

Abstract
Key points
The AMPA‐type ionotropic glutamate receptors (AMPARs) mediate the majority of excitatory synaptic transmission and their function impacts learning, cognition and behaviour.
The gating of AMPARs occurs in milliseconds, precisely controlled by a variety of auxiliary subunits that are expressed differentially in the brain, but the difference in mechanisms underlying AMPAR gating modulation by auxiliary subunits remains elusive and is investigated.
The elements of the AMPAR that are functionally recruited by auxiliary subunits, stargazin and cornichon 3, are located not only in the extracellular domains but also in the lipid‐accessible surface of the AMPAR.
We reveal that the two auxiliary subunits require a shared surface on the transmembrane domain of the AMPAR for their function, but the gating is influenced by this surface in opposing directions for each auxiliary subunit.
Our results provide new insights into the mechanistic difference of AMPAR modulation by auxiliary subunits and a conceptual framework for functional engineering of the complex.
Abstract
During excitatory synaptic transmission, various structurally unrelated transmembrane auxiliary subunits control the function of AMPA receptors (AMPARs), but the underlying mechanisms remain unclear. We identified lipid‐exposed residues in the transmembrane domain (TMD) of the GluA2 subunit of AMPARs that are critical for the function of AMPAR auxiliary subunits, stargazin (Stg) and cornichon 3 (CNIH3). These residues are essential for stabilizing the AMPAR–CNIH3 complex in detergents and overlap with the contacts made between GluA2 TMD and Stg in the cryoEM structures. Mutating these residues had opposite effects on gating modulation and complex stability when Stg‐ and CNIH3‐bound AMPARs were compared. Specifically, in detergent the GluA2‐A793F formed an unstable complex with CNIIH3 but in the membrane the GluA2‐A793F–CNIH3 complex expressed a gain of function. In contrast, the GluA2‐A793F–Stg complex was stable, but had diminished gating modulation. GluA2‐C528L destabilized the AMPAR–CNIH3 complex but stabilized the AMPAR–Stg complex, with overall loss of function in gating modulation. Furthermore, loss‐of‐function mutations in this TMD region cancelled the effects of a gain‐of‐function Stg carrying mutation in its extracellular loop, demonstrating that both the extracellular and the TMD elements contribute independently to gating modulation. The elements of AMPAR functionally recruited by auxiliary subunits are, therefore, located not only in the extracellular domains but also in the lipid accessible surface of the AMPAR. The TMD surface we defined is a potential target for auxiliary subunit‐specific compounds, because engineering of this hotspot induces opposing functional outcomes by Stg and CNIH3. The collection of mutant‐phenotype mapping provides a framework for engineering AMPAR gating using auxiliary subunits.
Keywords: inotropic glutamate receptor, auxiliary subunit, excitatory synaptic transmission, AMPA receptor, ion channel modulation, receptor engineering, ion channel complexes, post synaptic function
Key points
The AMPA‐type ionotropic glutamate receptors (AMPARs) mediate the majority of excitatory synaptic transmission and their function impacts learning, cognition and behaviour.
The gating of AMPARs occurs in milliseconds, precisely controlled by a variety of auxiliary subunits that are expressed differentially in the brain, but the difference in mechanisms underlying AMPAR gating modulation by auxiliary subunits remains elusive and is investigated.
The elements of the AMPAR that are functionally recruited by auxiliary subunits, stargazin and cornichon 3, are located not only in the extracellular domains but also in the lipid‐accessible surface of the AMPAR.
We reveal that the two auxiliary subunits require a shared surface on the transmembrane domain of the AMPAR for their function, but the gating is influenced by this surface in opposing directions for each auxiliary subunit.
Our results provide new insights into the mechanistic difference of AMPAR modulation by auxiliary subunits and a conceptual framework for functional engineering of the complex.
