

Abstract
Developmental glaucoma can occur as an isolated or syndromic condition and is genetically heterogeneous. We describe a three-generation family affected with developmental glaucoma, myopia, and/or retinal defects associated with variable craniofacial/dental, auditory, brain, renal, and limb anomalies. Whole exome sequencing identified a heterozygous c.124T>C, p.(Trp42Arg) allele in ADAMTSL1; co-segregation analysis confirmed the presence of this allele in four affected family members. The mutation affects a highly conserved residue and is strongly predicted to have a deleterious effect on protein function. Trp42 is normally modified by protein C-mannosylation, an unusual post-translational modification. Comparison of ADAMTSL1-WT (also known as punctin-1) and ADAMTSL1-p.Trp42Arg in vitro demonstrated that the latter was not secreted from transfected cells but retained intracellularly. Moreover, ADAMTSL1-p.Trp42Arg reduced secretion of co-transfected wild-type ADAMTSL1 suggesting a dominant negative effect for this mutation. These data imply a multi-system role for ADAMTSL1 and present the first disease-associated variant affecting a C-mannosylation motif.
Keywords: ADAMTSL1, C-mannosylation motif, Glaucoma, Myopia
Glaucoma diagnosed in infancy is typically designated as primary congenital glaucoma (PCG) in the absence of visible structural ocular anomalies or developmental glaucoma if associated with anterior segment dysgenesis (ASD) (Reis and Semina 2011). Variants in several genes including CYP1B1, LTBP2, MYOC, PAX6, FOXC1 and, most recently, TEK can cause non-syndromic congenital glaucoma with or without anterior segment dysgenesis (Wang and Wiggs 2014; Souma et al. 2016). When accompanied by systemic anomalies, congenital or developmental glaucoma can be part of Axenfeld-Rieger syndrome (MIM#180500 and 602482), Peters plus syndrome (MIM# 261540), SHORT syndrome (MIM# 269880), and other conditions. While causative genetic factors have been identified for many of these conditions, a large number of families remain undiagnosed due to the extreme genetic heterogeneity and possible involvement of additional unknown genes.
A three-generation family affected with a variable phenotype involving glaucoma (Figure 1, Table 1) was enrolled into this study approved by the Institutional Review Board of the Children’s Hospital of Wisconsin. The pedigree included ten affected individuals with a highly variable phenotype of ocular anomalies (congenital glaucoma, myopia, retinal detachment, and/or Axenfeld-Rieger anomaly), congenital hypothyroidism, hearing loss, microcephaly, dental defects, kidney anomalies, vascular anomalies in the brain, and distal limb anomalies. The overall clinical phenotype observed in this family, while showing some overlap with previously reported ocular syndromes, did not completely fit into any recognized condition.
Figure 1. A–F. Patient clinical photographs and images.
