

Abstract
Proteostasis is one of the seven “pillars of aging research” identified by the Trans‐NIH Geroscience Initiative and loss of proteostasis is associated with aging and age‐related chronic disease. Accumulated protein damage and resultant cellular dysfunction are consequences of limited protein repair systems and slowed protein turnover. When relatively high rates of protein turnover are maintained despite advancing age, damaged proteins are more quickly degraded and replaced, maintaining proteome fidelity. Therefore, maintenance of protein turnover represents an important proteostatic mechanism. However, measurement of protein synthesis without consideration for cell proliferation can result in an incomplete picture, devoid of information about how new proteins are being allocated. Simultaneous measurement of protein and DNA synthesis provides necessary mechanistic insight about proteins apportioned for newly proliferating cells versus for somatic maintenance. Using this approach with a number of murine models of slowed aging shows that, compared to controls, energetic resources are directed more toward somatic maintenance and proteostasis, and away from cell growth and proliferation. In particular, slowed aging models are associated with heightened mechanisms of mitochondrial proteostatic maintenance. Taking cell proliferation into account may explain the paradoxical findings that aging itself and slowed aging interventions can both be characterized by slower rates of protein synthesis.
Keywords: aging, mitochondria, proteostasis, somatic maintenance
Introduction
People aged 65 years and older represented about 14.5% of the United States population in 2014 and this number is expected to grow to 21.7% of the population by 2040 (US Census Bureau), whereas in the United Kingdom the population aged 65 and over had grown by 47% since mid‐1974 to make up nearly 18% of the total population in 2014 (Office for National Statistics). Despite the well‐recognized fact that the world population is aging, defining aging is not entirely straightforward. Medawar, for example, defined aging as the collection of changes that render human beings progressively more likely to die (Medawar, 1952). The definition of aging from Rose's book on the evolution of aging (Rose, 1991) is ‘a persistent decline in the age‐specific fitness components of an organism due to internal physiological degeneration’, which seems well suited to the discussion in this Symposium Review from Experimental Biology 2017.
Healthspan is a term used to describe the ‘health life expectancy’ or period of life spent free of chronic disease (Kennedy et al. 2014). A trans‐National Institutes of Health (NIH) initiative called the Geroscience Initiative has focused on strategies to increase healthspan. The premise of the initiative is that since aging is a major risk factor and driver of chronic diseases, understanding the molecular and cellular mechanisms responsible for aging holds promise for simultaneously decreasing all age‐related chronic diseases. Members of the Geroscience Initiative identified seven highly interrelated ‘pillars of aging’ that are critical for understanding and treating the aging process (Kennedy et al. 2014). Protein homeostasis (proteostasis) was one of the seven pillars of aging identified because protein dyshomeostasis has emerged is a common feature of aging and chronic disease as summarized in a number of excellent reviews (Balch et al. 2008; Labbadia & Morimoto, 2014; Kaushik & Cuervo, 2015; Labbadia & Morimoto, 2015). In this Symposium Review, we briefly summarize evidence that improved mechanisms of proteostatic maintenance – in particular mechanisms promoting mitochondrial proteostasis – are shared among experimental models of extended healthspan. Additionally, we will address what we believe are important considerations for designing studies to interrogate the potential for targeting mitochondrial proteostatic maintenance for interventions to increase healthspan. Specifically, we discuss the importance of assessing protein turnover in the context of cell proliferation, the impact of growth compared to somatic maintenance on proteostatic assessment, and how model systems can influence the interpretation of proteostasis.
Aging, proteostasis, and protein turnover: the influence of cell proliferation
Proteostasis is a term used to refer to a network of dynamic processes contributing to maintenance of proteome fidelity (Balch et al. 2008; Labbadia & Morimoto, 2014; Kaushik & Cuervo, 2015; Labbadia & Morimoto, 2015). Regulation of protein biogenesis, folding, targeting, quality control and degradation must be orchestrated in response to rapidly changing intrinsic and extrinsic signals. In light of the dynamic network of processes required to maintain a functional proteome, it seems more appropriate to refer to the process of maintaining proteome fidelity as proteodynamics (Basaiawmoit & Rattan, 2010). A challenge to studying protein maintenance with aging and aging interventions is the complex and dynamic nature of the proteostatic network since it is difficult to simultaneously assay all components of the network. Here we provide evidence that protein turnover is a critical proteostatic mechanism and that cell proliferation is a key consideration when assessing protein turnover as a mechanism for maintaining the proteome.