Abbreviations
- AMPAR
AMPA receptor
- CNIH
cornichon
- CNIH3
cornichon 3
- co‐IP
co‐immunoprecipitation
- DDM
n‐dodecyl‐β‐d‐maltoside
- DM
n‐decyl‐β‐d‐maltoside
- IP
immunoprecipitation
- KAR
kainate receptor
- LBD
ligand‐binding domain
- Lp1
loop 1
- NTD
amino terminal domain
- SS/P
steady state to peak
- Stg
stargazin
- TARP
transmembrane AMPA receptor regulatory protein
- TMD
transmembrane domain
- WT
wild‐type
Introduction
The ligand‐gated ion channels belonging to the AMPA type ionotropic glutamate receptors (AMPARs) play a central role in synaptic plasticity and cognition. In the brain, AMPARs are made of pore‐forming core subunits (GluA1–4) and transmembrane auxiliary subunits. The extracellular domains of a pore‐forming subunit consist of the amino terminal domain (NTD) and the ligand‐binding domain (LBD). The transmembrane domain (TMD) forms the channel pore whose gating is triggered by the binding of the neurotransmitter, glutamate, to the LBD. The ion channel function and trafficking of AMPARs are modulated by a variety of structurally unrelated transmembrane auxiliary subunits, transmembrane AMPA receptor regulatory proteins (TARPs; Chen et al. 2000; Tomita et al. 2003, 2005), cornichons (CNIHs; Schwenk et al. 2009), CKAMP44 (von Engelhardt et al. 2010), Shisa6 (Klaassen et al. 2016), synDIG1 (Kalashnikova et al. 2010) and GSG1L (Schwenk et al. 2012; Shanks et al. 2012). Stargazin (Stg; or TARP γ‐2) is the most extensively studied AMPAR auxiliary subunit, whose loss of function impacts AMPAR‐mediated synaptic transmission and cerebellar motor function (Jackson & Nicoll, 2011). However, the mechanisms for AMPAR gating modulation by Stg and other auxiliary subunits in general are poorly understood. Here, we investigate, at a single residue precision, the similarity and dissimilarity of the mechanisms for Stg and cornichon 3 (CNIH3) function.
As a member of the claudin superfamily, Stg consists of four transmembrane segments, two extracellular loops, and a C‐terminus with a PDZ domain ligand. The extracellular domain of Stg is critical for AMPAR gating modulation (Tomita et al. 2005), but the underlying mechanisms remain elusive. The cryo‐EM structures of the GluA2–Stg complex (Twomey et al. 2016; Zhao et al. 2016b) suggest that the basic residues ‘KGK’ at the D2 lobe of LBD (Dawe et al. 2016) and the linker connecting the LBD to the transmembrane helices in AMPARs could potentially interact with the acidic residues ‘EDADYEADTAE’ in the extracellular loop 1 (Lp1) of Stg. However, no direct contacts were found between the LBD and Lp1 of Stg, while an extensive interaction was observed between the transmembrane helices. It is plausible that transmembrane segments of the two proteins participate not only in complex formation but also play a key role in gating modulation. In fact, the M1 and M4 segments of the TMD from adjacent subunits in a tetrameric AMPAR are critical for TARP‐dependent modulation (Ben‐Yaacov et al. 2017). Similarly, in NMDARs, the GluN2C/D‐specific modulator CIQ requires for its action surface residues in the transmembrane helix M1 and pre‐M1 (Ogden & Traynelis, 2013). Furthermore, binding sites for modulators of Cys‐loop ion channel families were found at the TMD surfaces (Gill et al. 2011; Du et al. 2015). However, it is still unclear whether the residues facing outward from the TMD helices on the surface of AMPARs participate in gating modulation. In particular, if the mechanisms in the membrane contribute significantly to AMPAR gating modulation by auxiliary subunits, it is unclear how they relate to the known critical roles for the AMPAR LBD in this process (Dawe et al. 2016).
