

Abstract
Key points
A reduction in Kindlin‐2 levels in endothelial cells compromises vascular barrier function.
Kindlin‐2 is a previously unrecognized component of endothelial adherens junctions.
By interacting directly and simultaneously with β‐ or γ‐catenin and cortical actin filaments, Kindlin‐2 stabilizes adherens junctions.
The Kindlin‐2 binding sites for β‐ and γ‐catenin reside within its F1 and F3 subdomains. Although Kindlin‐2 does not associate directly with tight junctions, its downregulation also destabilizes these junctions. Thus, impairment of both adherens and tight junctions may contribute to enhanced leakiness of vasculature in Kindlin‐2 +/− mice.
Abstract
Endothelial cells (EC) establish a physical barrier between the blood and surrounding tissue. Impairment of this barrier can occur during inflammation, ischaemia or sepsis and cause severe organ dysfunction. Kindlin‐2, which is primarily recognized as a focal adhesion protein in EC, was not anticipated to have a role in vascular barrier. We tested the role of Kindlin‐2 in regulating vascular integrity using several different approaches to decrease Kindlin‐2 levels in EC. Reduced levels of Kindlin‐2 in Kindlin‐2+/– mice aortic endothelial cells (MAECs) from these mice, and human umbilical ECs (HUVEC) treated with Kindlin‐2 siRNA showed enhanced basal and platelet‐activating factor (PAF) or lipopolysaccharide‐stimulated vascular leakage compared to wild‐type (WT) counterparts. PAF preferentially disrupted the Kindlin‐2 +/− MAECs barrier to BSA and dextran and reduced transendothelial resistance compared to WT cells. Kindlin‐2 co‐localized and co‐immunoprecipitated with vascular endothelial cadherin‐based complexes, including β‐ and γ‐catenin and actin, components of adherens junctions (AJ). Direct interaction of Kindlin‐2 with β‐ and γ‐catenin and actin was demonstrated in co‐immunoprecipitation and surface plasmon resonance experiments. In thrombin‐stimulated HUVECs, Kindlin‐2 and cortical actin dissociated from stable AJs and redistributed to radial actin stress fibres of remodelling focal AJs. The β‐ and γ‐catenin binding site resides within the F1 and F3 subdomains of Kindlin‐2 but not the integrin binding site in F3. These results establish a previously unrecognized and vital role of Kindlin‐2 with respect to maintaining the vascular barrier by linking Vascuar endothelial cadherin‐based complexes to cortical actin and thereby stabilizing AJ.
Keywords: adherens junctions, kindlin‐2, vascular barrier
Key points
A reduction in Kindlin‐2 levels in endothelial cells compromises vascular barrier function.
Kindlin‐2 is a previously unrecognized component of endothelial adherens junctions.
By interacting directly and simultaneously with β‐ or γ‐catenin and cortical actin filaments, Kindlin‐2 stabilizes adherens junctions.
The Kindlin‐2 binding sites for β‐ and γ‐catenin reside within its F1 and F3 subdomains. Although Kindlin‐2 does not associate directly with tight junctions, its downregulation also destabilizes these junctions. Thus, impairment of both adherens and tight junctions may contribute to enhanced leakiness of vasculature in Kindlin‐2 +/− mice.
Abbreviations
- Abs
antibodies
- AJs
adherens junctions
- CT
cytoplasmic tail
- EC
endothelial cell
- EGFP
enhanced green fluorescent protein
- GST
glutathione S‐transferase
- HEK293 cells
human epithelial kidney 293 cells
- HUVEC
human umbilical endothelial cell
- ICG
indocyanine green dye
- LPS
lipopolysaccharide
- MAEC
mouse aortic endothelial cell
- PAF
platelet‐activating factor
- PFA
paraformaldehyde
- siRNA
small interfering RNA
- SPR
surface plasmon resonance
- TEER
transendothelial electrical resistance
- TJs
tight junctions
- TXINS
Triton X‐insoluble
- TXS
Triton X‐soluble
- VE‐cadherin
vascular endothelial cadherin
- WT
wild‐type
Introduction
Endothelial cells (ECs) serve as gatekeepers of the vasculature because they control the infiltration of cells and plasma constituents from blood into the vessel wall and surrounding tissues. Loss or impairment of this function occurs in many pathological conditions, such as inflammation, ischaemia or sepsis, and can lead to severe organ dysfunction. To date, three major types of interendothelial cell junctions have been distinguished: adherens junctions (AJs), tight junctions (TJs) and gap junctions (Niessen, 2007; Dejana et al. 2008). Of these, the AJs are particularly crucial regulators of vascular integrity because they not only control paracellular transport, but also are required for proper assembly of TJs (Bazzoni & Dejana, 2004). AJs are formed by cadherins, transmembrane glycoproteins that mediate cell‐cell adhesion through homotypic interactions of their extracellular domains. Vascular endothelial cadherin (VE‐cadherin), a homodimeric transmembrane protein, is prominent in ECs and its cytoplasmic domain binds to several intracellular partners, including p120, β‐catenin and γ‐catenin (plakoglobin) (Dejana et al. 2008). β‐catenin and γ‐catenin bind to α‐catenin, which interacts with the actin cytoskeleton either directly or indirectly through other actin‐binding proteins, such as α‐actinin or vinculin (Niessen, 2007; Dejana et al. 2008). Although α‐catenin cannot interact with β‐catenin and actin simultaneously in vitro, it serves as a molecular switch that regulates the balance between linear and branched actin fibres depending on its association with ancillary proteins (Drees et al. 2005; Yamada et al. 2005). Buckley et al. (2014) have demonstrated an interaction of E‐cadherin, β‐catenin and α‐catenin into a complex with F‐actin under force. Connection of VE‐cadherin to cortical actin fibres does appear to be critical for stability of mature AJs in quiescent EC monolayers and for vascular integrity. Also, VE‐cadherin regulates the actin network because AJ dynamic remodelling into active focal AJs leads to the appearance of contractile actin stress fibres (Millan et al. 2010; Huveneers et al. 2012). Thus, the molecular interactions linking VE‐cadherin to the actin cytoskeleton are complex and our understanding of these interactions is still evolving.
Kindlins are cytoskeleton‐associated proteins that are crucial regulators of integrin function (Larjava et al. 2008; Moser et al. 2009). There are three kindlins in mammals (Kindlin‐1, ‐2,‐3), which are quite homologous (Plow et al. 2009; Malinin et al. 2010). However, they do not compensate for one another; deficiencies of each kindlin in mice and/or humans result in distinct phenotypes, indicative of their unique functions(Larjava et al. 2008; Dowling et al. 2008; Ussar et al. 2008; Malinin et al. 2009; Plow, 2009). Kindlin‐2 is expressed in most non‐haematopoietic cells and is the most widely distributed of the three kindlin family members. Inactivation of the Kindlin‐2 gene in mice is peri‐implantation lethal by embryonic day 7.5 (Dowling et al. 2008; Montanez et al. 2008). In ECs, Kindlin‐2 directly interacts with clathrin and regulates clathrin‐dependent endocytosis, which is independent of its integrin binding function (Pluskota et al. 2013). Another integrin independent function of Kindlin‐2 is its interaction with β‐catenin and involvement in Wnt signalling in cancer cells (Yu et al. 2012). Enhanced leakiness of neovasculature formed in Kindlin‐2 +/− mice (Pluskota et al. 2011) and the Kindlin‐2 interaction with β‐catenin suggested that Kindlin‐2 might be a component and/or modulator of AJs, although this possibility has never been assessed directly. The present study tests this possibility and ultimately identifies a previously unrecognized function of Kindlin‐2 with respect to controlling interendothelial junctions.
Methods
Ethical approval
All procedures were performed under a protocol approved by the Clinic Institutional Animal Care and Use Committee of the Cleveland Clinic and conform with the principles and regulations as described by Grundy (2015). Mice were housed in AAALAC‐accredited facility under a 12:12 h light/dark cycle with ad libitum access to standard laboratory chow (Harlan Teklad 2918; Envigo, Dublin, VA, USA) and water. The Kindlin‐2 +/− mice have been characterized previously (Dowling et al. 2008; Pluskota et al. 2011, 2013). The mice showed an ∼50% reduction in Kindlin‐2 levels in ECs. The Kindlin‐2 +/− mice do not show any overt phenotype or spontaneous edema; however, if challenged, they show impaired angiogenesis in tumour and Matrigel implant models and enhanced leakage of neovasculature (Pluskota et al. 2011). We used 8–12‐week‐old mice of both sexes and Kindlin‐2 +/+ wild‐type (WT) littermates served as controls. In all experiments, depth of anaesthesia was evaluated by testing the pedal withdrawal reflex (toe and foot pad pinch).
Barrier function of tracheal blood vessels
WT and Kindlin‐2 +/− mice were killed by an anaesthetic overdose of ketamine/xylazine (280 mg kg−1; 18 mg kg−1) injected i.p. When deep anaesthesia was established, the mice were injected i.v. with green fluorescent polymer microspheres (500 nm) in sterile saline with or without platelet‐activating factor (PAF) (20 μg kg−1). After 4 min, to ensure death, cervical dislocation was performed and blood vessels were perfused with 1% paraformaldehyde (PFA)/PBS. Tracheas were removed, fixed in 1% PFA/PBS for 2 h, then immunostained with rat (EMD Millipore, Temecula, CA, USA) and hamster (BD Biosciences, San Jose, CA, USA) anti‐CD31 antibodies (Abs), followed by Alexa‐Fluor 647 goat ant‐rat and anti‐hamster Abs to visualize endothelial cells. Trachea were flat mounted with the luminal surface facing up and examined using fluorescence imaging confocal microscopy (Leica, Wetzlar, Germany) and ImagePro Plus Capture and Analysis software (Media Cybernetics, Warrendale, PA, USA). Microsphere positive area was quantified with Image ProPlus Capture and Analysis software. Six mice were used per group.
Integrity of ear skin vasculature
WT and Kindlin‐2 +/− mice were anaesthetized by isoflurane inhalation in a chamber system (Caliper Life Sciences, Waltham, MA, USA) and injected via the tail vein with indocyanine green dye (ICG) (2 mg kg−1) (Pulsion Medical Inc., Irving, TX, USA) in sterile saline with or without low‐dose of PAF (6 μg kg−1). Mice were immediately placed in the chamber of the IVIS Spectrum CT Imaging System (Perkin Elmer, Waltham, MA, USA) equipped with a scavenger system (isoflurane inhalation) and were maintained under anaesthesia for 40 min. During this time, leakage and accumulation of ICG from vasculature into ear tissue was monitored by capturing one image every 1 min. After the experiment, mice were put back into their cages and monitored until they woke. Fluorescence intensity in the ear was quantified at each time point using Living Image software (Caliper Life Sciences) and expressed as total radiant efficiency. Eight mice were used per group.
Barrier function of the lung vasculature
WT and Kindlin‐2 +/− mice were injected i.p. with Escherichia coli 0111:B4 lipopolysaccharide (LPS) (Sigma, St Louis, MO, USA) (5 mg kg−1) or with an equivalent volume of PBS, and pulmonary vascular barrier function was measured 24 h after injection. Mice were killed by isoflurane inhalation followed by cervical dislocation. Lungs were excised and their wet weight was measured immediately, followed by drying at 80°C for 48 h to obtain dry weight measurements. The ratio of wet to dry lung tissue weights was calculated for each mouse. As an independent approach, the mice were injected with Evans blue dye (100 μl of 1% sterile solution in PBS) via the tail vein and, after 30 min, the lungs were collected and weighed. Evans blue dye was extracted from the lung by incubation with 1 ml of formamide (55°C for 24 h) with constant shaking and the absorbance of extracted dye was measured at 610 nm. The amount of extravasated Evans blue per mg tissue was calculated as described previously (Chen et al. 2005). Eight mice per group were used.
