

Abstract
Biallelic GLDN mutations have recently been identified among infants with lethal congenital contracture syndrome 11 (LCCS11). GLDN encodes gliomedin, a protein required for the formation of the nodes of Ranvier and development of the human peripheral nervous system. We report 6 infants and children from 4 unrelated families with biallelic GLDN mutations, 4 of whom survived beyond the neonatal period into infancy, childhood, and late adolescence with intensive care and chronic respiratory and nutritional support. Our findings expand the genotypic and phenotypic spectrum of LCCS11 and demonstrate that the condition may not necessarily be lethal in the neonatal period.
Keywords: GLDN, gliomedin, arthrogryposis multiplex congenital, AMC
Main Text
Arthrogryposis multiplex congenita (AMC) is a disorder characterized by multiple joint contractures (limbs, jaw, spine) and limited movements at birth. AMC results from diverse mechanisms, including any condition that decreases or limits fetal movements. AMC is present in 1/3000 births (Lowry, et al., 2010) and can be associated with craniofacial abnormalities, pulmonary hypoplasia, shortened umbilical cord, and/or polyhydramnios (Hall, 2009). Severity of the AMC phenotype is heterogeneous ranging from limited limb involvement to more extensive neurologic deficits including central nervous system impairment. AMC has been associated with chromosomal abnormalities (Reed, et al., 1985), mitochondrial disorders (von Kleist-Retzow, et al., 2003) and sporadic and inherited mutations in more than 220 genes, including genes that regulate central nervous system development (neuronal axonogenesis, Schwann and glial cell development), skeletal muscle, synaptic transmission, and glycoprotein metabolism (Bayram, et al., 2016; Hall and Kiefer, 2016; Narkis, et al., 2007). Recently, the first association of biallelic GLDN (MIM #608603) mutations with a lethal form of AMC (LCCS11; MIM# 617194) was reported in 4 unrelated families (Maluenda, et al., 2016). Here we report 6 additional infants and children from 4 unrelated families with AMC and biallelic GLDN mutations. Four of the affected infants survived beyond the neonatal period with intensive care, chronic ventilatory and nutritional support, and rehabilitation therapy. Our findings confirm the association of GLDN mutations with AMC and extend the LCCS11 phenotype by describing long-term survival with chronic respiratory and nutritional support.
Patients and exome sequencing
The parents/caregivers of all infants and children provided informed consent for participation. These studies were reviewed and approved by the Institutional Review Boards at each university. Sanger sequencing was used to confirm putative variants identified by exome sequencing. We used Genematcher (Sobreira, et al., 2015) to identify additional investigators with interest in GLDN. Variants were annotated in GenBank RefSeq transcript NM_181789.2, and were submitted to the Leiden Open Variation Database 3.0 at http://www.lovd.nl/GLDN.
Family1
After clinical exome sequencing of peripheral blood-derived DNA from the affected daughter and both parents using the Agilent Clinical Exome kit did not identify pathogenic variants, we performed research analysis of available variant call format (VCF) and BAM files. Specifically, variants were annotated with Annovar (version (ver) 2015Dec14) http://annovar.openbioinformatics.org/en/latest/)(Wang, et al., 2010) and pathogenicity of novel or rare (minor allele frequency <0.01 in the Exome Aggregation Consortium (ExAC) database (exac.broadinstitute.org) (Lek, et al., 2016) coding region variants was predicted using functional programs in dbNSFP (ver 3.3a, https://sites.google.com/site/jpopgen/dbNSFP) (Liu, et al., 2011; Liu, et al., 2016). Pathogenicity of variants in splicing consensus regions was assessed using the dbscSNV database (Liu, et al., 2016). Candidate genes were reviewed manually for possible associations with the infants’ phenotypes.
Family 2
Exome sequencing of DNA isolated from peripheral blood of the affected child was performed as described previously (Kremer, et al., 2016) using the SureSelect Human All Exon V5 kit (Agilent). Reads were aligned to GRCh37/hg19 using the Burrows-Wheeler Aligner (ver 0.7.5 a) (Li and Durbin, 2009). Single nucleotide variants and small insertions and deletions (indels) were detected with SAMtools (ver 0.1.19) (Li, et al., 2009).