CNIHs, another category of AMPAR auxiliary subunits, have three membrane spanning helices with the amino terminus in the cytoplasm. The first transmembrane domain and the extracellular loop were mapped to be critical for increasing AMPAR‐mediated currents in HEK cells (Kato et al. 2010). Peptide array screening, combined with mutational analysis, revealed that critical residues in CNIH3 that destabilize the GluA2–CNIH3 complex in detergent are localized to the CNIH2/3‐specific extracellular segment and near the cytoplasmic side of the α‐helices that span the membrane (Shanks et al. 2014). Electrophysiological studies suggest that CNIH2/3 and TARPs may compete for binding to AMPAR, where GluA1‐containing heteromeric AMPARs preferentially associate with TARP γ‐8 over CNIH2/3 (Herring et al. 2013). However, information on exact binding sites and/or structural elements involved in the complex assembly in each AMPAR–auxiliary subunit complex is necessary to substantiate this hypothesis. Here, we identify that the surface residues in the TMD of the AMPAR that play key roles in gating modulation by Stg and CNIH3 are shared, but the mechanisms utilized by each auxiliary subunit differ.
Methods
Generating point mutations of GluA2
Crystal structures (PDB: 3KG2 and 4U4G) were displayed using PyMol to identify TMD residues at the surface.
Plasmid construction procedures
The rat GluA2 flip splice variant was used for all GluA2 experiments. The RNA editing isoform of the pore (Q/R) residue is indicated in each experiment. For example, GluA2‐flip (R) is abbreviated as GluA2iR. The FLAG tag was inserted near the C‐terminal domain (FATDYKDDDKEGYNVYGIESVKI, FLAG epitope in bold). pTREtVa‐GluA2flip‐FLAG vector was used as starting material. This vector contains SmaI at the 5′‐untranslated region (UTR), the full open reading frame (ORF) of GluA2 flip with a FLAG tag, and a HindIII site in the 3′‐UTR. GluA2flip has unique BstEII, EcoRI and BamHI sites in the ORF in this order from 5′ to 3′. Site directed mutagenesis and ORFs were confirmed and validated by DNA sequencing.
Single TMD4 mutants
The GluA2CT vector in pTREtVa (which contains only the C‐terminal BamHI–HindIII fragment of GluA2) was mutated via the QuikChange site‐directed mutagenesis protocol (Agilent Technologies, Santa Clara, CA, USA) and DNA sequences were verified. The mutants were then cloned into the GluA2 flip vector.
Single TMD1–3 Mutants
The GluA2 flip vector was digested at the BstEII and EcoRI sites. The resulting fragment containing the M1–3 region was then subcloned into pBS. The mutations were created using the QuikChange site‐directed mutagenesis protocol and DNA sequences were verified. The mutated M1–3 region was then cloned back into the pTREtVa GluA2 flip vector.
GluK2 construct
A C‐terminally FLAG tagged rat GluK2 was cloned into empty pTREtVa vector. The FLAG tag was inserted (LKHKPQDYKDDDDKAPVIVK, FLAG epitope in bold) near the very C‐terminus. Single, double and triple mutants of M1 and M4 were generated by the QuikChange site‐directed mutagenesis protocol and DNA sequences were verified.
Auxiliary subunit constructs
Mouse CNIH1, 2, 3 and stargazin (also known as TARP γ‐2, CACNG2) were tagged at their C‐terminus with the 1D4 epitope and cloned into the pTREtVb vector. Mutations in stargazin were introduced using the QuikChange site‐directed mutagenesis protocol. CNIH1, 2 and 3 were tagged with HA epitope and cloned into the pBOSS vector.
Intein‐mediated trans‐splicing
The following intein sequences of NpuDnaE encoded by humanized codons were synthesized. The ‘/’ indicates the cleavage site, forming a junction sequence GSCFNGT after intein trans‐splicing.