Isolation of mice aortic endothelial cells (MAECs)
WT and Kindlin‐2 +/− mice were killed by i.p. injection of an overdose of ketamine/xylazine mixture (280 mg kg−1; 18 mg kg−1). After deep anaesthesia was achieved, cervical dislocation was performed. Aortas were removed using a dissecting microscope and MAECs were isolated as described previously (Mahabeleshwar et al. 2006).
Modulation of Kindlin‐2 expression in human umbilical ECs (HUVEC) and human epithelial kidney 293 (HEK293) cells
To reduce the expression of Kindlin‐2, HUVECs were transfected with siGenome SMART pool human PLEKHC1 small interfering RNA (siRNAs) or non‐targeting (control) siRNA#2 (Thermo Scientific Dharmacon, Lafayette, CO, USA) (50–150 nm) using Targefect HUVEC reagents (Targeting Systems, El Cajon, CA, USA) in accordance with the manufacturer's instructions. After 48 h, the confluent EC monolayers were subjected to immunoprecipitation, immunofluorescence, assessment of endothelial barrier function, real‐time quantitative RT‐PCR and western blotting with antibodies to connexin 43 (Cell Signaling, Danvers, MA, USA), ZO‐1 (EMD Millipore, Temecula, CA, USA), claudin 5 (EMD Millipore), occludin (BD Biosciences, San Jose, CA, USA), β‐actin (Cell Signaling), Kindlin‐2 (clone 3A3; Millipore), Kindlin‐3 (2F3‐2, prepared in the laboratory), VE‐cadherin XP (Cell Signaling), α‐catenin (Cell Signaling), β‐catenin (BD Biosciences) and γ‐catenin (Cell Signaling). Similar approaches were used to reduce expression of Kindlin‐3 in HUVEC using the siGenome SMART pool human FERMT3 siRNA (Thermo Scientific Dharmacon) and Targefect HUVEC kit (Targeting Systems) in accordance with the manufacturer's instructions.
To assess specificity of Kindlin‐2 downregulation by Kindlin‐2‐specific siRNA, as well as to map the crucial Kindlin‐2 subdomains implicated in AJ stabilization, rescue experiments were performed. In these experiments HUVECs were treated with siGenome SMART pool human PLEKHC1 siRNAs and non‐targeting siRNA#2 (100 nm) and, after 24 h, the cells were nucleofected using HUVEC nucleofector kit (Lonza, Allendale, NJ, USA) with WT or previously described deletion mutants of Kindlin‐2 (Ma et al. 2008) as pEGFP‐C2 constructs (Clontech, Mountain View, CA, USA). Twentyy‐four hours post‐nucleofection, HUVECs were subjected to transendothelial electrical resistance (TEER) assays or were lysed to assess expression of full‐length and mutant Kindlin‐2‐enhanced green fluorescent protein (EGFP) by western blotting with anti‐EGFP. HEK293 cells were transfected with WT or deletion mutants of Kindlin‐2 using Lipofectamine 2000 (Thermofisher Scientific, Waltham, MA, USA) in accordance with the manufacturer's instructions. Forty‐eight hours post‐transfection, confluent monolayers were subjected to immunoprecipitation.
Quantification of disrupted AJs and TJs
Resting HUVEC monolayers were untreated or transfected with control or Kindlin‐2‐targeting siRNA as described and stained for AJs with rabbit anti‐VE‐cadherin XP Ab (dilution 1:200) or for TJs with rabbit anti‐ZO‐1 Ab (dilution 1:200) followed with Alexa‐488‐labelled anti‐rabbit Ab. The cells were examined using confocal microscopy and the numbers of disrupted junctions (intercellular gaps) were quantified in random 10 fields from each coverslip and three independent experiments were performed.
Endothelial cell barrier function assays
WT or Kindlin‐2 +/− MAECs or HUVECs were grown till confluence on fibronectin‐coated transwell inserts. Dextran 10 kDa fluorescein isothiocyanate (FITC) or BSA‐AlexaFluor 488 was added to the upper chamber in the absence or presence of PAF (500 ng ml−1) or thrombin (4 U ml−1) and incubated for 0–120 min at 37 °C. At various time points, fluorescence of the media in the lower chambers was measured at λex = 485 nm and λem = 530 nm using a fluorimeter (CytoFluor II; PerSeptive Biosystems, Framingham, MA, USA).
TEER assay
WT or Kindlin‐2 +/− MAECs or HUVECs were seeded onto fibronectin‐coated transwell inserts (diameter 6.5 mm, pore size 3 μm; Transwell, Corning, Cambridge, MA, USA) and grown until TEER stabilized at values >20 Ω × cm2. In the rescue experiments performed on HUVECs, 0.5 × 106 cells were added per transwell immediately after nucleofection and the assay was performed after 24 h with starting TEER values of ∼55–70 Ω × cm2. Changes in TEER were measured upon stimulation of the cells with PAF or thrombin at time 0 and measured over 0–10 min using a volt‐ohmmeter (EVOM; World Precision Instruments, Sarasota, FL, USA). At the indicated times, resistance readings (Ω) were obtained from each insert and multiplied by the membrane area (Ω × cm2). The resistance value of an empty fibronectin‐coated insert (without cells) was subtracted from each measurement.
Immunofluorescence
Confluent HUVEC monolayers grown on fibronectin (10 μg ml−1), treated with thrombin (4 U ml−1 for 10 min) or siRNAs as specified, were fixed in 4% PFA/PBS for 20 min at room temperature, permeabilized with 0.1% TritonX‐100 for 5 min at room temperature, blocked with 1% horse serum and stained with rabbit anti‐VE‐cadherin XP, mouse anti‐Kindlin‐2, anti‐Kindlin‐3, ZO‐1, connexin 43, β‐catenin or γ‐catenin for 16 h at 4°C. Next, the slides were incubated with goat anti‐mouse and anti‐rabbit Abs conjugated to Alexa 488 or Alexa 568 and with Alexa Fluor 647‐phalloidin (to visualize actin filaments) for 30 min at 22 °C. The slides were mounted using Immuno‐Fluore mounting medium (MP Biomedicals, Solon, OH, USA). Images were taken using a Leica confocal microscope and processed with Photoshop, version 7.0 (Adobe Systems, San Jose, CA, USA).
Immunoprecipitation and western blotting
Confluent HUVECs or MAECs were treated with PAF (500 ng ml−1) or thrombin (4 U ml−1) for 0–15 min at 37 °C. Triton X‐soluble (TXS) extraction buffer was added to the dishes (10 mm Tris‐Cl, pH 7.5, 150 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 1% TritonX‐100, 1% NP‐40, 0.1 mm sodium orthovanadate, 1 mm phenylmethane sulphonyl fluoride, one tablet of complete protease inhibitor cocktail; Roche Diagnostics, Mannheim, Germany). The cells were flash‐frozen in Triton X‐soluble (TXS) buffer on dry‐ice for 10 min and subsequently thawed on ice for 20 min. The cell lysates were scraped into Eppendorf tubes and centrifuged at 17 000 g for 10 min at 4°C. The supernatants (Triton X‐soluble fraction) were separated and the pellets were dissolved in Triton X‐insoluble (TXINS) extraction buffer (10 mm Tris‐Cl, pH 7.5, 150 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 0.5% SDS, 1% NP‐40, 0.1 mm sodium orthovanadate, 1 mm phenylmethane sulphonyl fluoride, one tablet of complete protease inhibitor cocktail). Aliquots of TXS and TXINS fractions were set aside for further western blot analysis. TXS fractions were precleared for 30 min at 4°C with Protein‐A/G Plus‐Sepharose (Santa Cruz Biotechnology, Dallas, TX, USA), previously blocked with 1% BSA at 4°C for 16 h. The Protein‐A/G Plus‐Sepharose was pelleted at 1500 g at 4 °C and the supernatants were incubated with rabbit anti‐VE‐cadherin Ab or with anti‐Kindlin‐2 mAb or non‐immune respective Ab overnight at 4 °C followed by Protein‐A/G Plus‐Sepharose (Santa Cruz Biotechnology) for 2 h at 4°C. After four washings with TXS extraction buffer, the VE‐cadherin‐containing complexes and Kindlin‐2‐associated proteins were analysed by western blotting with specific antibodies. To purify EGFP‐tagged Kindlin‐2‐associated proteins, HEK293 cell lysates prepared from confluent cultures were incubated with Chromatik EGFP‐Trap Sepharose (Allele Biotech Innovative Technology, San Diego, CA, USA) for 2 h at 4°C. Sepharose‐captured immune complexes were analysed by western blot using Abs to N‐cadherin (Thermofisher Scientific), actin, α‐catenin, β‐catenin and γ‐catenin and EGFP. The band densities were analysed using ImageJ (NIH, Bethesda, MD, USA).
Expression and preparation of recombinant proteins
Constructs expressing α‐catenin, β‐catenin and γ‐catenin in pcDNA3 vector were purchased from Addgene Inc. (Cambridge, MA, USA) and recloned into pGEX‐5X‐1 vector using SalI‐NotI, BamHI‐NotI and SalI‐NotI restriction sites, respectively. The VE‐cadherin‐EGFP pcDNA3 construct was a generous gift from Dr Elisabetta Dejana (IFOM, Milan, Italy). VE‐cadherin cytoplasmic tail (CT) (621–784) was amplified by PCR and inserted into pGEX‐5X‐1 vector using BamHI and NotI restriction sites. Glutathione S‐transferase (GST)‐fused VE‐cadherin‐CT, catenins and Kindlin‐2 were purified on glutathione‐sepharose. For selected assays, GST tags were cleaved from the proteins with FXa and free GST was removed on glutathione‐sepharose.
Surface plasmon resonance (SPR)
Real time protein–protein interactions were analysed using a Biacore3000 instrument (Biacore, Uppsala, Sweden). GST‐tagged VE‐cadherin‐CT, β‐, α‐ and γ‐catenin and control GST were immobilized on CM5 biosensor chips. Experiments were performed at 22°C in 10 mm Hepes buffer, pH 7.4, containing 150 mm NaCl, 0.005% surfactant P20 and 3 mm MgCl2 at flow rate 25 μl min−1. SPR sensograms were obtained by injecting various concentrations of recombinant full‐length Kindlin‐2 (upon removal of its GST‐tag) over immobilized proteins and reference flow cells. Association/dissociation curves were determined after subtraction of the reference surface values and buffer control. Sensograms were analysed using BIAevaluation software, version 4.01 (GE Healthcare, Little Chalfont, UK).
Actin spin‐down assay
Actin binding to Kindlin‐2 and β‐catenin or γ‐catenin was evaluated using Protein Spin‐Down Biochem kits (Cytoskeleton Inc., Denver, CO, USA). Actin was resuspended to 1 mg ml−1 in general actin buffer and left on ice for 30 min. Actin was polymerized by the addition of 1:10 actin polymerization buffer, 50 mm KCl, 2 mm MgCl2 and 1 mm ATP for 1 h at room temperature, producing 21 μm F‐actin stock. Test proteins were centrifuged prior to use at 150 000 g at 4°C for 90 min, incubated with the polymerized F‐actin for 30 min and centrifuged at 150 000 g at 24°C for 90 min. The supernatants and pellets were resuspended and boiled in reducing sample loading buffer and analysed on 10% SDS‐PAGE. Gels were stained with Gel code blue stain reagent (Thermo Fisher Scientific).
