacol Pharmacother. 2011;2:74. doi: 10.4103/0976-500X.81895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard S, Goineau S, Guillaume P, Henry J, Hanouz JL, Rouet R. Supplemental studies for cardiovascular risk assessment in safety pharmacology: a critical overview. Cardiovasc Toxicol. 2011;11:285–307. doi: 10.1007/s12012-011-9133-z. [DOI] [PubMed] [Google Scholar]
- Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Schulte PA, McKernan LT, Heidel DS, Okun AH, Dotson GS, Lentz TJ, Geraci CL, Heckel PE, Branche CM. Occupational safety and health, green chemistry, and sustainability: a review of areas of convergence. Environ Heal. 2013;12:31. doi: 10.1186/1476-069X-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa-Polezzi RC, de Bicudo HEMC. Effect of phenobarbital on inducing insecticide tolerance and esterase changes in Aedes aegypti (Diptera: Culicidae) Genet Mol Biol. 2004;27:275–283. doi: 10.1590/S1415-47572004000200024. [DOI] [Google Scholar]
- Sushko I, Novotarskyi S, Körner R, Pandey AK, Cherkasov A, Li J, Gramatica P, Hansen K, Schroeter T, Müller KR, Xi L, Liu H, Yao X, Öberg T, Hormozdiari F, Dao P, Sahinalp C, Todeschini R, Polishchuk P, Artemenko A, Kuz’min V, Martin TM, Young DM, Fourches D, Muratov E, Tropsha A, Baskin I, Horvath D, Marcou G, Muller C, Varnek A, Prokopenko VV, Tetko IV. Applicability domains for classification problems: Benchmarking of distance to models for Ames mutagenicity set. J Chem Inf Model. 2010;50:2094–2111. doi: 10.1021/ci100253r. [DOI] [PubMed] [Google Scholar]
- Talete SRL. Dragon for Windows (Software for Molecular Descriptor Calculations) 2007. [Google Scholar]
- Tice RR, Austin CP, Kavlock RJ, Bucher JR. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect. 2013;121:756–765. doi: 10.1289/ehp.1205784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inform. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- Tropsha A, Golbraikh A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr Pharm Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- Vandebriel RJ, van Loveren H. Non-animal sensitization testing: state-of-the-art. Crit Rev Toxicol. 2010;40:389–404. doi: 10.3109/10408440903524262. [DOI] [PubMed] [Google Scholar]
- Varnek A, Fourches D, Horvath D, Klimchuk O, Gaudin C, Vayer P, Solov’ev V, Hoonakker F, Tetko I, Marcou G. ISIDA - Platform for Virtual Screening Based on Fragment and Pharmacophoric Descriptors. Curr Comput Aided-Drug Des. 2008;4:191–198. doi: 10.2174/157340908785747465. [DOI] [Google Scholar]
- Vityuk N, Voskresenskaja E, Kuz’min V. The Synergism of Methods Barycentric Coordinates and Trend-vector for Solution —Structure-Property Tasks. Pattern Recognit Image Anal. 1999;3:521–528. [Google Scholar]
- Zakharov AV, Peach ML, Sitzmann M, Nicklaus MC. A new approach to radial basis function approximation and its application to QSAR. J Chem Inf Model. 2014;54:713–719. doi: 10.1021/ci400704f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, Oberg T, Dao P, Cherkasov A, Tetko IV. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J Chem Inf Model. 2008;48:766–784. doi: 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.