IntN:
GS/CLSYETEILTVEYGLLPIGKIVEKRIECTVYSVDNNGNIYTQPVAQWHDRGEQEVFEYCLEDGSLIRATKDHKFMTVDGQMLPIDEIFERELDLMRVDNLPN
IntC:
IKIATRKYLGKQNVYDIGVERDHNFALKNGFIASN/CFNGT
The IntN was fused immediately after the C‐terminus of GluA2. The IntC was fused immediately before the ORF without the first methionine of CNIH3 and stargazin. Necessary mutations were introduced into the intein fusion constructs by QuikChange and DNA sequences were verified.
Co‐immunoprecipitation experiments
All transfections in HEK cells in the following were conducted using the calcium phosphate method.
Co‐immunoprecipitation in n‐dodecyl‐β‐d‐maltoside
The mouse cDNA of auxiliary subunits fused with 1D4 tag at the very C‐terminus were cloned into pTREtVb vector. Stable HEK TetON cells (Clontech, Mountain View, CA, USA) that doxycycline (DOX)‐dependently express either CNIH3–1D4 or Stg–1D4 were generated and maintained in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics (Pen/Strep, 150 μg ml−1 G418, and 120 μg ml−1 hygromycin). Each cell line was grown to 40–50% confluence, then transfected with pTREtVa‐GluA2flip‐FLAG (wild‐type or TMD mutants). Five hours after transfection cells were rinsed twice with Dulbecco's Phosphate Buffered Saline (D‐PBS), and fresh growth medium containing 7.5 μg ml−1 DOX and 30 μm 2,3‐dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo[f]quinoxaline‐2,3‐dione (NBQX) was added. Twenty‐four hours after induction, cells were washed with D‐PBS and cell pellets were flash‐frozen and kept at −80°C until use. The next day cells were resuspended in 500 μl of lysis buffer (50 mm Na‐HEPES pH 7.4, 85 mm NaCl, 15 mm KCl, 1 mm phenylmethylsulfonyl fluoride (PMSF), 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, 1 μg ml−1 pepstatin A, and 500 μm benzamidine) and 0.25% n‐dodecyl‐β‐d‐maltoside (DDM; Sol grade; Antrace, Maumee, OH, USA). Cells were incubated in lysis buffer for 3 h at 4°C and lysates were ultracentrifuged at 75,000 g (TLA‐55; Beckman Coulter, Brea, CA, USA) for 15 min at 4°C. For immunoprecipitation 420 μl of supernatant were incubated with 20 μl of FLAG‐beads (Sigma‐Aldrich, St Louis, MO, USA) overnight at 4°C. Beads were washed 3 times with 500 μl of lysis buffer containing 0.1% DDM and proteins were eluted off the beads by incubation with 60 μl of 0.3 μg ml−1 FLAG(M2) peptide for 20 min on ice. The co‐immunoprecipitation (co‐IP) samples were analysed by Western blotting. Anit‐GluA2‐CT (Shanks et al. 2010) and anti‐Rho1D4 (Wellspring Worldwide, Chicago, IL, USA) monoclonal antibodies were used to detect GluA2 and auxiliary subunits, respectively.
Co‐IP in n‐decyl‐β‐d‐maltoside
Same procedure as above but with 0.6% n‐decyl‐β‐d‐maltoside (DM).
Co‐IP in digitonin
Cells were resuspended in 500 μl of lysis buffer containing 20 mm Tris, pH 8.0, 150 mm NaCl, protease inhibitors (1 mm PMSF, 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, 1 μg ml−1 pepstatin A and 500 μm benzamidine) and 0.5% digitonin (Millipore, Billerica, MA, USA). Lysates were incubated on a rotator for 3 h at 4°C, and then centrifuged at 12,000 g for 3 min at 4°C. The supernatant was ultracentrifuged at 75,000 g for 15 min at 4°C. Supernatants (420 μl) were incubated with 20 μl of FLAG M2‐beads (Sigma‐Aldrich) equilibrated with wash buffer (20 mm Tris, pH 8.0, 150 mm NaCl, protease inhibitors, 0.25% digitonin). The antibody‐conjugated beads and lysates were incubated for 2 h at 4°C with gentle rotation. Beads were washed 3 times with 500 μl of wash buffer and eluted using wash buffer containing epitope competing FLAG(M2) peptide at 0.3 μg ml−1.