Glutathione bead binding assay
Selected combinations of VE‐cadherin‐CT‐GST and full‐length Kindlin‐2, β‐catenin or γ‐catenin (upon removal of GST) at concentrations of 8 μm were incubated in the binding buffer (20 mm Tris, pH 7.8, 100 mm KCl, 10 mm MgCl2 and 1 mm dithiothreitol) in a total volume of 60 μl for 1 h at room temperature. Next, the protein mix was incubated with 60 μl of glutathione‐agarose for 1 h. After centrifugation and removal of the supernatant, the beads were washed four times, and the bead pellets were resuspended in 60 μl of sample loading buffer. The eluted complexes were analysed on 9% acrylamide gels, and gels were stained with Gel code blue stain reagent (Thermo Fisher Scientific).
Real‐time qRT‐PCR
HUVECs were treated with siGenome SMART pool human PLEKHC1 siRNAs or non‐targeting siRNA as described above. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer's instructions. cDNA was generated using an iScript cDNA Synthesis Kit and 0.25 μg of the RNA template in a total volume of 20 μl (Bio‐Rad, Berkeley, CA, USA). Quantitative RT‐PCR was performed using the respective gene‐specific primers (RT2 qPCR Primer Assay; Qiagen, Germantown, MD, USA) and iQ SYBR Green Supermix on an iCycler PCR system (Bio‐Rad) in accordance with the manufacturer's instructions. The PCR contained 150 ng of cDNA, 10 μm forward and reverse primers and 10 μl of 2 × iQ SYBR Green Supermix in a total volume of 20 μl. The cycle threshold (Ct) values were calculated with SDS, version 1.4 (Bio‐Rad). Results were calculated as expression of the target gene relative to expression of the reference gene (GAPDH).
Statistical analysis
Data are presented as the mean ± SEM. Statistical analyses were performed using a Kolmogorov–Smirnov normality test and a between‐group comparison of means was performed by one‐way ANOVA. To avoid an increased risk of a type 1 error arising from the use of multiple comparisons, a Holm–Sidak test was applied for all comparisons. All statistical analyses were performed using Sigma Plot, version 10.0 (Systat Software Inc., San Jose, CA, USA). P < 0.05 was considered statistically significant.
Results
Kindlin‐2 maintains vascular barrier in vivo
We had previously implicated Kindlin‐2 in pathological and developmental angiogenesis and found that new blood vessels formed in Kindlin‐2 +/− mice were leaky (Pluskota et al. 2011). However, the integrity of neovasculature and mature vasculature is subject to different regulatory cues (Wong et al. 1997), prompting us to examine the barrier function of pre‐existing blood vessels in Kindlin‐2 +/− mice.
As an initial step, we examined the escape of fluorescent ICG from ear vasculature of WT and Kindlin‐2 +/− mice without and upon stimulation with low‐dose PAF, a known inducer of vascular permeability. After i.v. injections of the two mouse strains with ICG ± PAF, we obtained series of vital images over a 40 min period using an in vivo imaging system (IVIS) to track the vascular leakage as accumulation of the fluorescent dye in the ear tissue (Fig. 1 A). The total radiant efficiency at 1 min time intervals throughout the 40 min observational period was quantified (Fig. 1 A, left). In the absence of PAF, in WT mice, the fluorescence intensities of the accumulated dye were low, and then increased slightly during the first 5 min, remaining constant until 40 min. By contrast, in Kindlin‐2 +/− mice, fluorescence intensities increased during the entire observational period and, at 40 min, they were 2‐fold higher than in WT mice (Fig. 1 A, left; see also the Supporting information, Video S1). Injection of low dose PAF (6 μg kg−1) significantly enhanced accumulation of ICG in both mouse strains but was much more extensive in the Kindlin‐2 +/− mice (Fig. 1 A, left; see also Supporting information, Video S2). Figure 1 A (middle) shows representative images of mice at the end‐point (40 min) and the colour scale indicates dye accumulation in the ears (red: lowest dye content; blue/purple: highest dye content). At this time point, the total dye accumulation (radiant efficiency) in Kindlin‐2 +/− mice was 2‐ to 2.5‐fold higher compared to WT littermates either in the absence or presence of PAF (P < 0.05, n = 8 mice per group) (Fig. 1 A, right).
Figure 1. Kindlin‐2 maintains the vascular barrier.
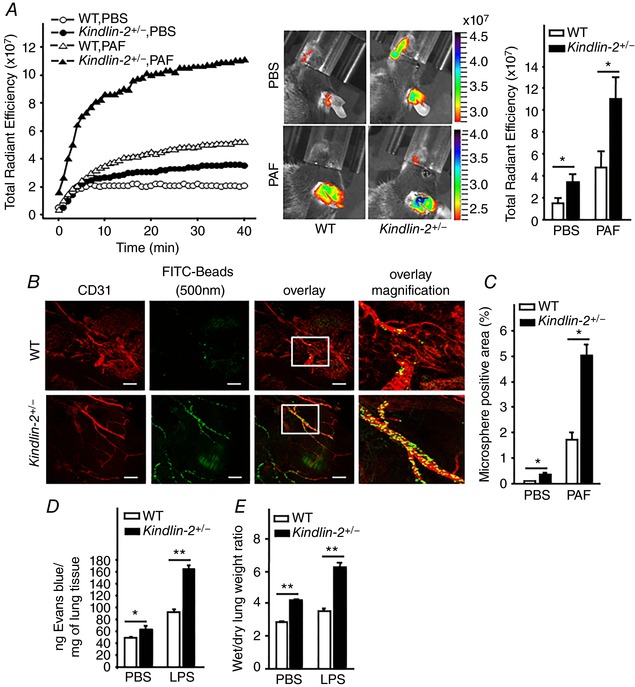
A, enhanced permeability in ear skin vasculature in Kindlin‐2 +/− mice. Left: kinetics of fluorescent dye ICG accumulation in the ear of one representative mouse per group monitored by IVIS expressed as total radiant efficiency [(p s−1 cm−2 sr−1)/(μW cm−2)]. Middle: representative images of leaked and accumulated ICG in mouse ears at 40 min. The colour scale associates with the middle images and indicates total radiant efficiency: the upper blue/purple colour indicates maximum, whereas the lower red colour indicates minimum total radiant efficiency. Right: statistical comparison of quantified total radiant efficiencies of all images at the 40 min time point. Data are expressed as the mean ± SEM *, P < 0.05 by one‐way ANOVA, (n = 8). B, enhanced PAF‐induced permeability of tracheal vasculature (anti‐CD31, red fluorescence) for FITC‐labelled beads (500 nm, green fluorescence) in Kindlin‐2 +/−mice. Scale bar = 60 μm. C, quantification of the area positive of green microspheres in low‐power images of tracheas from untreated and PAF‐treated mice. Data are the mean ± SEM. * P < 0.01 (n = 6). D and E, enhanced LPS‐induced pulmonary oedema in Kindlin‐2 +/− mice. D, leakage of Evans blue dye from pulmonary vasculature, E, ratio of lung tissue wet to dry weights (indicator of fluid accumulation). Data are expressed as the mean ± SEM. * P < 0.05, ** P < 0.001 by one‐way ANOVA (n = 8).
As a second approach to assess vascular barrier function, we measured extravasation of 500 nm fluorescence‐labelled microspheres via interendothelial cell gaps in tracheal vasculature with or without PAF challenge. No extravasation was observed in the absence of PAF (not shown). Upon administration of PAF, microspheres accumulated in the walls of venules of both WT and Kindlin‐2 +/− mice. However, microspheres were more numerous and extensive in Kindlin‐2 +/− mice than in WT littermates (Fig. 1 B). The area occupied by microspheres in low‐power images of tracheas from PAF‐treated Kindlin‐2 +/− mice (determined using ImagePro Plus Capture and Analysis software) was 3 ± 0.5‐fold higher than that in PAF‐treated WT mice (n = 6 mice per group, P < 0.01) (Fig. 1 C). In addition, Kindlin‐2 +/− mice also showed significantly increased Evans blue dye leakage into the lungs both under baseline and LPS‐induced conditions (Fig. 1 D).To further corroborate these results, we compared the wet to dry lung weight ratios as an indication of pulmonary oedema in WT and Kindlin‐2 +/− mice injected i.p. with control PBS or LPS. At baseline, the Kindlin‐2 +/− mice showed a 35% increase in the lung wet‐to‐dry weight ratios compared to WT mice, indicating fluid accumulation. Upon LPS challenge, the oedema was enhanced by 70% in Kindlin‐2 +/− mice compared to WT mice (Fig. 1 E).
Next, we compared changes in TEER of confluent monolayers of aortic endothelial cells isolated from WT and Kindlin‐2 +/− mice. In the absence of PAF, TEER of WT and Kindlin‐2 +/− ECs was constant but lower in Kindlin‐2 +/− ECs. PAF induced a temporary reduction in TEER in both WT and Kindlin‐2 +/− ECs, consistent with the opening of inter‐endothelial junctions. After 2–3 min, PAF‐triggered an 85–90% reduction in TEER of the Kindlin‐2 +/− ECs compared to untreated cells (Fig. 2 A). With WT ECs, PAF induced only a 50% reduction in TEER. For both WT and Kindlin‐2 +/− ECs, TEER values returned to the baseline levels after 7–8 min (Fig. 2 A).
Figure 2. Kindlin‐2 stabilizes interendothelial junctions.
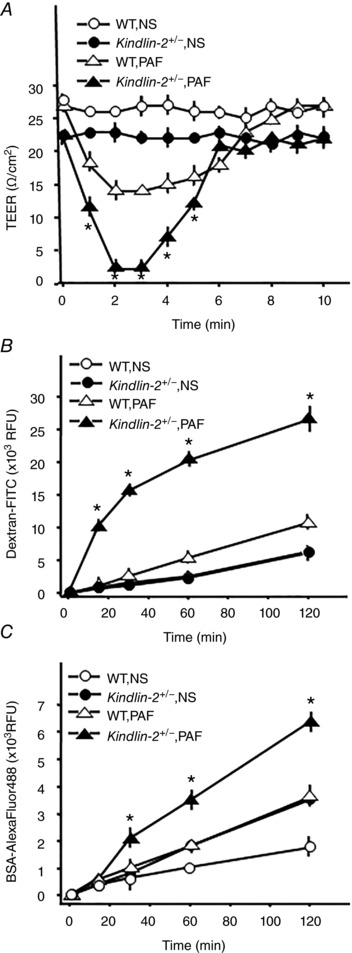
A, effect of PAF (500 ng ml−1) on TEER of WT and Kindlin‐2+/− MAECs. Data are the mean ± SEM. * P < 0.01 (n = 6) Kindlin‐2+/− vs. WT. Enhanced flux of FITC‐dextran 10 kDa (B) and Alexa Fluor 488‐BSA (C) through Kindlin‐2+/− MAECs compared to WT cells in the absence or presence of PAF (500 ng ml−1). Data are the mean ± SEM, (n = 6); * P < 0.001 WT vs. Kindlin‐2+/− MAECs.