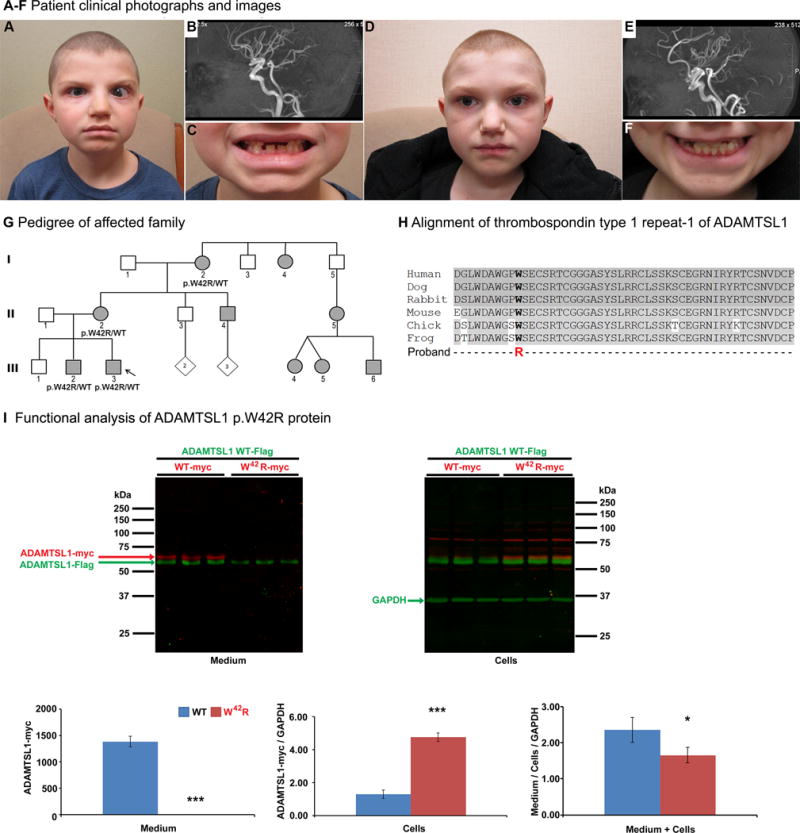
A. Facial photograph of proband at ~9½ years of age shows buphthalmos with enlarged cornea and esotropia of the left eye, brachycephaly and microcephaly, cupid’s bow upper lip, short philtrum, broad flat nasal bridge, slightly large and prominent ears, and square face with prominent jaw. B. Head MRA of the proband showing mild arterial tortuosity within the vertebrobasilar system. C. Delayed dental eruption can be seen in missing central upper incisors and retained primary upper lateral incisors which typically erupt/shed at 7–8 years of age (ADA 2005; 2006). D. Facial photograph of affected brother at ~11½ years of age. Please note brachycephaly and microcephaly, prominent cupid’s bow upper lip, mild midface hypoplasia, slightly large and prominent ears, and square face with prominent jaw. E. Head MRA of the affected brother showing marked tortuosity of the proximal arteries in the circle of Willis. F. Delayed dental eruption can be seen in missing upper secondary lateral incisors and lower right canine which typically erupt at 8–9 and 9–10 years of age, respectively (ADA 2006). G. Pedigree of affected family. Proband is indicated with an arrow. ADAMTSL1 genotype is indicated for tested individuals. Filled individuals indicate a variable phenotype of glaucoma, anterior segment dysgenesis, myopia, and/or retinal defects associated with microcephaly, dental defects, hearing loss, hypothyroidism, renal, vasculature, and/or limb anomalies (see Table 1 for details). H. Alignment of thrombospondin type 1 repeat-1 of ADAMTSL1 in different species. ADAMTSL1 sequences with the following UNIPROT IDs were utilized: human, Q8N6G6; canine, F1PC42; rabbit, XP_008252660; mouse, Q8BLI0; chick, E1C7J4; Xenopus tropicalis, F6YFJ8. I. Functional analysis of ADAMTSL1 p.W42R protein. Images of Western blots of medium and cell layer from triplicate HEK293T cell cultures expressing myc-His6 tagged wild-type or p.Trp42Arg ADAMTSL1 and co-transfected wild-type FLAG-tagged ADAMTSL1 are shown at the top. A two-color western blot system shows myc-tagged proteins in red and the FLAG-tagged wild type protein in green. The myc-His6 tag makes the corresponding mutant and wild-type proteins slightly larger than FLAG-tagged punctin-1. Note that p.Trp42Arg ADAMTSL1 is absent in the medium of the transfected cells and accumulates intracellularly (red). Note the visible reduction of FLAG-tagged ADAMTSL1 in the presence of p.Trp42Arg ADAMTSL1. Western blotting with a GAPDH antibody (green) was used for normalization of the cellular levels of transfected proteins for quantitation. At the bottom, the quantitative analysis of myc-tagged p.Trp42Arg ADAMTSL1 levels is presented. The data show absence of myc-tagged p.Trp42Arg in the medium (left-hand panel, ***p<0.0001) and accumulation in cells (center panel, ***p<0.0001). Quantitation of co-transfected wild-type ADAMTSL1, shown as the ratio of observed signal in medium/cells/GAPDH indicates that it is secreted at lower levels in the presence of p.Trp42Arg ADAMTSL1 than wild-type, supporting a dominant negative effect for the mutant protein (*p=0.038).