Family 3
Targeted capture of coding regions (Illumina TruSight One panel) was performed on DNA isolated from the affected fetus. Reads were aligned to GRCh37/hg19 using the Burrows-Wheeler Aligner (ver 0.7.5 a) (Li and Durbin, 2009). Variants were called using GATK (DePristo, et al., 2011) and annotated using vtools (San Lucas, et al., 2012) and ANNOVAR (Wang, et al., 2010). Variant pathogenicity was predicted using dbNSFP (Liu, et al., 2013). Arthrogryposis and contractural syndrome genes were analyzed in a targeted manner but were non-diagnostic. Homozygosity mapping (www.homozygositymapper.com) of the exome sequencing data revealed a segment of homozygosity on chromosome 15. Given the familial recurrence and suspicion for an autosomal recessive disorder, whole exome sequencing (Illumina Nextera Coding Exome) was performed on DNA samples from the affected fetus, affected living child, and parents.
Family 4
Exome sequencing of the second affected child and parents and variant annotation were performed as described previously (Bahamonde, et al., 2015) using whole exome capture (Nimblegene SeqCap EZ Human Exome version 3). Variants were annotated using Annovar (Wang, et al., 2010) and an in-house annotation database. Variants in coding regions (nonsense, missense, frameshift and in-frame insertions or deletions) and near exon-intron junctions were prioritized using both functional annotations (e.g., population allele frequency, predicted pathogenicity, and conservation) and possible modes of inheritance (recessive, compound heterozygous, X-linked and dominant de novo) with the eDiVA pipeline (ver 1.0, http://ediva.crg.es).
Clinical Findings
Family 1
Three affected infants were consecutively born to unrelated, healthy, European-descent parents. The pregnancy of the first infant was uncomplicated except for breech presentation and decreased fetal movements. The infant was delivered at 38 weeks gestation via cesarean section for non-reassuring antenatal fetal surveillance (birth weight 2440g (5th percentile)). He required extensive resuscitation at birth including intubation, ventilation, chest compressions, and epinephrine administration, developed progressive respiratory failure, pneumothoraces, and pulmonary hemorrhage, and died at 40 hours of age (Table 1). His physical examination was consistent with AMC. Post-mortem magnetic resonance imaging (MRI) showed pulmonary hypoplasia and bilateral hip dislocations. Autopsy showed a mildly concave chest with severe pulmonary hypoplasia, a fistula from the left anterior descending artery to right ventricle, bilateral small kidneys with calcifications, an ectopic right ureter without signs of obstruction, and intraventricular hemorrhage. The skeletal muscle fibers were small for age and central nuclei suggested centronuclear myopathy. Electron microscopy of the spinal roots (lumbosacral level) demonstrated lengthening of the node of Ranvier (Figure 1A). Chromosomal microarray did not identify any clinically significant deletions (>200kb) or duplications (>500kb).
Table 1.
Clinical Characteristics of Infants and Children with Biallelic GLDN Variants and Arthrogryposis Multiplex Congenita (AMC)
| Family 1 | Family 2 | Family 3 | Family 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
| Current status | Died at 2 days from pulmonary hypoplasia and pulmonary hemorrhage | Alive at 22 months, tracheostomy and home ventilation | Alive at 7 months, tracheostomy and home ventilation | 17 years old, requires intermittent non-invasive mask ventilation | Pregnancy terminated at 31 weeks gestation | Alive at 28 months without ventilatory support | Pregnancy terminated at 27 weeks gestation | Died at 12 hours from respiratory failure |
| Pregnancy/delivery | Born at 38 weeks, intubated at birth for respiratory failure | Born at 37 weeks, intrauterine growth restriction, polyhydramnios, intubated at birth for respiratory failure | Born at 39 weeks, breech, intrauterine growth restriction, polyhydramnios, intubated at birth for respiratory failure | Born at 33 weeks, polyhydramnios, intubated at birth for respiratory failure | Fetal akinesia, polyhydramnios, skin edema | Born at 41 weeks, intubated at birth for respiratory failure | Fetal akinesia | Born at 36 weeks, polyhydramnios, intubated at birth for respiratory failure |