Finally, we measured functional barrier of WT and Kindlin‐2 +/− EC confluent monolayers to BSA or 10 kDa dextran. In the absence of PAF, passage of both reagents through the monolayers was similar in both genotypes. However, upon cell treatment with PAF, the flux of 10 kDa dextran and BSA through the Kindlin‐2 +/− EC monolayers was increased by ∼5‐fold and 2‐fold, respectively, compared to WT ECs (n = 6 mice per group, P < 0.001) (Fig. 2 B and C).
Kindlin‐2 is a constituent of adherens junction complexes in ECs
To test whether Kindlin‐2 might be present in inter‐endothelial junctions, we double‐stained confluent monolayers of resting HUVECs with antibodies to Kindlin‐2 and to markers of various intercellular junctions. Confocal microscopy of resting HUVECs revealed that Kindlin‐2 was present not only within the cytoplasm, but also was enriched along cell–cell junctions and strongly colocalized with VE‐cadherin, the major component of AJs (Fig. 3 A). In addition, Kindlin‐2 co‐immunoprecipitated with components of AJs, VE‐cadherin, β‐catenin, γ‐catenin and α‐catenin, from lysates of confluent, resting HUVECs (Fig. 3 B). Because AJs are stabilized by actin filaments and we recently demonstrated direct interaction of actin with Kindlin‐2 (Bledzka et al. 2016), we considered that Kindlin‐2 helps to maintain vascular barrier by bridging AJ to actin. Confluent HUVECs were treated with thrombin, a potent inducer of vascular permeability (van Nieuw Amerongen et al. 1998) and stained for VE‐cadherin and Kindlin‐2 with specific antibodies and for F‐actin with Alexa Fluor 647‐phalloidin. In resting HUVECs, Kindlin‐2 strongly colocalized with VE‐cadherin and cortical actin filaments in stable AJs along the boundaries of adjacent cells (Fig. 3 C, upper and insert). Thrombin triggered the disassembly of stable AJs and the formation of multiple intercellular gaps (Fig. 3 C, lower, white asterisks). Under these conditions, only minor colocalization of Kindlin‐2 with VE‐cadherin and cortical actin was detected. Instead, Kindlin‐2 mostly colocalized with actin stress fibres that formed in the thrombin treated cells. In rarer cases, Kindlin‐2 was also detected in some AJs of thrombin‐stimulated cells, where it overlapped with perpendicular radial actin bundles that linked to VE‐cadherin (Fig. 3 C, upper insert, yellow arrows). These latter structures may represent focal AJs that are actively remodelling. This interpretation is similar to that reported previously (Fig. 3 C, yellow arrows) (Millan et al. 2010; Huveneers et al. 2012). Interestingly, in thrombin‐treated HUVECs, many VE‐cadherin‐positive AJs that are missing Kindlin‐2 are not connected to actin (Fig. 3 C, lower insert, white arrows). These data were corroborated by western blots of VE‐cadherin‐based complexes immunoprecipitated with anti‐VE‐cadherin and containing β‐ and γ‐catenin. Densitometric analysis of the western blots showed that association of Kindlin‐2 and actin within these complexes was substantially reduced by thrombin (Fig. 3 D), although their total cellular expression levels did not change (Fig. 3 D, input TritonX‐100‐soluble). This redistribution of Kindlin‐2, actin and other AJ proteins was not a result of loss into the insoluble cytoskeleton because these molecules were minimal in TritonX‐100 insoluble fractions compared to soluble fractions (Fig. 3 D, input insoluble). By contrast, Kindlin‐3 was absent in adherens, tight or gap junctions in HUVECs based on co‐localization and co‐immunoprecipitation studies (Fig. 4 A–C). Furthermore, a 70% reduction in Kindlin‐3 expression (siRNA treatment) (Bialkowska et al. 2010) did not enhance BSA penetration through the endothelial barrier (Fig. 4 D and E). Taken together, Kindlin‐2 is found to be a novel component of the stable AJs in resting ECs and probably assists in linking them to cortical actin bundles.
Figure 3. Kindlin‐2 is present in AJs.
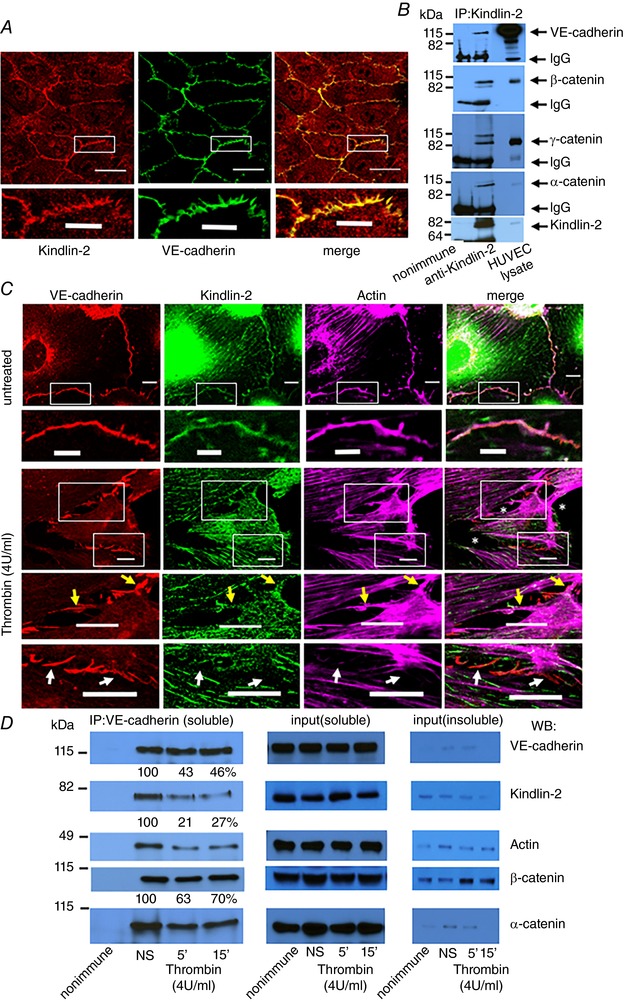
A, colocalization of Kindlin‐2 (red) and VE‐cadherin (green) in resting HUVECs within AJs. Scale bar = 35 μm. Higher magnification images of the cell junctions are shown (lower). Scale bar = 11 μm. Data are representative of three independent experiments. B, Kindlin‐2 co‐immunoprecipitates with the VE‐cadherin‐based complexes. Kindlin‐2 was immunoprecipitated from lysates of confluent resting HUVECs. Immune complexes were analysed by western blotting with Abs to Kindlin‐2 and to proteins of VE‐cadherin complexes, α‐, β‐ and γ‐catenin. C, images of resting or thrombin‐treated HUVEC monolayers stained with anti‐Kindlin‐2 (green), anti‐VE‐cadherin Abs (red) and Alexa Fluor 647‐phalloidin for actin (magenta). Thrombin (lower) caused stable AJs to disassemble and disconnect Kindlin‐2 and actin from VE‐cadherin (lower insert, white arrows), leading to the formation of multiple intercellular gaps (white asterisks). Remodelling VE‐cadherin‐positive focal AJs with axial actin fibres (magenta) still remaining in complex with Kindlin‐2 are also present (upper insert, yellow arrows). Untreated EC, scale bar = 12 μm, insert scale bar = 6 μm. Thrombin‐treated EC, scale bar = 23 μm. D, Kindlin‐2 and actin dissociate from AJs during thrombin‐induced AJ disassembly in HUVECs. Western blots of VE‐cadherin immunocomplexes from TritonX‐100‐soluble HUVEC fractions probed with Abs to Kindlin‐2, actin and AJ proteins. Band densities are expressed as a percentage of untreated cells, which was assigned a value of 100%. Western blot analysis of TritonX‐100‐soluble (5% input) and insoluble (25% input) HUVEC fractions demonstrated minimal expression of Kindlin‐2 and AJ proteins in insoluble fractions.
Figure 4. Kindlin‐3 is not implicated in the maintenance of endothelial barrier function.
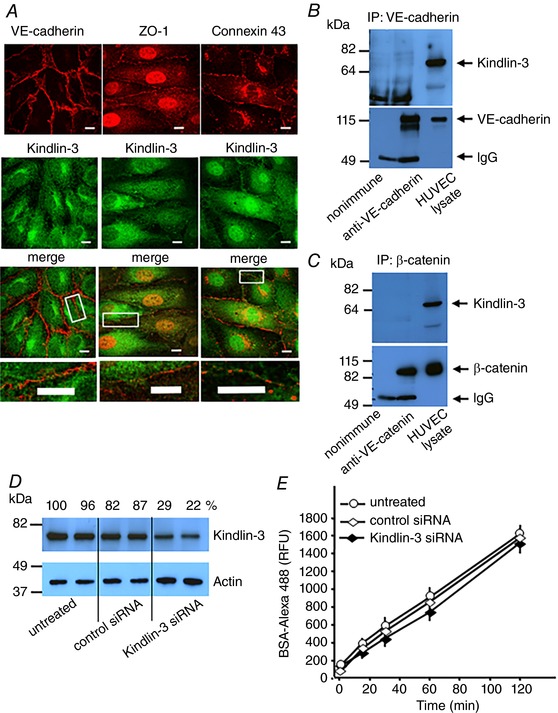
A, images of resting HUVECs stained with anti‐Kindlin‐3 (green) and anti‐VE‐cadherin (AJs), ZO‐1 (TJs) or connexin 43 (gap junctions) (red) Abs. Scale bar = 30 μm. Higher magnification images of the cell junctions are shown (lower). Scale bar = 11 μm. VE‐cadherin (B) and β‐catenin (C) were immunoprecipitated from lysates of resting confluent HUVECs and analysed on western blots with an anti‐Kindlin‐3 Ab. D, densitometric analysis of western blots shows 70–80% reduced expression of Kindlin‐3 in HUVECs by Kindlin‐3‐targeting siRNA (100 nm) compared to non‐treated cells or cells treated with non‐targeting siRNA (100 nm). E, reduction in Kindlin‐3 expression with Kindlin‐3 siRNA did not enhance flux of Alexa 488‐BSA via the HUVEC monolayer. Data are expressed as the mean ± SEM. P > 0.05 by one‐way ANOVA, (n = 8).