Table 1.
Phenotypic features in affected family members.
| Features | III-2 | III-3* | II-2 | I-2 | II-4 | I-4 | II-5 | III-4 | III-5 | III-6 |
|---|---|---|---|---|---|---|---|---|---|---|
| Ocular: | ||||||||||
| ARA and/or glaucoma | − | + | − | + | − | − | + | + | + | + |
| Myopia | + | + | + | + | + | U | U | U | U | U |
| Retinal detachment/degeneration | − | − | − | + | + | + | U | U | U | U |
| Craniofacial: | ||||||||||
| Microcephaly | + | + | + | + | U | U | + | + | + | + |
| Dysmorphic facial features | + | + | + | U | U | U | + | + | + | + |
| Square face with prominent jaw | + | + | + | U | U | U | U | + | + | U |
| Hearing loss | + | + | + | + | U | U | − | + | − | − |
| Delayed dental eruption | + | + | + | U | U | U | U | U | U | U |
| Premature loss of permanent teeth | U | U | + | + | U | U | U | U | U | U |
| Other: | ||||||||||
| Hypothyroidism | + | + | − | − | U | U | − | + | + | + |
| Brain vascular anomalies | + | + | U | U | U | U | U | U | U | U |
| Limb anomalies | + | + | − | U | U | U | + | + | + | + |
| Kidney anomalies | − | − | + | + | U | U | U | U | U | U |
| ADAMTSL1 variant | + | + | + | + | U | U | U | U | U | U |
+ feature present; − feature absent; U unknown,
indicates proband
The proband is a 9-year-old Caucasian male with Axenfeld-Rieger anomaly (ARA), congenital glaucoma with buphthalmos (left worse than right; Figure 1A), myopia, high frequency sensorineural hearing loss, migraines, congenital hypothyroidism due to thyroid dysgenesis, and other anomalies. Head MRA (Magnetic Resonance Angiogram) showed mild arterial tortuosity within the vertebrobasilar system (Figure 1B) and MRI (Magnetic Resonance Imaging) showed areas of delayed myelination/demyelination in the periventricular white matter and subcortical white matter of the frontal lobes (possibly related to the congenital hypothyroidism). Craniofacial features included brachycephaly and microcephaly with a head circumference of 48.5 cm (<3rd centile), prominent cupid’s bow upper lip, short philtrum, broad flat nasal bridge, slightly large and prominent ears, square face with prominent jaw, and delayed eruption of permanent teeth (Figure 1A and C). Limb anomalies included calcaneovalgus deformity of his feet treated with casting, femoral retroversion, and long tapered fingers with parallel palmar creases. He had a fractured clavicle at birth. Height and weight were near the 10th centile. Chromosomal microarray was normal.
The proband’s brother, age 11, has myopia but is not reported to have other ocular anomalies, has high-frequency sensorineural hearing loss, migraines, congenital hypothyroidism and other systemic abnormalities (Figure 1D). Head MRA showed marked tortuosity of the proximal arteries in the circle of Willis, which is an arterial anastomosis that connects the internal carotid and vertebrobasilar arterial systems (Figure 1E). Brain MRI showed numerous subcortical T2 hyperintensities in the bifrontal white matter, the deep white matter of the corona radiata bilaterally, and the corpus callosum. Craniofacial features include brachycephaly and microcephaly (<3rd centile), mild metopic prominence, prominent cupid’s bow upper lip, mild midface hypoplasia, slightly large and prominent ears, square face with prominent jaw, and delayed eruption of permanent teeth (Figure 1D and F). Limb features comprised mild webbing between the fingers with slight spatulate enlargement to the distal fingertips. Height and weight were near the 10th centile.