| Respiratory | Pulmonary hypoplasia, bilateral pneumothoraces, pulmonary hemorrhage, concave chest | Progressive respiratory insufficiency, laryngotracheo-malacia with cyanotic episodes, tracheostomy performed at 3 months, chronic ventilation | Has required respiratory support since birth, tracheostomy performed at 2 months, chronic ventilation | Pulmonary hypoplasia, tracheostomy performed at 3 months (decannulated at 7 years), without respiratory support from 7 to 14 years, since then intermittent non-invasive mask ventilation | No information | Paresis of right vocal cord and right side of the soft palate, tracheostomy performed at 6 weeks (decannulated at 24 months), without ventilatory support since 9 months | Pulmonary hypoplasia | Pulmonary hypoplasia and respiratory failure |
| Joint Abnormalities | Extension contractures of bilateral knees and ankles, bilateral clubfeet | Contractures of hips, knees fixed in extension, bilateral clubfeet, flexion contracture of left long finger, bilateral hip dislocation | Contractures of hips, knees, bilateral clubfeet, bilateral hyperextension of thumbs to radii | At birth: bilateral hip dislocation, contractures of knees and wrists, bilateral club feet. At age 17 years: bilateral hip luxation, mild contractures of shoulders, elbows, wrists, knees, ankles |
Lower limb extension contractures, finger flexion and wrist contractures | Bilateral hip flexion contractures with dislocated hips, extension contractures of knees, calcaneovalgus deformity of feet, normal upper extremities at birth and at 28 months | Extension contractures of hips and knees, flexion contractures of fingers | Extension contractures of hips and knees, flexion contractures of elbows, wrists and fingers, bilateral vertical talus |
| Neuromuscular | Skeletal muscle hypoplasia, decreased muscle bulk (especially of gastrocnemius, buttocks), fibrosis, fiber atrophy | Axial and appendicular hypotonia | Axial and appendicular hypotonia | Very weak or absent deep tendon reflexes, mild distal muscular atrophy, weakness affecting more the upper (MRC 3–4) than the lower limbs (MRC 4–5) | No information | Severe axial and appendicular hypotonia, distal muscular atrophy of lower limbs, weakness more pronounced in lower extremities than upper extremities, deep tendon reflexes very weak or absent in lower extremities, febrile seizure at 26 months | No information | Diffuse muscle atrophy/hypoplasia |
| Other congenital anomalies | Bilateral undescended testes, bilateral small kidneys, ectopic right ureter | Widened calvarium on radiograph | Widened anterior fontanelle, bilateral undescended testes | High palate, low set ears | No information | Right-sided cryptorchidism | No information | No additional anomalies on physical examination |
| Other clinical features | Microscopic calcifications in kidneys | Feeding difficulties, poor weight gain, gastrostomy tube placed at 2 months, normal echocardiogram | Feeding difficulties, poor weight gain, gastrostomy tube placed at 1 month | Progressive scoliosis, diaphragm paralysis, borderline intellectual functioning (IQ 74) | Narrow feet, deep longitudinal plantar crease | Feeding difficulties, gastrostomy tube placed at 4 months, delayed motor and language development | No information | Autopsy with diffuse hemorrhage involving the lungs, liver, kidney, pancreas, and adrenals |
| Muscle Biopsy Results | Abnormal muscle fibers (small for age), scattered muscle fibers with central nuclei suggestive of centronuclear myopathy, type II muscle fiber predominance | Type IIC muscle fibers estimated at less than 5% of total fibers, otherwise non-diagnostic | Not performed | Abnormal muscle with unspecific changes including increase of the variation of fiber diameter, few atrophic and some triangular muscle fibers | Not performed | Abnormal fibers with nonspecific changes | Not performed | Not performed |