Reduction of Kindlin‐2 expression disrupts AJs
To substantiate and gain insight into the mechanisms by which Kindlin‐2 regulates the vascular barrier, we knocked down Kindlin‐2 in confluent HUVEC cultures using siRNA. Densitometry of western blots of siRNA‐treated HUVECs demonstrated a ∼80% decrease of Kindlin‐2 expression compared to ECs treated with non‐targeting siRNA (Fig. 5 A). A greater reduction in Kindlin‐2 levels led to dissociation of cells. HUVEC staining with anti‐VE‐cadherin Ab and image analyses showed that the 80% reduction in Kindlin‐2 levels led to a 3.5‐fold increase in the formation of gaps in EC monolayer and a decrease in stable, continuous AJs compared to untreated cells or HUVEC treated with non‐targeting siRNA (Fig. 5 B and C). Disruption of the interendothelial junctions was specifically attributed to reduced Kindlin‐2 expression because mRNA and protein levels of the components of AJs, TJs and gap junctions were not altered upon Kindlin‐2 reduction (data not shown). Consistent with these results, we observed a 2‐fold enhancement of PAF‐induced BSA and dextran flux through the HUVEC cultures treated with Kindlin‐2‐targeting siRNA (Fig. 5 D and E). Thus, Kindlin‐2 prevents the increase of permeability in endothelial cells. In addition, this reduction of Kindlin‐2 in HUVECs not only decreased baseline TEER by 30%, but also potentiated thrombin‐dependent decrease of TEER (50% in control siRNA vs. 75% in Kindlin‐2‐siRNA‐treated cells) (Fig. 5 F). Similar TEER changes were obtained when HUVECs were stimulated with PAF (data not shown). Triple staining of Kindlin‐2‐siRNA‐treated HUVECs for VE‐cadherin, Kindlin‐2 and actin revealed not only fewer stable AJs linked to cortical actin (Fig. 6 A, left vs. right), but also loss of VE‐cadherin connections with axial actin fibres within remodelling focal AJs (Fig. 6 A, right, insert, yellow arrows). We characterized VE‐cadherin‐based complexes in HUVECs treated with Kindlin‐2 siRNA by immunoprecipitation of VE‐cadherin from TritonX‐100‐soluble fractions and western blotting with antibodies to the catenins, actin and Kindlin‐2. Densitometry of band intensity revealed that reduction of Kindlin‐2 expression in HUVECs led to a 50–70% decrease in the association not only of Kindlin‐2, but also of actin and α‐catenin with the VE‐cadherin complexes (Fig. 6 B). By contrast, association of β‐ and γ‐catenin with VE‐cadherin was not altered. Western blots of HUVEC TritonX‐100‐soluble fractions showed that the decrease of Kindlin‐2 expression by Kindlin‐2‐specific siRNA did not affect the expression levels of any of the AJ proteins (Fig. 6 B). Their mRNA levels were also unchanged (data not shown). Kindlin‐3 remained unchanged in Kindlin‐2‐siRNA‐treated HUVECs, indicating specificity (Fig. 6 B, soluble input). Next, we immunopurified VE‐cadherin from resting and PAF‐ or thrombin‐stimulated WT and Kindlin‐2 +/− MAECs. Compared with WT ECs, resting Kindlin‐2 +/− ECs showed a reduced association of Kindlin‐2, actin and α‐catenin but not of β‐ or γ‐catenin with VE‐cadherin. These differences were even more pronounced upon treatment of cells with PAF or thrombin, which induced AJ disassembly and reorganization (Fig. 6 C). Expression levels of AJ proteins and actin were similar in WT and Kindlin‐2 +/− ECs and unaffected by thrombin or PAF treatment (Fig. 6 C, input TritonX‐100 soluble fraction).
Figure 5. Decrease of Kindlin‐2 expression disturbs the endothelial barrier.
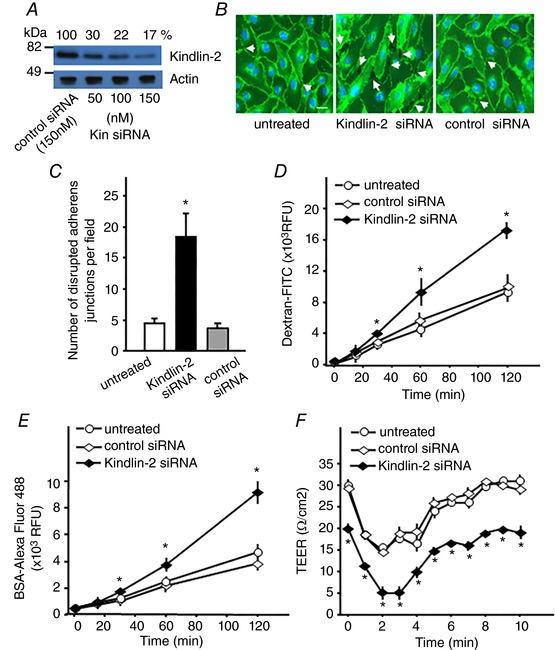
A, western blot analysis of HUVECs treated with non‐targeting or Kindlin‐2 siRNA probed with anti‐Kindlin‐2 or anti‐actin Abs as a loading control. Band densitometric quantification is expressed as the percentage of Kindlin‐2 expression in Kindlin‐2 siRNA‐treated HUVECs compared to EC treated with non‐targeting siRNA (100%). B, representative images of confluent HUVEC monolayers treated with non‐targeting or Kindlin‐2 siRNA (100 nm) and stained with an anti‐VE‐cadherin Ab to visualize AJs. White arrows indicate the prominent gaps formed between cells as a result of AJ disruption. Scale bar = 30 μm. Data are representative of three independent experiments. C, quantification of disrupted AJs in HUVEC monolayers from (B). Data are the number of disrupted AJs (intercellular gaps) per field and are expressed as the mean ± SEM. * P < 0.05, (n = 3 experiments, 10 fields per group were quantified in each experiment), non‐treated or control siRNA‐treated vs. Kindlin‐2 siRNA‐treated HUVECs. D–F, siRNA‐mediated (100 nm) suppression of Kindlin‐2 expression in HUVECs enhances PAF‐induced (500 ng ml−1) flux of FITC‐dextran 10 kDa (D) and Alexa Fluor 488‐BSA (E) and reduces TEER (F) in the absence or presence of thrombin (4 U ml−1). Data are the mean ± SEM (* P < 0.05, n = 9 in each case, non‐treated or control siRNA‐treated vs. Kindlin‐2 siRNA‐treated HUVECs.)
Figure 6. Reduction of Kindlin‐2 expression disrupts AJs.
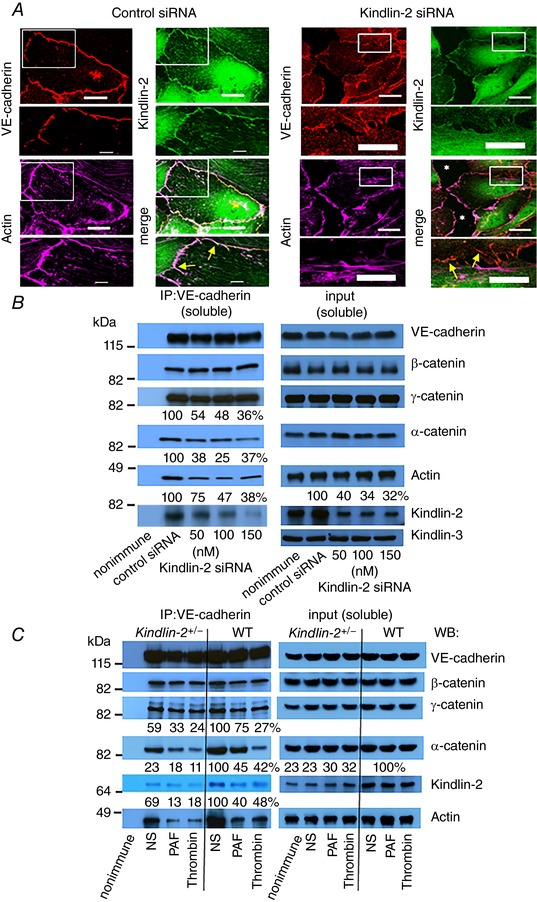
A, HUVECs treated with control or Kindlin‐2 siRNA were stained for VE‐cadherin (red), Kindlin‐2 (green) and F‐actin (magenta). HUVECs treated with control siRNA show continuous stable AJs colocalizing with VE‐cadherin, Kindlin‐2 and F‐actin (left, yellow arrows in the insert). Scale bar = 23 μm, insert scale bar = 6 μm. HUVECs treated with the Kindlin‐2 siRNA show multiple disassembling and/or remodelling AJs, as well as intercellular gaps (white asterisks, right). Scale bar = 23 μm and insert scale bar = 12 μm. When Kindlin‐2 levels are reduced by siRNA, VE‐cadherin is still present at the junctions, although its connection to actin fibres is reduced or lost (right, yellow arrows in the insert). Images are representative of three independent experiments. B, reduced Kindlin‐2 expression in HUVECs results in a decreased association of actin and α‐catenin with VE‐cadherin‐based complexes. Confluent HUVEC monolayers were treated with respective siRNAs. Western blot analysis of VE‐cadherin immunocomplexes from TritonX‐100‐soluble cell fractions with the indicated antibodies. Western blots of 5% input from TritonX‐100 soluble fractions are shown (right). Band intensities are expressed as a percentage of control‐siRNA‐treated cells, which were assigned a value of 100%. Data are representative of three independent experiments. C, reduction of Kindlin‐2 expression disrupts AJs in MAECs. Confluent monolayers of WT or Kindlin‐2+/− MAECs were treated with PAF (500 ng ml−1) or thrombin (4 U ml−1) for 15 min. VE‐cadherin immunocomplexes from TritonX‐100‐soluble cell fractions were analysed by western blotting with the indicated Abs. The 5% input from TritonX‐100 soluble fractions was analysed (right). The numbers indicate band densities expressed as a percentage of control WT, untreated cells, which was assigned a value of 100%. Three independent experiments were performed.
Kindlin‐2 is crucial to tight junction integrity
Next, we aimed to determine whether Kindlin‐2 is a component of endothelial tight or gap‐junctions. Immunofluorescence of confluent, resting HUVECs revealed that Kindlin‐2 did not colocalize or co‐immunoprecipitate with ZO‐1 or occludin, components of tight junctions (Fig. 7 A and B). In addition, Kindlin‐2 did not colocalize with connexin 43, a marker of endothelial gap junctions (Fig. 7 C). Thus, Kindlin‐2 appears to be a previously unrecognized component of AJs but not of tight or gap junctions. Although Kindlin‐2 is absent from TJs, its 80% decrease in HUVECs after Kindlin‐2‐ siRNA treatment also disrupted TJs, as revealed by ZO‐1 staining (Fig. 7 D and E). Such interdependence of AJ and TJ has been observed in epithelial cells upon disruption of E‐cadherin‐based AJ with blocking antibodies or in E‐cadherin‐null embryos (Ohsugi et al. 1997; Bazzoni & Dejana, 2004).
Figure 7. Kindlin‐2 is absent in tight and gap junctions, although reduction of its expression disrupts tight junctions in HUVECs.
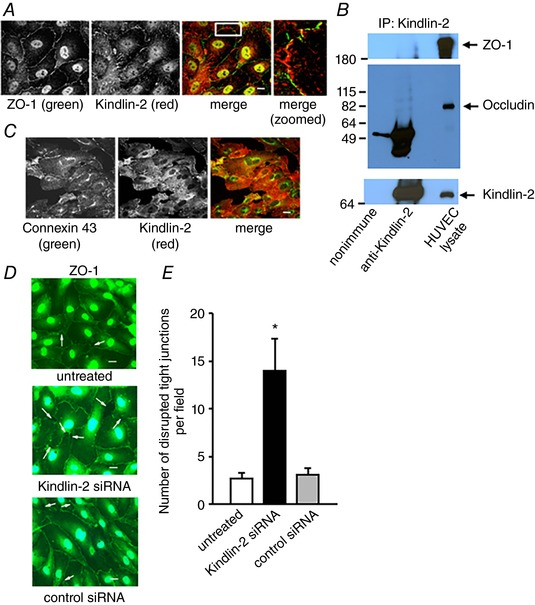
A, Kindlin‐2 does not co‐localize with ZO‐1 and (B) does not co‐immunoprecipitate with the components of tight junctions: ZO‐1 and occludin. C, Kindlin‐2 does not colocalize with Connexin 43 of gap junctions in resting HUVECs. D, representative images of confluent HUVEC monolayers treated with control or Kindlin‐2 siRNA (100 nm) and stained with anti‐ZO‐1, a marker of tight junctions. White arrows indicate intercellular gaps formed in cell cultures. Scale bar = 30 μm. E, quantification of disrupted TJs in HUVECs upon treatment with the indicated siRNAs. Data are the numbers of disrupted TJs (intercellular gaps) per field and are expressed as the mean ± SEM. * P < 0.05, Kindlin‐2 siRNA‐treated HUVECs vs. untreated or treated with non‐targeting siRNA (n = 3 experiments, 10 fields per group were examined in each experiment).