The proband’s mother has moderate-to-high myopia (−7.00), blue scleral hue and a history of strabismus surgery at age 13, migraines, microcephaly, hearing loss, medullary sponge kidney, and delayed dental eruption followed by early loss of permanent teeth (20–30 years of age). A maternal uncle to the proband is reported to have similar ocular features and retinal detachment but was not available for review. The proband’s grandmother has a history of bilateral extreme myopia (−25.00), congenital glaucoma requiring multiple surgeries, cataracts requiring surgery at 38 years of age, and myopic degeneration. Iris anomalies included an abnormal pupil and synechiae in the left eye and neovascularization of the iris in the right. She also has microcephaly (52 cm), hearing loss, nephrolithiasis, migraines, and early loss of permanent teeth (20–30 years of age). A sister to the grandmother is reported to have retinal detachment but no other details are available.
In addition, a niece to the grandmother (through a reportedly unaffected brother) is reported to have congenital glaucoma, microcephaly, and clinodactyly. This woman’s three children are similarly affected with congenital glaucoma, ARA, microcephaly, congenital hypothyroidism, developmental delay, flat mid-face, and thin upper lip. One of the three also has hearing loss and digital hypoplasia and one has nail hypoplasia (Bitrian et al. 2013).
Targeted gene sequencing and copy number variation analyses (performed in the laboratory and/or commercially) excluded causative mutations in major genes associated with ARA and congenital glaucoma, namely PITX2, CYP1B1, and FOXC1 (Reis and Semina 2011). Thus, whole exome sequencing of DNA samples from the proband, brother, and grandmother was undertaken. Library capture was completed using the Agilent Sure Select v4 capture kit (Santa Clara, CA) and 100 base pair paired end-sequencing was performed using the Illumina HiSeq 2000 at Axeq Technologies (Rockville, MD). The obtained data were aligned using the Burrows-Wheeler Aligner (BWA) and variants were called using the SAMTOOLS analysis pipeline available through Axeq. Exome data were analyzed using the SNP & Variation Suite (Golden Helix, Bozeman, MT) to identify variants that are shared between the affected individuals and then prioritized based on their absence/rarity in the general population (as reported in publicly available databases dbSNP (http://www.ncbi.nlm.nih.gov/snp), ExAC Browser (http://exac.broadinstitute.org/), and gnomAD (http://gnomad.broadinstitute.org/)) and possible effect on protein function; for missense variants, functional profiling was performed through the Golden Helix dbNSFP Functional Predictions 2.3 track using SIFT, Polyphen2, Mutation Taster, MutationAssessor, FATHMM, and RadialSVM as well as nucleotide conservation score GERP++ RS. This analysis identified several rare variants (Supp. Table S1) with a heterozygous ADAMTSL1 (MIM# 609198) missense variant, c.124T>C, p.(Trp42Arg) (Supp. Figure S1A), determined to represent the strongest candidate for the affected phenotype because it: 1) was predicted to be damaging by five out of six effect prediction programs listed above; 2) demonstrated high conservation at both nucleotide and protein level (Supp. Table 1 and Figure 1H); 3) represented an ultra-rare allele observed only in a single case out of 245,358 general population alleles in gnomAD; 4) involved a gene with a robust expression profile showing enrichment in relevant mouse ocular tissues such as the iris, cornea, ciliary body, retina and eyecup (BioGPS portal http://biogps.org; Supp. Figure S1B); and 5) involved a family with two related genes, namely ADAMTSL2 and ADAMTSL4, being involved in human connective tissue and ocular anterior segment disorders; specifically, ADAMTSL2 mutations lead to the generalized connective tissue disorder geleophysic dysplasia, which includes dysmorphic facial features (Le Goff and Cormier-Daire 2011), and ADAMTSL4 mutations are the most common genetic cause of recessive ectopia lentis (MIM #225100) and ectopia lentis et pupillae (MIM #225200), which include iris/pupil anomalies, cataract, myopia, glaucoma, and retinal detachment in some cases (Ahram et al. 2009; Christensen et al. 2010).