Co‐IP of CNIH1–3 with GluA2 wild‐type and TMD mutants
pTREtVa‐GluA2flip‐FLAG (wild‐type, C528L, L789F and A793F mutants) and pBOSS‐CNIH1, 2, 3‐HA were co‐transfected into TetON HEK cells. Five hours after transfection, DOX was added at 7.5 μg ml−1 for 24 h prior to harvesting for co‐IP using anti‐FLAG(M2) antibody‐conjugated beads using procedures described above.
Analysis of co‐IP efficiency
Analysis was performed in NIH ImageJ by inverting colours of film images and analysing the plot profile. The area for each band was recorded. For analysis of co‐IP, the elution band of the co‐immunoprecipitated protein (CNIH3 or Stg) was normalized to the input band to account for any potential differences in expression level. This value was then normalized to the elution band of the immunoprecipitated GluA2 protein to account for any differences in immunoprecipitation efficiency. The values for different constructs were then normalized to the co‐immunoprecipitation with GluA2 wild‐type (WT) set at 1, which would be the normalized co‐IP efficiency. The statistical significance of normalized co‐IP efficiency was evaluated using one‐way ANOVA with Dunnett's post hoc multiple comparison test. Due to the limited number of samples loadable on a single gel, in the data shown in Figs 2 and 5, the experiments were divided into four to five groups. Experiments were repeated 3–6 times in each group. Each gel contained its own positive control, the wild‐type GluA2iR or GluA2iQ, so as not to re‐use the positive controls across groups during the statistical analyses.
Figure 2. Binding efficiency of CNIH3 to GluA2 varies with point mutations to the GluA2 TMD.
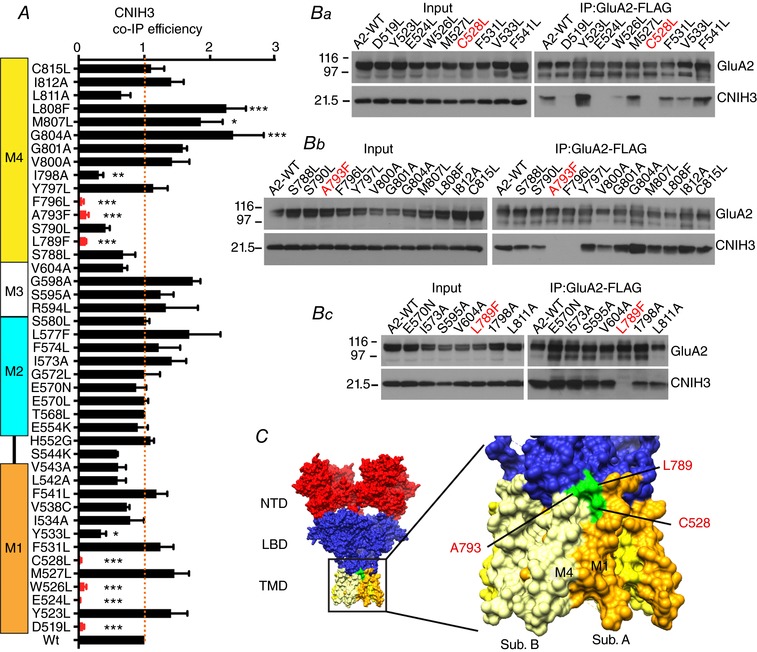
A, bar graph summarizing the co‐IP efficiency of CNIH3 upon IP of GluA2iR WT and mutants. Efficiency is calculated by dividing co‐IP signal by input signal by IP signal and normalized to the WT efficiency. M1–4 indicate the locations of TMD helices. Statistical significance against WT was determined by one‐way ANOVA with post hoc Dunnett's multiple comparison test (* P < 0.05; ** P < 0.01; *** P < 0.001; n = 3–6; mean ± SEM). Ba–c, representative western blots of input and immunoprecipitate (IP). A stable HEK cell line expressing CNIH3–1D4 was transfected with plasmids expressing GluA2iR–FLAG WT and mutants. Anti‐FLAG M2 antibody was used for IP. Western blots for GluA2 and CNIH3 were probed with anti‐GluA2CT (αGluA2CT) and anti‐Rho1D4 (αRho1D4), respectively. Molecular mass markers are on the left (kDa). C, surface representation of the GluA2 structure (PDB: 4U4G). Locations of the residues that are critical for CNIH3 interaction and studied in detail are in green. The M4 of subunit B and M1 of subunit A are shown in light yellow and orange, respectively.