Kindlin‐2 interacts directly and simultaneously with β‐catenin or γ‐catenin and actin linking the VE‐cadherin complex to the actin cytoskeleton
To determine whether Kindlin‐2 directly interacts with VE‐cadherin‐CT or indirectly through the catenins, we incubated VE‐cadherin‐CT‐GST with Kindlin‐2 in the presence or absence of the β‐ or γ‐catenin (GST tags were removed from the catenins and Kindlin‐2). Next, VE‐cadherin‐CT‐GST was pulled down from these mixtures using glutathione agarose beads and, after thorough washing, complexes were eluted and analysed by SDS‐PAGE. Analysis of the gels showed that Kindlin‐2 did not interact with VE‐cadherin‐CT, whereas β‐catenin did, as reported previously (Lampugnani et al. 1995; Dejana et al. 2008). However, in the presence of β‐catenin, Kindlin‐2 bound to the VE‐cadherin‐CT, suggesting that β‐catenin bridged Kindlin‐2 to VE‐cadherin‐CT‐GST (Fig. 8 A, left). Similar results were obtained when γ‐catenin, a homologue of β‐catenin, which also binds VE‐cadherin, was used to replace β‐catenin (Fig. 8 A, right). Direct interaction between Kindlin‐2 and VE‐cadherin‐CT‐GST and GST‐tagged α‐, β‐ and γ‐catenin was explored using SPR. Kindlin‐2 interacted with GST‐tagged β‐ and γ‐catenin immobilized on biosensor chips in a concentration‐dependent manner (Fig. 8 B and C). The interaction between GST alone and Kindlin‐2 was negligible (Fig. 8 D). Using BiaEvaluation software to analyse the SPR progress curves, Kindlin‐2 bound to β‐catenin with an estimated K d of 8.5 × 10−7 m and to γ‐catenin with a K d of 4.57 × 10−7 m. These values were derived by fitting the kinetic data to a 1:1 global Langmuir model. By contrast, Kindlin‐2 had negligible binding to VE‐cadherin‐CT and α‐catenin (data not shown). These results were corroborated when HUVECs were stained for Kindlin‐2 (green) and β‐catenin or γ‐catenin (red). In resting cells, Kindlin‐2 strongly colocalized with both catenins at cell junctions (Fig. 8 E and F, upper). Thrombin triggered the disassembly of these complexes with catenins decreasing at the junctions and with Kindlin‐2 reorganizing into multiple stress fibres (Fig. 8 E and F, lower).
Figure 8. Kindlin‐2 interacts directly with β‐catenin and γ‐catenin.
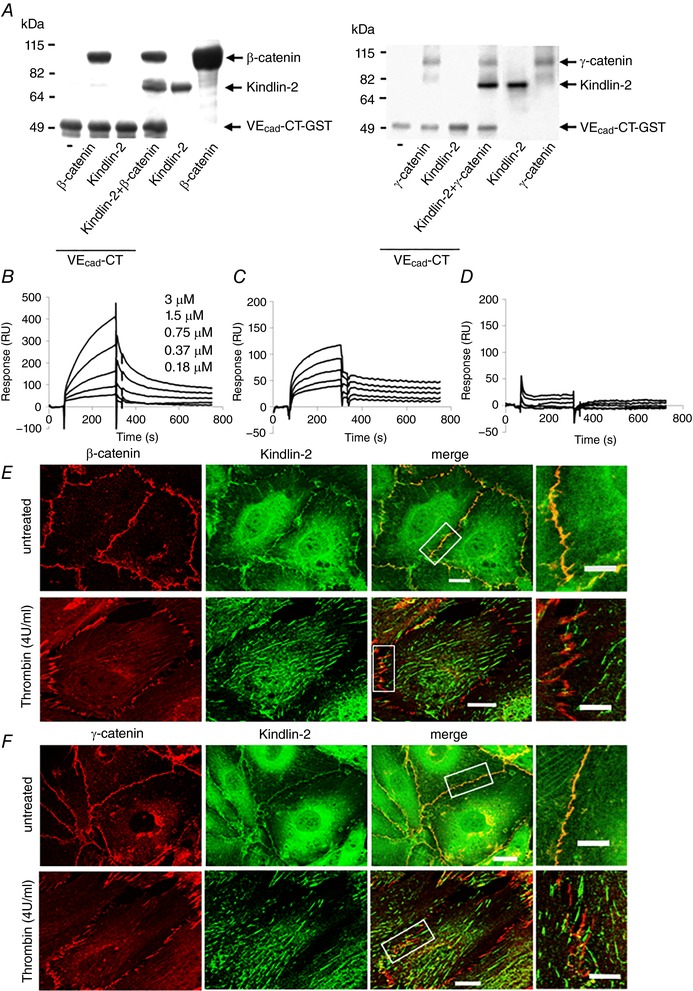
A, Kindlin‐2 does not bind directly to the cytoplasmic tail of VE‐cadherin (VEcad‐CT) but does bind to β‐catenin (left) and to γ‐catenin (right). Complexes of GST‐tagged VE‐cad‐CT with β‐catenin or γ‐catenin and Kindlin‐2 were captured on glutathione beads and analysed on 9% PAGE. Images are representative of two independent experiments. B–D, binding of Kindlin‐2 to β‐catenin (B), γ‐catenin (C) GST alone (D) by SPR. The progress curves shown have had mock runs (buffer only) and the empty flow cell controls subtracted. Three independent experiments were performed. E–F, colocalization of Kindlin‐2 (green) with β‐catenin (E) and γ‐catenin (red) (F) in resting and thrombin‐treated HUVECs. Higher magnification images of the cell junctions are shown (right). Scale bars = 35 μm.
We recently reported that Kindlin‐2 interacts with actin via its F0 subdomain (Bledzka et al. 2016) and we tested whether Kindlin‐2 can interact simultaneously with β‐catenin (or γ‐catenin) and F‐actin using actin spindown assays, in which actin‐bound proteins are recovered in an insoluble F‐actin pellet after ultracentrifugation. All proteins used in this assay had their GST‐tags removed. As determined by densitometric scanning of gels, ∼70% of Kindlin‐2 pelleted with actin filaments, whereas β‐catenin alone showed 0 % association (Fig. 9 A). However, β‐catenin did sediment with actin in the presence of Kindlin‐2, indicating that Kindlin‐2 can simultaneously interact with both proteins, creating a complex. Similar results were obtained for Kindlin‐2 binding to γ‐catenin and actin (Fig. 9 B).Taken together, these data show that Kindlin‐2 can directly and simultaneously interact with β‐catenin or γ‐catenin and actin, thereby connecting VE‐cadherin complexes to the actin cytoskeleton.
Figure 9. Kindlin‐2 connects β‐catenin or γ‐catenin with actin filaments.
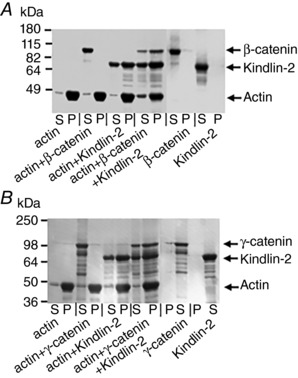
Association of the β‐catenin/Kindlin‐2 complex (A) and γ‐catenin/Kindlin‐2 complex (B) with F‐actin by co‐sedimentation. Kindlin‐2 and/or β‐catenin (γ‐catenin) were added to F‐actin followed by separation of supernatant (S) and pellet fractions (P) by ultracentrifugation and analysis by SDS‐PAGE. Two independent experiments were performed.
The F1 and F3 subdomains of Kindlin‐2 are essential for its interaction with β‐ and γ‐catenin
The kindlins are FERM‐containing proteins composed of N‐terminal F0 subdomain preceding the typical F1, F2 and F3 subdomains (Malinin et al. 2010).To determine binding site(s) in Kindlin‐2 for AJs components, we expressed full‐length Kindlin‐2 and deletion mutants lacking individual subdomains (ΔF0, ΔF1, ΔF2 and ΔF3) as EGFP fusion proteins in HEK293 cells capable of expressing these proteins at high levels. These mutant forms of Kindlin‐2 were immunoprecipitated with EGFP‐Trap Sepharose from lysates of confluent HEK 293 cells and analysed by western blotting with antibodies to N‐cadherin (predominant cadherin in HEK293 cells), β‐ and γ‐catenin, actin and EGFP. WT Kindlin‐2 and its mutants coimmunoprecipitated with endogenous N‐cadherin, β‐ and γ‐catenin. Densitometry revealed that mutants lacking either the F1 subdomain (Δ96–274) or the F3 subdomain (Δ567–652) showed markedly reduced (by 70–80%) interaction with β‐ and γ‐catenin, as well as with N‐cadherin (Fig. 6 A, left). Also, not only the Kindlin‐2 ΔF1 and ΔF3 mutants, but also the ΔF0 mutant showed dramatically reduced interaction with actin (Fig. 10 A, left), which is consistent with a recent study (Bledzka et al. 2016). Of note, overexpression of Kindlin‐2 mutants did not affect expression levels of actin or any tested AJ components in HEK 293 cells (Fig. 10 A, input, right). These data were corroborated with an alternative approach in which GST‐tagged β‐catenin was mixed with the lysates of HEK293 cells expressing WT and the deletion mutants of Kindlin‐2 and then complexes were subsequently purified with EGFP‐Trap Sepharose. Densitometric analysis of western blots of the captured complexes with anti‐GST Ab also showed a substantially reduced interaction of β‐catenin with Kindlin‐2 lacking the F1 or F3 domain (Fig. 10 B, upper). Control western blots with anti‐EGFP Ab showed that, although expression levels of Kindlin‐2 variants were not identical, expression levels of the deletion mutants were not lower than that of WT Kindlin‐2 (Fig. 10 A and B, lower). These results indicate that Kindlin‐2 interacts with β‐ and γ‐catenin via its F1 and F3 subdomains, whereas its interaction with actin also requires the F0 subdomain. Also, the Kindlin‐2 (QW/AA615) mutant, which has diminished interaction with the β‐integrin cytoplasmic tail, (Ma et al. 2008), still associated with AJ complexes, indicating the independence of the Kindlin‐2:β‐ and γ‐catenin interaction on integrin binding (Fig. 10 B).
Figure 10. The F0, F1 and F3 domains of Kindlin‐2 are essential for connecting the cadherin/catenin complexes to the actin cytoskeleton and AJ stabilization.