To verify the presence of the variant, DNA from all available family members was amplified using the following primers: exon 2_F, AGAATCTGATTGCGCGTTTT and exon 2_R, CAAATAAAAACACGTAAGAACACAAA (PCR product= 436 bp). PCR products were sequenced bidirectionally using Big Dye Terminator chemistry and the ABI 3730XL sequencer (Applied Biosystems/Life Technologies, Carlsbad, CA, USA). Sequences were reviewed manually and using Mutation Surveyor (SoftGenetics, State College, PA) and compared to the NM_001040272.5 transcript. The variant was found to be present in the affected brothers, mother, and grandmother; no other family members were available for testing (Figure 1G). This variant has been deposited to the LOVD ADAMTSL1 database (https://databases.lovd.nl/shared/genes/ADAMTSL1).
The ADAMTS (A Disintegrin-like and Metalloproteinase with Thrombospondin type 1 motifs) superfamily contains nineteen secreted metalloproteinases involved in various biological processes including extracellular matrix (ECM) degradation, matrix assembly, angiogenesis and cell migration; it also contains a family of seven ADAMTS-like proteins (ADAMTSL family) comprising an ADAMTS ancillary domain but lacking the protease domain and thus, enzymatic activity. Many of these genes are affected in human genetic disease (Dubail and Apte 2015; Le Goff and Cormier-Daire 2011). As mentioned above, ADAMTSLs are functionally associated with inherited connective tissue disorders, including ocular phenotypes, and have been shown to localize to or affect the formation of fibrillin microfibrils in ECM (Hubmacher and Apte 2015; Ahram et al. 2009; Christensen et al. 2010). Despite being the prototypic ADAMTSL, little is currently known about ADAMTSL1, which contains thirteen thrombospondin type 1 repeats (TSRs), four Immunoglobulin-like C2-type domains and a single PLAC (protease and lacunin) domain in its full-length form (1762 amino acids) and four TSRs in a short splice variant named punctin-1 (525 amino acids) (Apte 2009; Wang et al. 2007). TSRs have a highly conserved three-layered fold stabilized by three disulfide bonds and stacked side chains of Trp and Arg residues near the N-terminus. Moreover, they undergo two unusual post-translational modifications that can regulate secretion, termed C-mannosylation and O-fucosylation.
C-mannosylation occurs on Trp residues within the sequence motif W0XXW3 or W0XXC3. Previously, using ADAMTSL1 as a prototype for the protein family, we analyzed C-mannosylation of TSR1, which contains the sequence W36DAW39GPW42SECSRT48C (bold indicates Trp residues within C-mannosylation motifs, the underlined sequence is the adjacent consensus motif for O-fucosylation at Thr48). Trp42, according to the consensus sequence W0SXXC+3 serves as the W0 recipient for a mannose as well as the +3 residue in the context of C-mannosylation at Trp39. The Trp42 residue was previously demonstrated to be C-mannosylated using mass spectral glycoproteomics (Wang et al. 2009) and is highly conserved (Figure 1H). To analyze its significance, an engineered mutation, p.Trp42Ala, was previously generated in punctin-1. In contrast to wild-type punctin-1, which was efficiently secreted and detected in medium as a 60 kDa protein, p.Trp42Ala punctin-1 was poorly secreted and accumulated intracellularly (Wang et al. 2009). This suggested that C-mannosylation, like O-fucosylation, is potentially involved in protein folding.