Protein repair systems are limited and therefore proteins damaged by stresses associated with aging (i.e. reactive oxygen species, advanced glycation endproducts) accumulate over time and contribute to cellular, organ and organism dysfunction. Protein synthesis is a primary mechanism for maintaining quality control and proteome fidelity (Charmpilas et al. 2015), and a commonly held belief is that bulk protein synthesis rates decline with aging (Rattan, 2010). Accumulated protein damage and resultant cellular dysfunction is the ultimate consequence of limited protein repair systems and slowed protein synthesis (Hipkiss, 2006; Tavernarakis, 2008). Reciprocally, when relatively high rates of protein turnover are maintained despite advancing age, damaged proteins are more quickly degraded and replaced, maintaining proteome fidelity (Fig. 1). Given that normal aging is associated with a slowing of protein synthesis, it is interesting that long‐lived models also appear to have slower protein synthesis throughout the lifespan (Tavernarakis, 2008; Kapahi, 2010; Kapahi et al. 2010; Price et al. 2012; Dai et al. 2014; Karunadharma et al. 2015). The observation that both aging and slowed‐aging have apparent decreases in protein synthesis is a paradox that has largely escaped the notice of researchers in protein metabolism and aging.
Figure 1. Protein turnover is an important proteostatic mechanism.
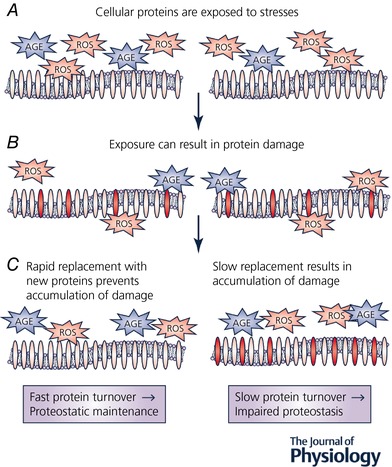
A, cells are exposed to a variety of stresses. Stresses that may be more unrelenting with increasing age include reactive oxygen species (ROS) and advanced glycation endproducts (AGE). B, prolonged exposure to these stresses can result in damage to proteins (and lipids and DNA), represented here as red ovals. C, when protein turnover is maintained at relatively high rates, damaged proteins are quickly degraded and replaced with newly synthesized proteins, preventing accumulation of damaged proteins despite continued exposure to stresses (left). However, when protein turnover rates are slow, it is more likely that protein damage will accumulate (right). Therefore, maintaining rates of protein turnover represents an important mechanism for supporting proteome fidelity.
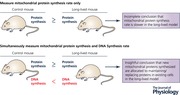
To help understand why both aging and slowed aging are associated with decreased protein turnover, robust approaches for assessing protein turnover must be used. The commonly used measurement of protein content does not provide this insight because both increases and decreases in protein turnover can occur without a change in protein content (Miller & Hamilton, 2012; Miller et al. 2014). Therefore, to capture changes in the important process of turnover, stable isotopically labelled amino acids are frequently used to provide a relatively acute measurement of protein synthesis. However, even these measurements require caution since a short labelling period can bias the measured synthesis rates toward rapidly turning over or abundant proteins (Miller et al. 2015). Therefore, we use a robust and insightful approach to measure rates of protein synthesis with a stable isotope of water, deuterium oxide (D2O).
The use of D2O allows for measurement of synthetic rates over prolonged periods of time in free living animal and human subjects provides information about the synthesis of both slowly and rapidly synthesized proteins of diverse abundances (Miller et al. 2015). Another major strength to using D2O is that rates of DNA synthesis can be measured simultaneously with the rates of protein synthesis (details of calculations available in Neese et al. 2002). Through the simultaneous assessment of protein and DNA it has become clear to us that cell proliferation, as measured by DNA synthesis, is an important outcome when considering the slowed‐aging effects of protein turnover. When cells proliferate (increased DNA synthesis) protein mass is doubled (increased protein synthesis) in the growth phases (G1 and G2), so that two daughter cells have the full complement of genetic material and protein machinery (Grebien et al. 2005). In post‐mitotic cells like skeletal muscle fibres, growth is accompanied by DNA synthesis in a supportive cell and donation of that nuclear material from supportive cells to the post‐mitotic cell (Collins et al. 2005) to minimize changes to the cytosolic volume to DNA ratio (Allen et al. 1999). In both cell types, the synthesis of new DNA is accompanied by the synthesis of new proteins. In contrast, when new proteins are synthesized for cellular remodelling or replacement of damaged proteins there is not a corresponding increase in DNA synthesis. Thus, when D2O is used to label newly synthesized proteins during growth/proliferation, the ratio of protein synthesis to DNA synthesis essentially stays the same, whereas during maintenance or remodelling, the ratio of protein synthesis to DNA synthesis increases (Fig. 2; Miller et al. 2014). The simultaneous assessment of protein and DNA synthesis has unexpectedly provided great insight into the paradoxical observation that both aged and long‐lived models have slowed protein turnover.