Figure 5. Binding efficiency of Stg to GluA2 TMD point mutants.
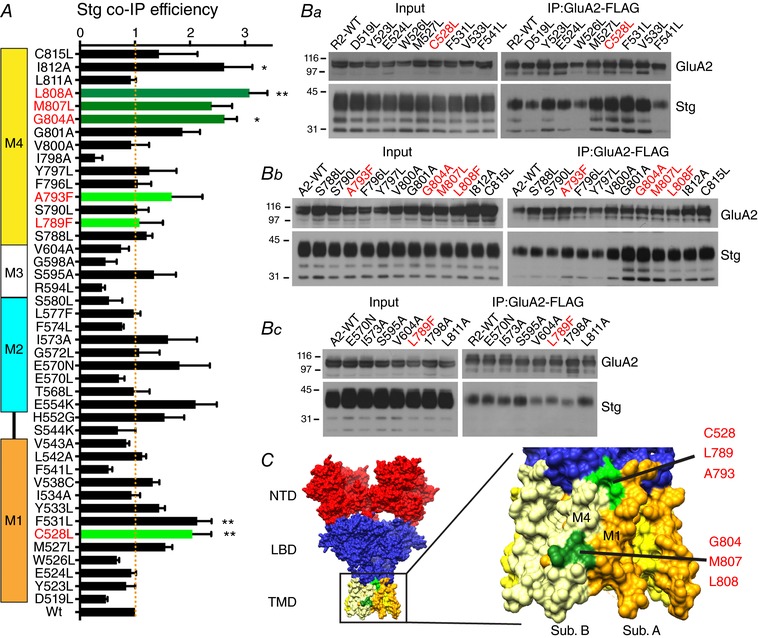
A, co‐IP efficiency of Stg upon IP of GluA2iR WT and mutants. Efficiency is calculated by dividing co‐IP signal by input signal by IP signal and normalized to the WT efficiency. M1–4 indicate the locations of TMD helices. Statistical significance was determined by one‐way ANOVA with post hoc Dunnett's comparison test against the GluA2wt (* P < 0.05; ** P < 0.01; n = 3; mean ± SEM). B, representative western blots of input and immunoprecipitate (IP). Stable HEK cell line expressing Stg–1D4 was transfected with plasmids expressing GluA2iR–FLAG WT and mutants. Anti‐FLAG M2 antibody was used for IP. Western blots for GluA2 and Stg were probed with anti‐GluA2CT (αGluA2CT) and anti‐Rho1D4 (αRho1D4), respectively. Molecular mass markers are on the left (kDa). The mutants studied by electrophysiology are in red. C, locations of residues G804, M807 and L808 are at the surface (green) of the TMD in the crystal structure PDB: 4U4G. The C528, L789 and A793 are shown in light green.