| Neurophysiology | Not performed | Not performed | Not performed | Normal motor and sensory nerve conduction velocity, reduced MAP, pathologic SSEP indicating disturbed conduction in CNS | Not performed | Nerve conduction studies showed axonal motor and sensory neuropathy at 1 week of age. Normal motor and sensory nerve conduction velocity age 7 months | Not performed | Not performed |
| GLDN Variants | c.927_930del: p.Asn309Lysfs*5/c.1436G>C: p.Arg479Pro | c.927_930del: p.Asn309Lysfs*5/c.1436G>C: p.Arg479Pro | c.927_930del: p.Asn309Lysfs*5/c.1436G>C: p.Arg479Pro | Homozygous c.1305G>A: p.Trp435* |
Homozygous c.1305G>A: p.Trp435* |
Homozygous c.1305G>A: p.Trp435* |
Not performed | c.1178G>A: p.Arg393Lys/c.1428C>A: p.Phe476Leu |
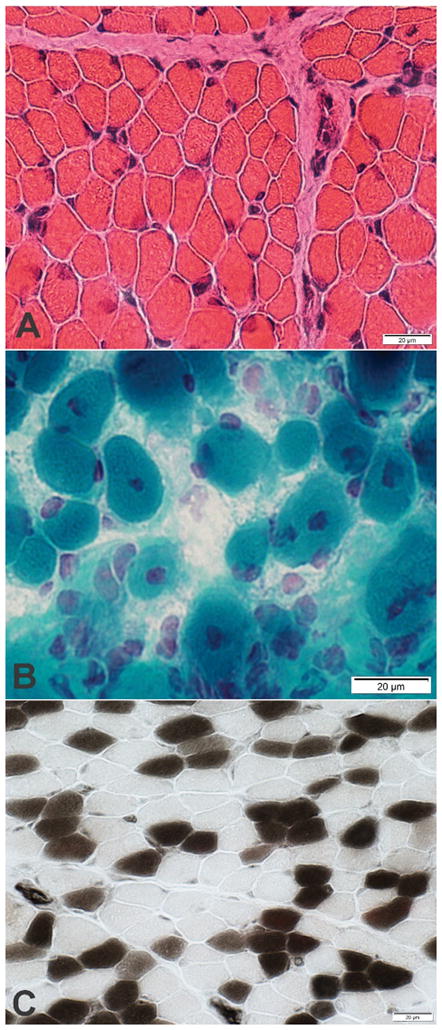
Figure 1.

Figure 1AB. Electron microscopy from spinal roots at the lumbosacral level from first affected infant in Family 1 (A) shows lengthening of the node of Ranvier (black arrows) as compared to control (B).
Figure 1C–E. Muscle biopsy from second affected infant in Family 1. C. Muscle fibers have varied sizes. Smaller fibers are intermediate sized and polygonal. (Hematoxylin & Eosin stain); D. More severely involved region of muscle. Fiber size is more varied. Some muscle fibers have central nuclei. (Gomori trichrome stain); E. Excessive numbers of immature, Type 2C, muscle fibers. The 2C fibers are small and explain some of the variation of muscle fiber size. There is type 2 muscle fiber predominance. (ATPase pH4.3 stain)
The second infant was born at 37 weeks gestation via repeat cesarean section (birth weight 2150g (3rd percentile)). Pregnancy was notable for intrauterine growth restriction, bilateral clubfeet, and polyhydramnios. The infant had minimal respiratory effort at birth, briefly required bag and mask ventilation, and then developed progressive hypoxemia and hypercarbia eventually requiring intubation and mechanical ventilation. Her physical exam was consistent with AMC (Table 1). Skeletal radiographs demonstrated widening of the calvarium. Laryngoscopy demonstrated laryngo- and tracheomalacia, floppy epiglottis, and pooling of oral secretions. She was extubated to CPAP at 3 days of life, and underwent chest computer tomography (CT) scan at 3 weeks which showed diffuse atelectasis. Gastrostomy tube was placed at 2 months for insufficient oral intake. Muscle biopsy demonstrated non-diagnostic decrease in type IIC muscle fibers (less than 5% of the population) (Figure 1C–E). After discharge from the neonatal intensive care unit on oxygen (1/8 liters per minute) at 2 months of age, she developed recurrent cyanotic events, attributed to dynamic airway collapse related to tracheomalacia, and polysomnography demonstrated central apnea. Brain MRI was normal. She continues to require chronic mechanical ventilation via tracheostomy at 22 months of age. She rolls independently, reaches for and throws toys, stacks blocks, and signs to communicate, but is unable to sit independently. She continues to have head lag with pull to sitting, slip-through with vertical suspension, decreased muscle tone, and decreased muscle strength in her upper (antigravity) and lower (non-antigravity) extremities.
The third infant was born at 39 weeks gestation via repeat cesarean section (birth weight 2420 grams (1st percentile)). Antenatal findings also demonstrated bilateral clubfeet, flexed wrists, extended knees, limited fetal movements, fetal weight less than 5th percentile, breech presentation, and polyhydramnios. The infant was intubated shortly after birth due to insufficient respiratory effort. Physical examination was consistent with AMC (Table 1). He was extubated to CPAP on the second day of life, but persistent respiratory failure prompted tracheostomy at 2 months of age. He is currently ventilator dependent at 7 months of age and has a gastrostomy tube for nutritional supplementation.