Figure 2. Considering cell proliferation influences the interpretation of protein synthesis.
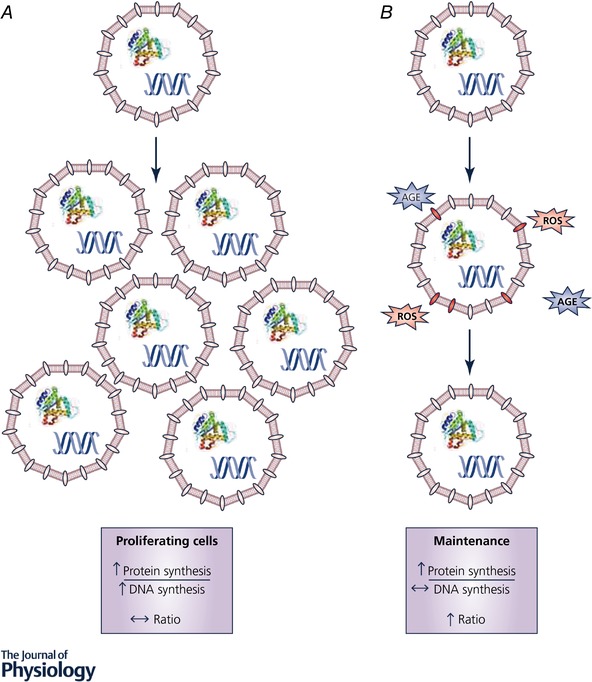
A, in proliferating cells (growth), measuring protein synthesis alone would lead to the conclusion that protein synthesis rates are increased. If protein and DNA synthesis are measured simultaneously, the rates of both protein and DNA synthesis are increased. Expressing rates of protein synthesis relative to DNA synthesis (ratio) leads to the conclusion that newly synthesized proteins are primarily being allocated to new cells and less so to maintaining existing cell structures. B, in contrast, when rates of protein synthesis are increased without much of a change in rates of DNA synthesis (proliferation), the ratio of protein:DNA synthesis rates is greater and the conclusion is that newly synthesized proteins are primarily being allocated to maintaining/replacing proteins in existing cells.
In 1977, Kirkwood hypothesized that in the wild predation results in evolutionary changes that favours the allocation of energetic resources toward rapid reproduction, which comes at the expense of maintaining somatic cells (Kirkwood, 1977). With this trade‐off, low energetic investment in maintaining somatic cells in favour of growth and reproduction results in accumulated protein damage and contributes to the aging process. However, relaxation of predation results in low energetic investment in growth and reproduction and is directed toward somatic maintenance in part through improved proteostasis. This concept has been supported by laboratory models that restrict energetic investment in reproduction and thus increase lifespan (Partridge et al. 2005). Simultaneous measurement of protein and DNA synthesis provides insight into the proteostatic mechanisms that give rise to somatic maintenance.
As mentioned, in multiple long‐lived models it appears that protein synthesis is decreased compared to controls (Tavernarakis, 2008; Kapahi, 2010; Kapahi et al. 2010; Price et al. 2012; Dai et al. 2014; Karunadharma et al. 2015), and by this measurement alone, the conclusion would be that decreased protein synthesis gives rise to slowed aging. Although the fact that protein synthesis decreases is indisputable, the conclusion does not consider what cellular processes contributed to that observed decrease. When DNA synthesis is measured simultaneously with protein synthesis, it is apparent that cell proliferation is slower in long‐lived models (Miller et al. 2012; Drake et al. 2013, 2014, 2015). Further, DNA synthesis decreases to a greater degree than protein synthesis thus indicating that a greater proportion of protein synthesis is directed toward maintaining existing cellular structures (somatic maintenance) and less toward proliferation (growth). Therefore, considering protein synthesis in the context of cell proliferation provides mechanistic insight into the directing of energetic resources toward somatic maintenance and proteostasis and away from cell growth and proliferation. Further, these assessments provide insight into the conundrum of why there is an apparent decrease in protein synthesis in long‐lived models since although overall protein synthesis is decreased, it is largely due to decreased proliferation concurrent with increased proteostatic mechanisms.