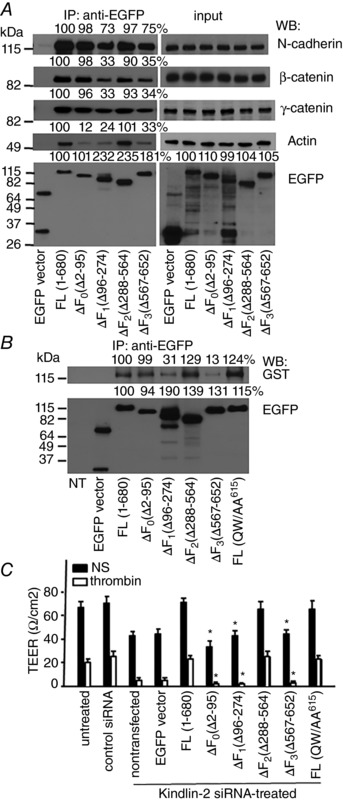
A, WT and various deletion mutants of Kindlin‐2 tagged to EGFP, as well as control EGFP, were expressed in HEK293 cells and immunopurified from resting confluent cell monolayers with EGFP‐Trap Sepharose. The immunoprecipitates were analysed on western blots with the indicated Abs. B, β‐catenin‐GST was incubated with lysates of the HEK293 cells expressing WT and Kindlin‐2 deletion mutants. Kindlin‐2 mutants were purified with EGFP‐Trap Sepharose and analysed on western blots with Abs to GST and EGFP. The values in the western blots indicate band intensities expressed as a percentage of control cells expressing full‐length Kindlin‐2, which were assigned a value of 100%. C, HUVECs were treated with non‐targeting or Kindlin‐2‐siRNA (100 nm) and, 24 h later, were transfected with full‐length Kindlin‐2‐EGFP or its mutants, as indicated. TEER changes were monitored in the absence or presence of thrombin (4 U ml−1) 24 h post‐transfection. Data are the mean ± SEM. * P < 0.01 (n = 6), ΔF0, ΔF1 or ΔF3‐Kindlin‐2‐transfected HUVEC vs. EC transfected with full‐length Kindlin‐2.
To confirm functional importance of the F0, F1 and F3 domains in Kindlin‐2‐dependent maintenance of the endothelial barrier, we reduced the expression of endogenous Kindlin‐2 in HUVECs and subsequently expressed WT and Kindlin‐2 deletion mutants in these cells. A representative image in Fig. 5 A and its densitometric analysis demonstrates that Kindlin‐2‐specific siRNA (added at a final concentration of 100 nm) decreased expression of endogenous Kindlin‐2 in HUVEC by 80% compared to 100 nm control siRNA. This knockdown led to a ∼45% decrease in TEER in the absence of thrombin compared to cells treated with non‐targeting siRNA. In addition, the Kindlin‐2‐siRNA‐treated cells became more sensitive to thrombin because it reduced their TEER by 80–90% compared to 55% in the control siRNA‐treated cells (Fig. 10 C). Importantly, expression of full‐length Kindlin‐2, ΔF2 and Kindlin‐2(QW/AA615) mutants reversed the effect of suppressed Kindlin‐2 expression in Kindlin‐2‐siRNA‐treated HUVECs and increased TEER to the levels observed in non‐treated and control‐siRNA‐treated cells (Fig. 10 C). However, the Kindlin‐2 deletion mutants lacking either F0, F1 or F3 domains failed to show increased TEER, indicating that each of these domains plays a critical role in stabilizing AJs (Fig. 10 C). Also, similar data were obtained when thrombin was replaced with PAF, implicating that the involvement of Kindlin‐2 in the stabilization of interendothelial junctions is stimuli independent (data not shown). A control western blot showed that expression levels of Kindlin‐2 mutants were not lower than that of WT Kindlin‐2 in Kindlin‐2 siRNA‐treated HUVECs (data not shown).
Discussion
Using down‐regulation of Kindlin‐2 expression in mice and in cellular systems, we demonstrate that Kindlin‐2 contributes to the maintenance of vascular barrier integrity. This previously unappreciated function of Kindlin‐2 arises from its capacity to directly and simultaneously interact with β‐ (or γ‐catenin) and actin filaments thereby linking VE‐cadherin‐based AJs to the actin cytoskeleton.
The link between actin cytoskeleton and AJs is crucial to their barrier function. In confluent EC monolayers, complexes of VE‐cadherin with β‐ (or γ‐catenin) and α‐catenin are linked to cortical‐actin fibres, which stabilizes the AJs and maintains the intercellular barrier. However, AJs are highly dynamic structures, and permeability enhancers, such as thrombin, induce cell retraction, opening and disassembly of stable AJs and dissociation of cortical actin fibres, leading to the formation of intercellular gaps. The cortical actin fibres reorganize into radial actin stress fibres connected to remodelling and active focal AJs. As with stable AJs, connection of focal AJs with radial actin fibres protects and stabilizes them and supports vascular integrity (Millan et al. 2010; Huveneers et al. 2012).
In AJs, the cytoplasmic domain of a cadherin interacts with β‐or γ‐catenin, which binds to α‐catenin, and α‐catenin interacts with actin either directly or through various actin‐binding proteins (Bazzoni & Dejana, 2004; Niessen, 2007; Dejana et al. 2008). Whether α‐catenin can simultaneously bind to β‐catenin and F‐actin is still not fully resolved (Drees et al. 2005; Yamada et al. 2005; Buckley et al. 2014). Our studies identify Kindlin‐2 as a protein that can directly and simultaneously interact with β‐ or γ‐catenin and actin both in stable and remodelling focal AJs (Fig. 11). Because Kindlin‐2 not only directly binds to G and F‐actin, but also promotes G‐actin polymerization (Bledzka et al. 2016), we propose that Kindlin‐2 may also participate in AJ formation and remodelling. Accordingly, β‐catenin and γ‐catenin interact simultaneously with VE‐cadherin‐CT and Kindlin‐2. β‐ and γ‐catenin are highly homologous, consisting of two N‐ and C‐terminal regions and a central armadillo repeat domain (Castano et al. 2002; Solanas et al. 2004). VE‐cadherin‐CT is recognized via the armadillo domain (Castano et al. 2002; Solanas et al. 2004), whereas Kindlin‐2 interacts with the N‐ and C‐termini (Yu et al. 2012). Kindlin‐2 does not compete with α‐catenin for β‐catenin binding (data not shown), although our immunoprecipitation data suggest that it enhances α‐catenin interaction with β‐catenin because there is less α‐catenin complexed with VE‐cadherin/β‐catenin when Kindlin‐2 is decreased in the complexes (Fig. 6 B and C). The regulatory N‐ and C‐tails of β‐catenin interact with its armadillo domain and inhibit its binding to E‐cadherin and α‐catenin (Castano et al. 2002; Solanas et al. 2004). Thus, Kindlin‐2 interaction with the N‐ and C‐termini of β‐catenin may not only link β‐catenin to F‐actin, but also facilitate or stabilize the cadherin‐based complexes (Gavard & Gutkind, 2006). These interactions are depicted in the model shown in Fig. 11.
Figure 11. Model depicting Kindlin‐2 connecting VE‐cadherin/catenin complexes to actin filaments and stabilizing AJs.
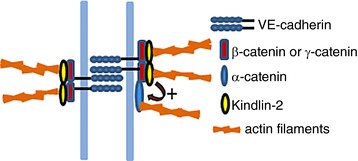
VE‐cadherin cytoplasmic tail binds to β‐ or γ‐catenin. Kindlin‐2 has the capacity to directly and simultaneously interact with β‐catenin or γ‐catenin and actin fibres bridging AJs to actin cytoskeleton. This linking function of Kindlin‐2 can also be indirect because Kindlin‐2 may also support and facilitate the interaction of β‐catenin with α‐catenin, the actin‐binding catenin. Altogether, Kindlin‐2 plays an important role in connecting VE‐cadherin/catenin complex to actin filaments, thereby not only stabilizing AJs, but also with respect to contributing to their remodelling.
Although Kindlin‐2 was not detected in TJs, reduction of Kindlin‐2 also resulted in disruption of TJ integrity. This effect may be a result of reduced expression of TJ proteins when Kindlin‐2 is downregulated. However, our results of the RT‐PCR and western blotting show similar expression levels of three TJ proteins: ZO‐1, occludin and claudin‐5 in HUVECs treated with either Kindlin‐2 +/− siRNA or control siRNA (data not shown). Thus, Kindlin‐2 does not regulate expression of TJ components. We cannot exclude the possibility that the accelerated degradation of TJs may occur upon reduction of Kindlin‐2 expression. Based on the observations by Bazzoni & Dejana (2004) and Ohsugi et al. (1997) demonstrating that AJs are required for proper assembly of TJs, we speculate that Kindlin‐2 may indirectly support TJ assembly; however, we cannot exclude the possibility that Kindlin‐2 may also protect TJs from degradation. Thus, impairment of both adherens and tight junctions may contribute to enhanced leakiness of vasculature in Kindlin‐2 +/− mice.
Because Kindlin‐2 interacts with β‐ and γ‐catenin, which interact with all cadherin family members, E‐, N‐ and P‐cadherins, the presence and stabilizing function of Kindlin‐2 may not be limited to the VE‐cadherin‐based AJs and vascular integrity. For example, the capacity of Kindlin‐2 to interact with actin and γ‐catenin (plakoglobin), the critical catenin of the AJs in the heart (Sheikh et al. 2009; Mezzano & Sheikh, 2012; Swope et al. 2013), may contribute to the abnormal cardiac development and function associated with Kindlin‐2 knockout in zebrafish (Dowling et al. 2008) and mice (Zhang et al. 2016).
Among the kindlins, the capacity to stabilize AJs in endothelial cells is unique to Kindlin‐2. Kindlin‐3 did not associate with any of the interendothelial junctions and the reduction of its expression failed to impair HUVEC barrier function for BSA (data not shown). We could not detect Kindlin‐1 in ECs by western blotting or by co‐immunoprecipitation with junctional proteins; its expression appears to be limited to epithelial cells (Ussar et al. 2008). However, Kindlin‐1‐deficient keratinocytes do show impaired cell–cell adhesion and disruption of subcortical actin network. Consistent with a potential role of Kindlin‐1 in the maintenance of intercellular junctions (Qu et al. 2012). This Kindlin‐1 function may contribute to the symptoms of Kindler syndrome, a skin disease caused by mutations in Kindlin‐1 (Jobard et al. 2003; D'Souza et al. 2010).
The first identified function of the kindlins in mammalian cells was their ability to activate the integrins (Ma et al. 2008; Montanez et al. 2008) and we demonstrated that Kindlin‐2 is essential for β3‐integrin activation and integrin‐dependent responses of ECs (Ma et al. 2008; Pluskota et al. 2011). Interestingly, the β3‐integrin knockout mice are characterized by increased vascular leakiness (Robinson et al. 2004). However, in the β3‐null mice, this increase has been attributed to enhanced expression of the VEGFR2 and enhanced VEGF‐dependent responses (Robinson et al. 2004), whereas, in Kindlin‐2 +/− ECs, VEGFR2 expression is unaltered (Pluskota et al. 2011). These previous results, along with our data demonstrating an unaltered association of β‐catenin with the Kindlin‐2 (QW/AA) variant with a mutated integrin binding site, strongly indicate that the role Kindlin‐2 in AJ stabilization is independent of its integrin activating function. This AJ stabilizing function appears to be dependent on the F1 and F3 domains of Kindlin‐2. Although the F3 domain is implicated in several functions of Kindlin‐2 (Pluskota et al. 2013), to our knowledge, this is the first assignment of a specific function to the F1 domain.
In summary, our studies identify Kindlin‐2 as a stabilizing linker of endothelial AJs with the actin cytoskeleton, thereby re‐enforcing the vascular barrier. These findings provide the basis for the consideration of new roles of kindlins in AJ‐dependent cellular functions, such as regulation of paracellular transport, contact inhibition of cell proliferation and cellular polarization.