Family 2
A male infant was born at 32 weeks gestation to parents of East-European Roma origin (birth weight 1780 g, (50th percentile)) and has lived with a foster family since 6 years of age. Information about his disease course, neurocognitive development during the first years of life, and family history including consanguinity is limited (Table 1). Pregnancy was complicated by polyhydramnios, and he was delivered by cesarean section due to non-reassuring fetal surveillance. He required resuscitation, intubation, and ventilation immediately after birth due to respiratory failure. Physical examination was consistent with AMC (Table 1). Due to persistent respiratory insufficiency associated with pulmonary hypoplasia and diaphragm paralysis, he underwent tracheostomy at 3 months of age.
He underwent surgical treatment for bilateral hip luxation and clubfeet (age 2 years) and scoliosis with Harrington rod insertion (age 9 years). Over his first decade of life, he successfully weaned from respiratory support including tracheostomy decannulation at 7 years of age. By 12 years of age, he was independent in his daily life activities, fully ambulant, able to dress himself, and ride a bicycle over more than 10 kilometers. His neurologic examination was notable for myopathic facies, very weak to absent deep tendon reflexes, and symmetric atrophy and weakness of his proximal muscles.
At 14 years of age, he rapidly deteriorated following an upper respiratory tract infection. He continues to require noninvasive mask ventilation during sleep and when supine now at age 17 years. He is wheelchair dependent due to reduction of muscle strength affecting upper and lower limbs with a slight proximal predominance. A muscle biopsy at the time of deterioration showed non-specific changes with an increase of the variation of fiber diameter, few atrophic fibers, and some triangular fibers. Nerve conduction velocity studies suggested axonopathy with reduced amplitudes, although some nerves demonstrated reduced conduction velocity. Somatosensory evoked potentials indicated abnormal conduction in the central nervous system. MRI of the brain was normal, and MRI of the spine showed narrowing of the cervical spinal canal without signs of myelopathy. Intensive rehabilitation measures modestly improved his motor abilities. The patient regained the ability to rise from his wheelchair, to walk a few steps without support, and lift his hands to his head. Neuropsychological evaluation at age 16 years using the Wechsler Intelligence Scale for Children IV revealed a full-scale score of 74 consistent with borderline intellectual functioning. He attends a school for physically and intellectually disabled children.
Family 3
After the birth of two unaffected females to healthy, unrelated parents of East European-descent, a third pregnancy was complicated by reduced fetal movements, multiple contractures, narrow thorax, polyhydramnios, and skin edema first noticed at 30 weeks of gestation and pregnancy was terminated at 31 weeks. Physical examination of the female fetus (body weight 1770 g (80th percentile)) was consistent with AMC (Table 1). Autopsy information is not available for this fetus.
The fourth child (male) was born at 41 weeks gestation by vaginal delivery after a reportedly normal pregnancy (birth weight 3560 g (30th percentile)). Due to respiratory distress at birth, the infant was intubated and mechanically ventilated. His physical examination at birth was consistent with AMC (Table 1). Due to persistent respiratory insufficiency, inability to cough, and insufficient swallowing, he underwent tracheostomy at 6 weeks of age. His respiratory function gradually improved, and he was successfully weaned from ventilatory support at nine months of age and subsequently decannulated at 19 months of age. Physiotherapy and non-surgical treatment including splinting led to an improvement of his contractures, increased range of motion, and improved muscular tone in his lower limbs. Distal muscular atrophy developed over time. Motor and language development was delayed; he was able to sit independently at age 13 months and started to crawl at age 24 months. At age 28 months, he uses 5 words and understands and follows simple commands. Insufficient swallowing aggravated by paresis of both the right vocal cord and right side of the soft palate prompted gastrostomy tube placement at 4 months of age. Oral feedings were initiated at 1 year of age and continue at age 28 months with most of his feedings by mouth and nutritional supplementation via gastrostomy tube. His clinical course was also notable for orchidopexy at 5 months of age and a febrile seizure at 26 months of age. Nerve conduction velocity studies performed at 1 week of age demonstrated axonal motor and sensory neuropathy but were normal when repeated at 7 months of age. Electromyography did not show any pathological finding. Muscle biopsy and MRI of the brain and spinal cord were normal.