The ADAMTSL1 allele identified here is the first natural disease-associated variant affecting a C-mannosylation motif and its functional impact was therefore analyzed in-depth (Figure 1I). For this analysis, myc-His6 tagged p.Trp42Arg punctin-1 or wild-type punctin-1 were co-transfected with FLAG-tagged wild-type punctin-1 in HEK293T cells to determine whether the variant affected secretion from cells and to quantify a possible impact of the mutant construct on the secretion of wild-type protein. Extracts of the cell layer and conditioned medium were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by western blotting. The western blots were performed using rabbit anti-myc polyclonal antibody (Sigma-Aldrich, St. Louis, MO; catalog no. C3956, diluted 1:2000), mouse anti-FLAG monoclonal antibody (Sigma-Aldrich, St. Louis, MO, catalog no. F3165 diluted 1:2000) and mouse anti-GAPDH monoclonal antibody (EMD Millipore, Billerica, MA; catalog no. MAB374, diluted 1:10,000), quantitatively imaged using the Odyssey CLx scanner (LI-COR Biosciences, Lincoln, NE) and, assuming normal distribution, compared using a parametric two-sample T-Test, equal variance (Microsoft Excel). These analyses illustrated the key effects of the mutation: 1) p.Trp42Arg punctin-1 was not secreted and accumulated in cells and 2) in the presence of the mutant construct, wild-type punctin-1 was secreted at reduced levels and also accumulated intracellularly (Fig. 1I). These findings suggest that the missense variant constitutes a de facto null allele and demonstrate a dominant negative effect on the wild-type protein.
Substitution of conserved Trp residues of TSRs may disrupt stacking with Arg residues present in the anti-parallel β-strand and thus interfere with protein folding and secretion. O-fucosylation occurs adjacent to C-mannosylation in TSRs and is already established as a modification necessary for the secretion of TSR-containing proteins (Wang et al. 2007). The basis for the dominant negative effect is not yet understood since little is known about possible intra- or inter-molecular complexes formed by ADAMTSL1 or its function, but we speculate that the mutant protein may aggregate with the wild-type or its normal binding partners within the secretory pathway. Although secretion of the wild-type protein was not completely blocked, it is worth noting that punctin-1 is the short form of ADAMTSL1 and there may be a greater impact on the secretion of full-length ADAMTSL1 since larger ADAMTS proteins are typically inefficiently secreted compared to shorter forms. A plasmid construct for full-length ADAMTSL1 is presently unavailable. The function of punctin-1 in tissues is currently unknown, however, by analogy with other family members (Apte 2009; Dubail and Apte 2015; Hubmacher and Apte 2015), there is a strong possibility that it mediates the assembly and turnover of extracellular matrix at the affected sites. Taken together with previous findings (Wang et al. 2009), the data obtained here suggest that C-mannosylation is potentially a second mechanism that ensures secretion of properly folded punctin and likely other isoforms including the full-length ADAMTSL1 protein. C-mannosylation is currently a relatively obscure post-translational modification and the enzyme mediating it was only recently cloned (Shcherbakova et al. 2017).
Most previously reported mutations in ADAMTS family members manifest as recessive phenotypes (Le Goff and Cormier-Daire 2011). The present data strongly suggests that the p.(Trp42Arg) mutation may have a dominant negative effect on protein secretion, thus manifesting dominant or semi-dominant effects. In part, together with potential modifier genes, this effect may provide an explanation for the variable clinical phenotypes seen in this pedigree. Studies of ADAMTSL1 in families with similar phenotypes will provide further insight into the possible function of this gene in embryonic development.
Supplementary Material
Supp. Figure S1. A. Sequencing chromatograms of ADAMTSL1 alleles. B. Expression of mouse Adamtsl1 (based on BioGPS, biogps.org). Please note enrichment in ocular tissues such as iris, ciliary body, retina, cornea and eyecup (red box).
Supp. Table S1. Summary of heterozygous rare damaging variants shared by affected family members.
Acknowledgments
We are grateful to the family for participating in this genetic study. We thank Dr. Eric Weh for assistance with variant analysis and Dr. Robert Haltiwanger for helpful comments on the draft manuscript.
GRANT SPONSOR: National Institutes of Health EY015518 (EVS), HL107147 (SSA) and EY024943 (SSA), funds provided by the Children’s Research Institute Foundation at Children’s Hospital of Wisconsin (EVS), as well as P30EY001931 NIH Core Grant.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflict of interest to disclose.