Limitations and future directions
In demonstrating the in vitro effects of kindlin‐2 on vascular barrier function, we utilized MAECs and HUVECs cultured on fibronectin. Whether EC from other anatomical origins, grown on other matrices or matrices of differing stiffness, will exhibit a similar dependency on kindlin‐2 with respect to barrier function and whether the integrin activating function of kindlin‐2 will come into play under altered conditions remains to be assessed. Our demonstration that Kindlin‐2 +/− mice display a diminished vascular barrier function in several in vivo models suggests that our in vitro observations do translate into the in vivo setting. We note that a reduction in kindlin‐2 levels affected both basal and PAF induced permeability. This pattern is distinct from the effects of endothelial cell nitric oxide synthase deficiency in mice which, similar to the kindlin2+/‐ mice, displayed enhanced PAF induced permeability, although they did not show altered basal permeability (Hatakeyama et al. 2006), suggesting that the regulation of barrier function by kindlin‐2 may not be entirely dependent on a NO‐dependent signalling pathway.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
EP and EFP contributed to the conception and design of the research. EP, KMB, KB, DS, DAS, SJ and DV performed the experiments and analysed the data. EP, KMB, KB, DAS and SJ interpreted the results. EP and EFP wrote the manuscript. KMB, KB, DS, DAS, SJ and DV edited and revised the manuscript. All authors approved the final version of the manuscript submitted for publication.
Funding
This work was supported by NIH grants from the Heart, Lung and Blood Institute (P01 HL 073311, 2 P01 HL076491 and R01 HL096062 to EFP). Imaging of vascular permeability using the IVIS system was supported by a NIH Shared Instrument Grant award S10OD018205. HUVECs were provided by a grant awarded to the Clinical and Translational Science Collaborative of Cleveland (UL1TR000439) from the National Institutes of Health and the National Institutes of Health Roadmap for Medical Research.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1. Mice were injected i.v. with a mixture of fluorescent dye ICG and PBS. Fluorescence intensities were monitored in ear tissue for 0–40 min using IVIS. Representative movies of leakage and accumulation of ICG in mouse ears during the 30 min observational period. The colour scale indicates total radiant efficiency: the upper blue/purple colour indicates maximum, whereas the lower red colour indicates minimum total radiant efficiency. Left: a WT mouse; right : a Kindlin‐2 +/– mouse.
Video S2. Mice were injected i.v. with a mixture of fluorescent dye ICG and PAF (6 μg kg−1) (PAF). Fluorescence intensities were monitored in ear tissue for 0–40 min using IVIS. Representative movies of leakage and accumulation of ICG in mouse ears during the 30 min observational period. The colour scale indicates total radiant efficiency: the upper blue/purple colour indicates maximum, whereas the lower red colour indicates minimum total radiant efficiency. Left: a WT mouse; right panel: a Kindlin‐2 +/– mouse.
Acknowledgements
We thank Dr Judy Drazba and Cleveland Clinic Imaging Core for their excellent assistance.
Linked articles This article is highlighted by a Perspective by Ye & Dora. To read this Perspective, visit https://doi.org/10.1113/JP275082.
This is an Editor's Choice article from the 15 October 2017 issue.
References
- Bazzoni G & Dejana E (2004). Endothelial cell‐to‐cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84, 869–901. [DOI] [PubMed] [Google Scholar]
- Bialkowska K, Ma YQ, Bledzka K, Sossey‐Alaoui K, Izem L, Zhang X, Malinin N, Qin J, Byzova T & Plow EF (2010). The integrin coactivator kindlin‐3 is expressed and functional in a non‐hematopoietic cell, the endothelial cell. J Biol Chem 285, 18640–18649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledzka K, Bialkowska K, Sossey‐Alaoui K, Vaynberg J, Pluskota E, Qin J & Plow EF (2016). Kindlin‐2 directly binds actin and regulates integrin outside‐in signaling. J Cell Biol 213, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ & Dunn AR (2014). Cell adhesion. The minimal cadherin‐catenin complex binds to actin filaments under force. Science 346, 1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano J, Raurell I, Piedra JA, Miravet S, Dunach M & Garcia de HA (2002). Beta‐catenin N‐ and C‐terminal tails modulate the coordinated binding of adherens junction proteins to beta‐catenin. J Biol Chem 277, 31541–31550. [DOI] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P & Byzova TV (2005). Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med 11, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza MA, Kimble RM & McMillan JR (2010). Kindler syndrome pathogenesis and fermitin family homologue 1 (kindlin‐1) function. Dermatol Clin 28, 115–118. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F & Lampugnani MG (2008). The role of adherens junctions and VE‐cadherin in the control of vascular permeability. J Cell Sci 121, 2115–2122. [DOI] [PubMed] [Google Scholar]
- Dowling JJ, Gibbs E, Russell M, Goldman D, Minarcik J, Golden JA & Feldman EL (2008). Kindlin‐2 Is an essential component of intercalated discs and is required for vertebrate cardiac structure and function. Circ Res 102, 423–431. [DOI] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ & Weis WI (2005). Alpha‐catenin is a molecular switch that binds E‐cadherin‐beta‐catenin and regulates actin‐filament assembly. Cell 123, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard J & Gutkind JS (2006). VEGF controls endothelial‐cell permeability by promoting the beta‐arrestin‐dependent endocytosis of VE‐cadherin. Nat Cell Biol 8, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama T, Pappas PJ, Hobson RW, Boric MP, Sessa WC & Duran WN (2006). Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol 574, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H & de RJ (2012). Vinculin associates with endothelial VE‐cadherin junctions to control force‐dependent remodeling. J Cell Biol 196, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobard F, Bouadjar B, Caux F, Hadj‐Rabia S, Has C, Matsuda F, Weissenbach J, Lathrop M, Prud'homme JF & Fischer J (2003). Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum Mol Genet 12, 925–935. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B & Dejana E (1995). The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, β‐catenin, and α‐catenin with vascular endothelial cadherin (VE‐cadherin). J Cell Biol 129, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larjava H, Plow EF & Wu C (2008). Kindlins: essential regulators of integrin signalling and cell‐matrix adhesion. EMBO Rep 9, 1203–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YQ, Qin J, Wu C & Plow EF (2008). Kindlin‐2 (Mig‐2): a co‐activator of beta3 integrins. J Cell Biol 181, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Somanath PR & Byzova TV (2006). Methods for isolation of endothelial and smooth muscle cells and in vitro proliferation assays. Methods Mol Med 129, 197–208. [DOI] [PubMed] [Google Scholar]
- Malinin NL, Plow EF & Byzova TV (2010). Kindlins in FERM adhesion. Blood 115, 4011–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma Y‐Q, Podrez EA, Tosi M, Lennon DP, Caplin AI, Shurin SB, Plow EF & Byzova TV (2009). A point mutation in kindlin‐3 ablates activation of three integrin subfamilies in humans. Nature Med 15, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano V & Sheikh F (2012). Cell‐cell junction remodeling in the heart: possible role in cardiac conduction system function and arrhythmias? Life Sci 90, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan J, Cain RJ, Reglero‐Real N, Bigarella C, Marcos‐Ramiro B, Fernandez‐Martin L, Correas I & Ridley AJ (2010). Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanez E, Ussar S, Schifferer M, Bosl M, Zent R, Moser M & Fassler R (2008). Kindlin‐2 controls bidirectional signaling of integrins. Genes Dev 22, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M, Legate KR, Zent R & Fassler R (2009). The tail of integrins, talin, and kindlins. Science 324, 895–899. [DOI] [PubMed] [Google Scholar]
- Niessen CM (2007). Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol 127, 2525–2532. [DOI] [PubMed] [Google Scholar]
- Ohsugi M, Larue L, Schwarz H & Kemler R (1997). Cell‐junctional and cytoskeletal organization in mouse blastocysts lacking E‐cadherin. Dev Biol 185, 261–271. [DOI] [PubMed] [Google Scholar]
- Plow EF, Qin J & Byzova T (2009). Kindling the flame of integrin activation and function with kindlins. Curr Opin Hematol 16, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskota E, Dowling JJ, Gordon N, Golden JA, Szpak D, West XZ, Nestor C, Ma YQ, Bialkowska K, Byzova T & Plow EF (2011). The integrin coactivator kindlin‐2 plays a critical role in angiogenesis in mice and zebrafish. Blood 117, 4978–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskota E, Ma Y, Bledzka KM, Bialkowska K, Soloviev DA, Szpak D, Podrez EA, Fox PL, Hazen SL, Dowling JJ, Ma YQ & Plow EF (2013). Kindlin‐2 regulates hemostasis by controlling endothelial cell‐surface expression of ADP/AMP catabolic enzymes via a clathrin‐dependent mechanism. Blood 122, 2491–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H, Wen T, Pesch M & Aumailley M (2012). Partial loss of epithelial phenotype in kindlin‐1‐deficient keratinocytes. Am J Pathol 180, 1581–1592. [DOI] [PubMed] [Google Scholar]
- Robinson SD, Reynolds LE, Wyder L, Hicklin DJ & Hodivala‐Dilke KM (2004). Beta3‐integrin regulates vascular endothelial growth factor‐A‐dependent permeability. Arterioscler Thromb Vasc Biol 24, 2108–2114. [DOI] [PubMed] [Google Scholar]
- Sheikh F, Ross RS & Chen J (2009). Cell‐cell connection to cardiac disease. Trends Cardiovasc Med 19, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanas G, Miravet S, Casagolda D, Castano J, Raurell I, Corrionero A, de Herreros AG & Dunach M (2004). beta‐Catenin and plakoglobin N‐ and C‐tails determine ligand specificity. J Biol Chem 279, 49849–49856. [DOI] [PubMed] [Google Scholar]
- Swope D, Li J & Radice GL (2013). Beyond cell adhesion: the role of armadillo proteins in the heart. Cell Signal 25, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Moser M, Widmaier M, Rognoni E, Harrer C, Genzel‐Boroviczeny O & Fassler R (2008). Loss of Kindlin‐1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet 4, e1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Draijer R, Vermeer MA & Van Hinsbergh VW (1998). Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res 83, 1115–1123. [DOI] [PubMed] [Google Scholar]
- Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS & Peters KG (1997). Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res 81, 567–574. [DOI] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI & Nelson WJ (2005). Deconstructing the cadherin‐catenin‐actin complex. Cell 123, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wu J, Wang Y, Zhao T, Ma B, Liu Y, Fang W, Zhu WG & Zhang H (2012). Kindlin 2 forms a transcriptional complex with beta‐catenin and TCF4 to enhance Wnt signalling. EMBO Rep 13, 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Mu Y, Veevers J, Peter AK, Manso AM, Bradford WH, Dalton ND, Peterson KL, Knowlton KU, Ross RS, Zhou X & Chen J (2016). Postnatal Loss of Kindlin‐2 Leads to Progressive Heart Failure. Circ Heart Fail 9, e003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1. Mice were injected i.v. with a mixture of fluorescent dye ICG and PBS. Fluorescence intensities were monitored in ear tissue for 0–40 min using IVIS. Representative movies of leakage and accumulation of ICG in mouse ears during the 30 min observational period. The colour scale indicates total radiant efficiency: the upper blue/purple colour indicates maximum, whereas the lower red colour indicates minimum total radiant efficiency. Left: a WT mouse; right : a Kindlin‐2 +/– mouse.
Video S2. Mice were injected i.v. with a mixture of fluorescent dye ICG and PAF (6 μg kg−1) (PAF). Fluorescence intensities were monitored in ear tissue for 0–40 min using IVIS. Representative movies of leakage and accumulation of ICG in mouse ears during the 30 min observational period. The colour scale indicates total radiant efficiency: the upper blue/purple colour indicates maximum, whereas the lower red colour indicates minimum total radiant efficiency. Left: a WT mouse; right panel: a Kindlin‐2 +/– mouse.