Family 4
After the birth of an unaffected son to healthy, unrelated parents of Spanish-Belarussian descent, a second pregnancy was complicated by antenatal findings of fetal akinesia with hyperextended knees, flexed arms, and closed hands and the pregnancy was terminated at 27 weeks gestation. The autopsy of this male fetus (weight 930g (45th percentile)) demonstrated pulmonary hypoplasia and physical findings consistent with AMC (Table 1). A subsequent pregnancy was notable for polyhydramnios and antenatal ultrasound findings of arthrogryposis and pulmonary hypoplasia at 26 weeks gestation. Fetal MRI performed at 32 weeks showed normal brain and spinal cord images. The female infant was born at 37 weeks gestation and died at 12 hours of age due to respiratory failure. Her physical examination was consistent with AMC (Table 1). Anatomical examination of the brain and spinal cord revealed edematous and friable tissue, but no obvious macroscopic or histopathologic abnormalities.
Exome Sequencing Results
In Family 1, all 3 affected infants had biallelic, novel GLDN mutations: a heterozygous frameshift deletion (c.927_930del) which results in a premature stop codon (p.Asn309Lysfs*5) and a pathogenic missense mutation c.1436G>C, p.Arg479Pro (CADD (Kircher, et al., 2014) score of 24, predicted damaging by majority of in silico functional prediction programs in ANNOVAR) (Table 1, Supplemental Figure S1).
In Family 2, the affected male infant was homozygous for c.1305G>A, p.Trp435* which results in a premature stop codon in the last exon of GLDN (p.Trp435*). This mutation is observed in 4 heterozygous individuals of non-Finnish European descent in ExAC (allele frequency 0.000033, dbSNP: rs775011495:G>A). The same homozygous mutation (c.1305G>A (p.Trp435*)) was also identified in the affected fetus and affected living child of Family 3.
In Family 4, the proband was compound heterozygous for extremely rare, missense GLDN mutations: c.1178G>A, p.Arg393Lys (ExAC: 8 heterozygous individuals, allele frequency 0.000068, dbSNP: rs147954907:G>A) and c.1428C>A, p.Phe476Leu (ExAC: 1 heterozygous individual, allele frequency 0.0000082, dbSNP: rs750803388:C>A). The c.1428C>A, p.Phe476Leu mutation is predicted to be highly damaging by in silico prediction programs in ANNOVAR (Wang, et al., 2010), while medium (“possibly damaging”) to low pathogenicity scores are predicted for the c.1178G>A, p.Arg393Lys mutation. Both mutations affect positions highly conserved in mammals (PhastCons = 1).
All five GLDN mutations (p.Asn309Lysfs*5, p.Arg479Pro, p.Trp435*, p.Arg393Lys, p.Phe476Leu) identified in these 8 patients are located within the conserved extracellular olfactomedin domain of gliomedin which binds axonal immunoglobulin cell adhesion molecules neurofascin 186 (NF186) and NrCAM to induce clustering of sodium channels at heminodes of myelinating Schwann cells (Eshed, et al., 2005; Han and Kursula, 2015). Functional studies of GLDN mutations within the olfactomedin domain performed in Chinese hamster ovary (CHO) cells demonstrate reduced surface localization of gliomedin and loss of NF186 binding compared to wild-type (Maluenda, et al., 2016). We speculate that the genetic variants identified in our series may have similar effects. It is possible that the p.Asn309Lysfs*5 variant may result in loss of protein function by nonsense mediated mRNA decay.
The p.Asn309Lysfs*5 and p.Arg479Pro variants are novel. A fetus with bilateral clubfeet, pulmonary hypoplasia, and distal arthrogryposis of the hands was found to be homozygous for a nonsense variant at codon 479 (c.1435C>T; p.Arg479*) (Maluenda, et al., 2016), and 8 individuals in ExAC are heterozygous for other variants at this codon (p.Arg479*, p.Arg479Gly, p.Arg479Gln). The p.Trp435* (c.1305G>A) variant was identified among 4 heterozygous individuals in ExAC, but no homozygous individuals are reported. The homozygous p.Trp435* variants in 2 unrelated infants of Eastern European-descent families suggest a possible founder effect among this ethnic group. Of note, a non-Finnish European descent individual heterozygous for c.1304G>A which also results in p.Trp435* is reported in ExAC. The two variants (p.Arg393Lys, p.Phe476Leu) identified in family 4 are both reported in ExAC, but extremely rare (minor allele frequency <0.0001) with no homozygous individuals reported. The collapsed frequency of GLDN variants predicted to be deleterious by the majority of in silico programs in ANNOVAR (Wang, et al., 2010) described above is 1.4% among non-Finnish European descent individuals and 1.0% among African-descent individuals, suggesting the incidence of AMC related to GLDN variants could be 2–5/100,000 births.