References
- ADA Division of Communications; Journal of the American Dental Association.; ADA Council on Scientific Affairs. For the dental patient. Tooth eruption: the primary teeth. J Am Dent Assoc. 2005;136(11):1619. doi: 10.14219/jada.archive.2005.0095. [DOI] [PubMed] [Google Scholar]
- ADA Division of Communications; Journal of the American Dental Association., ADA Council on Scientific Affairs. For the dental patient. Tooth eruption: the permanent teeth. J Am Dent Assoc. 2006;137(1):127. doi: 10.14219/jada.archive.2006.0031. [DOI] [PubMed] [Google Scholar]
- Ahram D, Sato TS, Kohilan A, Tayeh M, Chen S, Leal S, Al-Salem M, El-Shanti H. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J Hum Genet. 2009;84(2):274–8. doi: 10.1016/j.ajhg.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284(46):31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitrian E, McPherson E, Grajewski A. Poster presented at ARVO Annual Meeting IOVS. Vol. 54. Seattle, Washington: 2013. Expanded Phenotype of Axenfeld Spectrum: Congenital Hypothyroidism and Glaucoma; p. 6242. [Google Scholar]
- Christensen AE, Fiskerstrand T, Knappskog PM, Boman H, Rødahl E. A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Invest Ophthalmol Vis Sci. 2010;51(12):6369–73. doi: 10.1167/iovs.10-5597. [DOI] [PubMed] [Google Scholar]
- Dubail J, Apte SS. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol. 2015;44–46:24–37. doi: 10.1016/j.matbio.2015.03.001. [DOI] [PubMed] [Google Scholar]
- Hubmacher D, Apte SS. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015;47:34–43. doi: 10.1016/j.matbio.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Cormier-Daire V. The ADAMTS(L) family and human genetic disorders. Hum Mol Genet. 2011;20(R2):R163–167. doi: 10.1093/hmg/ddr361. [DOI] [PubMed] [Google Scholar]
- Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22(5):314–324. doi: 10.1097/ICU.0b013e328349412b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova A, Tiemann B, Buettner FF, Bakker H. Distinct C-mannosylation of netrin receptor thrombospondin type 1 repeats by mammalian DPY19L1 and DPY19L3. Proc Natl Acad Sci U S A. 2017;114(10):2574–2579. doi: 10.1073/pnas.1613165114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, Feng L, Limviphuvadh V, Whisenhunt KN, Maurer-Stroh S, Yanovitch TL, Kalaydjieva L, Azmanov DN, Finzi S, Mauri L, Javadiyan S, Souzeau E, Zhou T, Hewitt AW, Kloss B, Burdon KP, Mackey DA, Allen KF, Ruddle JB, Lim SH, Rozen S, Tran-Viet KN, Liu X, John S, Wiggs JL, Pasutto F, Craig JE, Jin J, Quaggin SE, Young TL. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126(7):2575–87. doi: 10.1172/JCI85830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LW, Dlugosz M, Somerville RP, Raed M, Haltiwanger RS, Apte SS. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J Biol Chem. 2007;282(23):17024–31. doi: 10.1074/jbc.M701065200. [DOI] [PubMed] [Google Scholar]
- Wang LW, Leonhard-Melief C, Haltiwanger RS, Apte SS. Post-translational Modification of Thrombospondin Type-1 Repeats in ADAMTS-like 1/Punctin-1 by C-Mannosylation of Tryptophan. J Biol Chem. 2009;284(44):30004–30015. doi: 10.1074/jbc.M109.038059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Wiggs JL. Common and rare genetic risk factors for glaucoma. Cold Spring Harb Perspect Med. 2014;4(12):a017244. doi: 10.1101/cshperspect.a017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Figure S1. A. Sequencing chromatograms of ADAMTSL1 alleles. B. Expression of mouse Adamtsl1 (based on BioGPS, biogps.org). Please note enrichment in ocular tissues such as iris, ciliary body, retina, cornea and eyecup (red box).
Supp. Table S1. Summary of heterozygous rare damaging variants shared by affected family members.