Maintenance of mitochondrial protein synthesis as a proteostatic mechanism in models of increased lifespan/healthspan
The mitochondrial reticulum is an important determinant of cellular energetics, and is now also known to participate in direct communication with other subcellular organelles (Rose et al. 2016; Filadi et al. 2017) and carry out important protein quality control activities such as the mitochondrial unfolded protein response (Carreras‐Sureda et al. 2017). Mitochondria are also an important site for production of reactive oxygen species important for signalling beneficial adaptations (Gonzalez‐Freire et al. 2015; Gomez‐Cabrera et al. 2016), but are also capable of contributing to macromolecular damage and dysfunction associated with aging and age‐related chronic diseases (Hohn et al. 2016). It is widely accepted that maintaining mitochondrial function contributes to metabolic health, while deterioration of mitochondrial function is at minimum a characteristic of, but more likely a contributor to, aging (Lanza & Nair, 2009; Gonzalez‐Freire et al. 2015) and age‐associated chronic diseases including insulin resistance/type II diabetes (Di Meo et al. 2017) and cardiovascular diseases (Paneni et al. 2017).
Mitochondrial adaptation to changes in cellular energetic demands and energetic stresses, such as occur with calorie restriction, involves rapid and highly dynamic remodelling involving fission, fusion and turnover of proteins that are encoded by both nuclear and mitochondrial genes. Therefore, approaches for assessing mitochondrial protein turnover must be capable of capturing these dynamics. As an example, we use data from studies examining mitochondrial biogenesis during caloric restriction for which there are discrepant findings. Increased mitochondrial biogenesis is reported in calorie‐restricted mice (Nisoli et al. 2005), rats (Lopez‐Lluch et al. 2006) and flies (Zid et al. 2009), while others report no change in mitochondrial biogenesis in calorie‐restricted rats (Hancock et al. 2011) and mice (Lanza et al. 2012). However, if one considers DNA synthesis, it appears that caloric restriction also decreases cellular proliferation (Miller et al. 2012), and when the ratio of protein synthesis to DNA is considered it is apparent that synthesis to maintain mitochondrial proteostasis actually increases while protein synthesis directed toward proliferation decreases (Miller et al. 2014).
Since our study in calorically restricted mice, we have made similar assessments in multiple long‐lived models (Drake et al. 2013, 2014, 2015; Miller et al. 2013, 2014). In each of these models, we found that the ratio of mitochondrial protein synthesis to DNA synthesis is greater in the long‐lived model compared to controls. Therefore, by considering protein synthesis and DNA synthesis together, the apparent paradox between why pro‐aging and slowed aging both appear to have decreased rates of protein synthesis becomes explainable. In the case of aging, the decrease in protein synthesis likely indicates a decrease in somatic maintenance. In the case of slowed aging, the overall rate of protein synthesis decreases, but this is due to a decrease in the amount of protein synthesis dedicated toward growth and proliferation and an increased amount of protein synthesis dedicated to proteostatic mechanisms for the purposes of somatic maintenance.
Conclusions and a look forward
One of the primary conclusions from this symposium is that protein turnover is a key proteostatic mechanism. Additionally, when assessing rates of protein synthesis, it is important to also consider rates of cell proliferation to provide insight about how newly synthesized proteins are being allocated – either to proliferating cells or to maintenance/replacement of existing structures. Using this approach with murine models of slowed aging led to the conclusion that mitochondrial proteostatic maintenance is a characteristic shared among these slowed aging models. Taking proliferation into account may explain the paradoxical findings that aging itself and slowed aging interventions can both seem to be associated with slower rates of protein synthesis.
Looking forward, there are a number of aspects of protein turnover as a mechanism of proteostasis maintenance that remain incompletely understood. For example, while it seems clear that turnover of mitochondrial proteins is a cellular priority for slowed aging, the mechanisms by which this selective translation occurs are unclear. Identifying the specific proteins that are selectively translated could also provide critical information for identifying targets for slowed aging interventions. While slower rates of tissue DNA synthesis have emerged as a consistent characteristic of slowed aging, whole tissues are complex and comprise many cell types. Identifying the specific cell types that have slower proliferation rates and understanding the phenotype of these more slowly proliferating cells could also help focus efforts to develop novel slowed aging interventions.