Homozygous gldn−/− mice do not have apparent neurologic deficits or features of arthrogryposis and demonstrate normal nerve conduction and peripheral nervous system myelin (Feinberg, et al., 2010). However, the Schwann cell microvilli from these mice appear disorganized with abnormal orientation to the axolemma, and the sciatic nerves lack sodium channel clustering at the heminodes (Feinberg, et al., 2010). Antibodies to gliomedin and neurofascin have been identified among patients with Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy, suggesting the node of Ranvier is the primary immunologic target (Devaux, 2012). Both active and passive immunity to gliomedin results in a progressive neuropathy among Lewis rats characterized by decreased nerve conduction velocity, nodal elongation, paranodal demyelination, and disruption of sodium channel clustering (Devaux, 2012). Spinal root ultrastructure of the first male infant of Family 1 demonstrates lengthening of the node of Ranvier (Figure 1A), similar to a previous report of an infant with recessive GLDN mutations (Maluenda, et al., 2016). Muscle biopsy from the female infant in Family 1 demonstrates muscle fibers of varied sizes including some with central nuclei and type 2 muscle fiber predominance with excessive numbers of immature type 2C muscle fibers (Figure 1C–E). The difference between murine gldn−/− and human GLDN biallelic mutation phenotypes may be attributable to differences between genetic abrogation of GLDN expression and expression of abnormal gliomedin protein.
Of the 6 previously reported infants with biallelic GLDN mutations and AMC, two live- born infants died within the first day of life likely due to pulmonary insufficiency (Maluenda, et al., 2016). Survival of 4 infants in our series (currently at ages 7 months, 22 months, 28 months and 17 years) suggests that pulmonary insufficiency in patients with AMC due to biallelic GLDN mutations may not necessarily be life-limiting. The three children surviving beyond infancy had considerably delayed milestones, but their motor development improved with chronologic age, therapy services, and aggressive nutritional and respiratory support. Even among infants with the same genotype (Family 1) there is variability in disease severity, suggesting that other genetic or environmental factors modify phenotype. Our findings confirm the previous report of GLDN mutations associated with AMC (LCCS 11) and extend the phenotypic spectrum of affected infants and children.
Supplementary Material
Acknowledgments
Grant Sponsor: This work was supported by grants from the National Institutes of Health (K08 HL105891 (JAW), K12 HL120002 (FSC), R21/33 HL 120760 (FSC)), the Children’s Discovery Institute (FSC, JAW), the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (grant #FKZ 01ZX1405C (TBH)), the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013–2017’ (SEV-2012-0208), the CERCA Programme/Generalitat de Catalunya and the “la Caixa” Foundation.
The authors thank the families for their participation in these research studies. The authors thank GeneDx© for sharing the VCF and BAM files for research analysis and the Exome Aggregation Consortium and the groups that provided exome variant data for comparison; a full list of contributing groups can be found at http://exac.broadinstitute.org/about. Author contributions and conflicts of interest: Conception and design: JAW, GMS, DJW, MS, DKG, DB, FSC, RS. Acquisition, analysis, or interpretation of data: JAW, GMS, TBH, KW, AS, AM, FM, SO, MB, DJW, DB, MS, BA, TMS, DKG, RT, EW, JC, BBW, RES, AP, FSC, RS. Drafting and revising the manuscript for important intellectual content: JAW, GMS, DJW, DB, MS, DKG, BBW, FSC, RS. All authors have approved the final version and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. None of the authors has conflicts of interest to declare. All of the authors have seen and approved the submission and take full responsibility for the manuscript.