Additional information
Competing interests
The authors have no competing interests or conflicts of interest to disclose.
Author contributions
Both authors participated in writing the manuscript and both authors approved the final manuscript. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors did not receive funding to support this review, other than the support from The Physiological Society for the symposium itself. The full Experimental Biology symposium, ‘The Modulation of Aging Through Altered Proteostasis’, was supported by The Physiological Society.
Acknowledgements
The authors wish to acknowledge all of the hard‐working members of the Translational Research on Aging and Chronic Disease Laboratory. We are fortunate to have had a dedicated group of talented trainees and professionals who have helped shape our current understanding of aging and proteostasis over the past eight years.
Biographies
The authors co‐direct the Translational Research on Aging and Chronic Disease laboratory in the Department of Health and Exercise Science at Colorado State University in Fort Collins, CO. Karyn Hamilton is a Professor and has been on faculty since 2004. She completed her PhD at the University of Florida and did post‐doctoral fellowships at Baylor College of Medicine and the University of Florida.

Benjamin Miller is an Associate Professor and has been on faculty since 2007. He completed his PhD at the University of California, Berkeley and a post‐doctoral fellowship at the Institute of Sports Medicine in Copenhagen.
This review was presented at the symposium ‘The Modulation of Aging through Altered Proteostasis’, which took place at Experimental Biology 2017, Chicago, USA, 22–26 April 2017.
References
- Allen DL, Roy RR & Edgerton VR (1999). Myonuclear domains in muscle adaptation and disease. Muscle Nerve 22, 1350–1360. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A & Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Basaiawmoit RV & Rattan SI (2010). Cellular stress and protein misfolding during aging. Methods Mol Biol 648, 107–117. [DOI] [PubMed] [Google Scholar]
- Carreras‐Sureda A, Pihan P & Hetz C (2017). The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charmpilas N, Daskalaki I, Papandreou ME & Tavernarakis N (2015). Protein synthesis as an integral quality control mechanism during ageing. Ageing Res Rev 23, 75–89. [DOI] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA & Morgan JE (2005). Stem cell function, self‐renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301. [DOI] [PubMed] [Google Scholar]
- Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ & Rabinovitch PS (2014). Altered proteome turnover and remodeling by short‐term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S, Iossa S & Venditti P (2017). Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 233, R15–R42. [DOI] [PubMed] [Google Scholar]
- Drake JC, Bruns DR, Peelor FF 3rd, Biela LM, Miller RA, Hamilton KL & Miller BF (2014). Long‐lived crowded‐litter mice have an age‐dependent increase in protein synthesis to DNA synthesis ratio and mTORC1 substrate phosphorylation. Am J Physiol Endocrinol Metab 307, E813–E821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JC, Bruns DR, Peelor FF 3rd, Biela LM, Miller RA, Miller BF & Hamilton KL (2015). Long‐lived Snell dwarf mice display increased proteostatic mechanisms that are not dependent on decreased mTORC1 activity. Aging Cell 14, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JC, Peelor FF 3rd, Biela LM, Watkins MK, Miller RA, Hamilton KL & Miller BF (2013). Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci 68, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Theurey P & Pizzo P (2017). The endoplasmic reticulum‐mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 62, 1–15. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Vina J & Ji LL (2016). Role of redox signaling and inflammation in skeletal muscle adaptations to training. Antioxidants (Basel) 5, E48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P & Ferrucci L (2015). Reconsidering the role of mitochondria in aging. J Gerontol A Biol Sci Med Sci 70, 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F, Dolznig H, Beug H & Mullner EW (2005). Cell size control: new evidence for a general mechanism. Cell Cycle 4, 418–421. [DOI] [PubMed] [Google Scholar]
- Hancock CR, Han DH, Higashida K, Kim SH & Holloszy JO (2011). Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J 25, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipkiss AR (2006). Accumulation of altered proteins and ageing: causes and effects. Exp Gerontol 41, 464–473. [DOI] [PubMed] [Google Scholar]