References
- Bahamonde MI, Serra SA, Drechsel O, Rahman R, Marce-Grau A, Prieto M, Ossowski S, Macaya A, Fernandez-Fernandez JM. A Single Amino Acid Deletion (DeltaF1502) in the S6 Segment of CaV2.1 Domain III Associated with Congenital Ataxia Increases Channel Activity and Promotes Ca2+ Influx. PLoS One. 2015;10(12):e0146035. doi: 10.1371/journal.pone.0146035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram Y, Karaca E, Coban Akdemir Z, Yilmaz EO, Tayfun GA, Aydin H, Torun D, Bozdogan ST, Gezdirici A, Isikay S, Atik MM, Gambin T, Harel T, El-Hattab AW, Charng WL, Pehlivan D, Jhangiani SN, Muzny DM, Karaman A, Celik T, Yuregir OO, Yildirim T, Bayhan IA, Boerwinkle E, Gibbs RA, Elcioglu N, Tuysuz B, Lupski JR. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016;126(2):762–78. doi: 10.1172/JCI84457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ. Antibodies to gliomedin cause peripheral demyelinating neuropathy and the dismantling of the nodes of Ranvier. Am J Pathol. 2012;181(4):1402–13. doi: 10.1016/j.ajpath.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed Y, Feinberg K, Poliak S, Sabanay H, Sarig-Nadir O, Spiegel I, Bermingham JR, Jr, Peles E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron. 2005;47(2):215–29. doi: 10.1016/j.neuron.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, Peles E. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65(4):490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JG. Pena-Shokeir phenotype (fetal akinesia deformation sequence) revisited. Birth Defects Res A Clin Mol Teratol. 2009;85(8):677–94. doi: 10.1002/bdra.20611. [DOI] [PubMed] [Google Scholar]
- Hall JG, Kiefer J. Arthrogryposis as a Syndrome: Gene Ontology Analysis. Mol Syndromol. 2016;7(3):101–9. doi: 10.1159/000446617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Kursula P. The olfactomedin domain from gliomedin is a beta-propeller with unique structural properties. J Biol Chem. 2015;290(6):3612–21. doi: 10.1074/jbc.M114.627547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer LS, Distelmaier F, Alhaddad B, Hempel M, Iuso A, Kupper C, Muhlhausen C, Kovacs-Nagy R, Satanovskij R, Graf E, Berutti R, Eckstein G, Durbin R, Sauer S, Hoffmann GF, Strom TM, Santer R, Meitinger T, Klopstock T, Prokisch H, Haack TB. Bi-allelic Truncating Mutations in TANGO2 Cause Infancy-Onset Recurrent Metabolic Crises with Encephalocardiomyopathy. Am J Hum Genet. 2016;98(2):358–62. doi: 10.1016/j.ajhg.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG Exome Aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32(8):894–9. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34(9):E2393–402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat. 2016;37(3):235–41. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry RB, Sibbald B, Bedard T, Hall JG. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res A Clin Mol Teratol. 2010;88(12):1057–61. doi: 10.1002/bdra.20738. [DOI] [PubMed] [Google Scholar]
- Maluenda J, Manso C, Quevarec L, Vivanti A, Marguet F, Gonzales M, Guimiot F, Petit F, Toutain A, Whalen S, Grigorescu R, Coeslier AD, Gut M, Gut I, Laquerriere A, Devaux J, Melki J. Mutations in GLDN, Encoding Gliomedin, a Critical Component of the Nodes of Ranvier, Are Responsible for Lethal Arthrogryposis. Am J Hum Genet. 2016;99(4):928–933. doi: 10.1016/j.ajhg.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkis G, Landau D, Manor E, Ofir R, Birk OS. Genetics of arthrogryposis: linkage analysis approach. Clin Orthop Relat Res. 2007;456:30–5. doi: 10.1097/BLO.0b013e3180312bee. [DOI] [PubMed] [Google Scholar]
- Reed SD, Hall JG, Riccardi VM, Aylsworth A, Timmons C. Chromosomal abnormalities associated with congenital contractures (arthrogryposis) Clin Genet. 1985;27(4):353–72. doi: 10.1111/j.1399-0004.1985.tb02278.x. [DOI] [PubMed] [Google Scholar]
- San Lucas FA, Wang G, Scheet P, Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics. 2012;28(3):421–2. doi: 10.1093/bioinformatics/btr667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–30. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist-Retzow JC, Cormier-Daire V, Viot G, Goldenberg A, Mardach B, Amiel J, Saada P, Dumez Y, Brunelle F, Saudubray JM, Chretien D, Rotig A, Rustin P, Munnich A, De Lonlay P. Antenatal manifestations of mitochondrial respiratory chain deficiency. J Pediatr. 2003;143(2):208–12. doi: 10.1067/S0022-3476(03)00130-6. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.