- Hohn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, Kehm R, Konig J, Grune T & Castro JP (2016). Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol 11, 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P (2010). Protein synthesis and the antagonistic pleiotropy hypothesis of aging. Adv Exp Med Biol 694, 30–37. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL & Kockel L (2010). With TOR, less is more: a key role for the conserved nutrient‐sensing TOR pathway in aging. Cell Metab 11, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunadharma PP, Basisty N, Dai DF, Chiao YA, Quarles EK, Hsieh EJ, Crispin D, Bielas JH, Ericson NG, Beyer RP, MacKay VL, MacCoss MJ & Rabinovitch PS (2015). Subacute calorie restriction and rapamycin discordantly alter mouse liver proteome homeostasis and reverse aging effects. Aging Cell 14, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S & Cuervo AM (2015). Proteostasis and aging. Nat Med 21, 1406–1415. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss‐Coray T & Sierra F (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB (1977). Evolution of ageing. Nature 270, 301–304. [DOI] [PubMed] [Google Scholar]
- Labbadia J & Morimoto RI (2014). Proteostasis and longevity: when does aging really begin? F1000Prime Rep 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J & Morimoto RI (2015). The biology of proteostasis in aging and disease. Annu Rev Biochem 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR & Nair KS (2009). Muscle mitochondrial changes with aging and exercise. Am J Clin Nutr 89, 467S–471S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, Zabielski P, Klaus KA, Morse DM, Heppelmann CJ, Bergen HR 3rd, Dasari S, Walrand S, Short KR, Johnson ML, Robinson MM, Schimke JM, Jakaitis DR, Asmann YW, Sun Z & Nair KS (2012). Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab 16, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P & de Cabo R (2006). Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci USA 103, 1768–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medawar PB (1952). An Unsolved Problem of Biology: An Inaugural Lecture Delivered at University College London, 6 December, 1951. H. K. Lewis & Co. Ltd, London. [Google Scholar]
- Miller BF, Drake JC, Naylor B, Price JC & Hamilton KL (2014). The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev 18, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF & Hamilton KL (2012). A perspective on the determination of mitochondrial biogenesis. Am J Physiol Endocrinol Metab 302, E496–E499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF, Robinson MM, Bruss MD, Hellerstein M & Hamilton KL (2012). A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF, Robinson MM, Reuland DJ, Drake JC, Peelor FF 3rd, Bruss MD, Hellerstein MK & Hamilton KL (2013). Calorie restriction does not increase short‐term or long‐term protein synthesis. J Gerontol A Biol Sci Med Sci 68, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF, Wolff CA, Peelor FF 3rd, Shipman PD & Hamilton KL (2015). Modeling the contribution of individual proteins to mixed skeletal muscle protein synthetic rates over increasing periods of label incorporation. J Appl Physiol (1985) 118, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neese RA, Misell LM, Turner S, Chu A, Kim J, Cesar D, Hoh R, Antelo F, Strawford A, McCune JM, Christiansen M & Hellerstein MK (2002). Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci USA 99, 15345–15350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S & Carruba MO (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317. [DOI] [PubMed] [Google Scholar]
- Paneni F, Diaz Canestro C, Libby P, Luscher TF & Camici GG (2017). The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol 69, 1952–1967. [DOI] [PubMed] [Google Scholar]
- Partridge L, Gems D & Withers DJ (2005). Sex and death: what is the connection? Cell 120, 461–472. [DOI] [PubMed] [Google Scholar]
- Price JC, Khambatta CF, Li KW, Bruss MD, Shankaran M, Dalidd M, Floreani NA, Roberts LS, Turner SM, Holmes WE & Hellerstein MK (2012). The effect of long term calorie restriction on in vivo hepatic proteostatis: a novel combination of dynamic and quantitative proteomics. Mol Cell Proteomics 11, 1801–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan SI (2010). Synthesis, modification and turnover of proteins during aging. Adv Exp Med Biol 694, 1–13. [DOI] [PubMed] [Google Scholar]
- Rose G, Santoro A & Salvioli S (2016). Mitochondria and mitochondria‐induced signalling molecules as longevity determinants. Mech Ageing Dev 165, 115–128. [DOI] [PubMed] [Google Scholar]
- Rose MR (1991). Evolutionary Biology of Aging. Oxford University Press, New York. [Google Scholar]
- Tavernarakis N (2008). Ageing and the regulation of protein synthesis: a balancing act? Trends Cell Biol 18, 228–235. [DOI] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S & Kapahi P (2009). 4E‐BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila . Cell 139, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]