Generation of DualTetON expression vectors
DualTetON expression vectors were generated using a previously reported approach (Farina et al. 2011). In brief, the cDNAs of epitope (or intein) tagged GluA2 and auxiliary subunit were cloned into pTREtVa and pTREtVb, respectively. The AscI and PacI site, if they existed within the open reading frame, were destroyed in each cDNA by site directed mutagenesis in order to facilitate the next step of combining two plasmids. Three‐body ligation of (1) PacI, AscI, CIP treated pTREtVa‐GluA2, (2) PacI and AscI digested pTREtVb‐auxiliary subunit, and (3) stuffer fragment with AscI sites at both ends were carried out. For electrophysiological recordings, GluA2 and auxiliary subunit (CNIH3 and Stg) were co‐expressed using the DualTetON vector.
Electrophysiology
The TetON HEK cells were plated on a coverglass coated with poly‐d‐lysine (37.5 μg ml−1 in H2O) for 15 min. Excess coating material was removed by washing in D‐PBS three times. Cells were plated and incubated on a coverglass until they were adherent (typically within 12 h). Transfection was done 12–18 h after plating using the calcium phosphate method. DualTetON plasmid that co‐expresses variants of both GluA2 and auxiliary subunit (either CNIH3 or Stg) was co‐transfected with pTREt‐mVenus that expresses mVenus at a ratio of 15:1, in order to ensure that the majority of mVenus‐positive cells also express the GluA2 and CNIH3. Immediately after transfection, 30 μm NBQX and 5 μg ml−1 DOX was added. Cells were used for recording 24–36 h after induction. Ligand (1 mm glutamate) was applied via theta tubing glass capillary mounted on a piezo actuator (P‐830.30; Physik Instrumente, Karlsruhe, Germany) controlled by an LVPZT amplifier (E‐505, Physik Instrumente), DAQ device (NI USB‐6221; National Instruments, Austin, TX, USA), and custom written programs in LabView software (National Instruments). Recording was done using a single channel of a Multiclamp700B amplifier (Molecular Devices, Sunnyvale, CA, USA) operated by pCLAMP10 software. Signals were low pass filtered at 2 kHz and digitized using Digidata1440A (Molecular Devices) at a sampling rate of 50 kHz. Borosilicate glass capillaries (o.d. 1.5 mm, i.d. 0.86 mm; Sutter Instrument Co., Novato, CA, USA) were pulled to manufacture electrodes with pipette resistances of 4–7 MΩ. Internal solution was (in mm) 110 NaCl, 10 NaF, 5 EGTA, 0.5 CaCl2, 1 MgCl2, 10 Na2ATP, 5 HEPES, adjusted to pH 7.3 with CsOH and 295 mosmol l−1. External solution was (in mm): 145 NaCl, 2.5 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 10 glucose, adjusted to pH7.3 with NaOH and 301 mosmol l−1. Standard solution without ligand was the external solution. The ligand solution contained 1 mm glutamate in the external solution, supplemented with 2 mm glucose and 3 mm NaCl to facilitate visualizing the interface of the two solutions and recording liquid junction potential after breaking the patch. The open tip response was recorded after each experiment. Only those recording obtained from solution exchange rate with 20–80% rise time smaller than 300 μs were used. Decay of AMPAR‐mediated current was fit to double exponential decay (using IgorPro; Wavemetrics, Lake Oswego, OR, USA). The weighted decay time constant (τw,des) was defined as previously reported (Soto et al. 2014). The 200 ms window after the peak was used to calculate τw,des. The steady state/peak amplitude ratio was defined by the amplitude at 200 ms after the peak divided by the peak amplitude. The statistical significance of each data set in Table 2 was evaluated in software Prism (GraphPad Software, La Jolla, CA, USA) using one‐ or two‐way ANOVA with Dunnett's or Tukey's post hoc test.
Table 2.
Summary of electrophysiological recordings
| GluA2iQ constructs | Auxiliary subunit constructs | n | Holding potential (mV) | τ (weighted) (ms) | SS/P | ||
|---|---|---|---|---|---|---|---|
| Data set 1 (dual expression without using intein) | |||||||
| GluA2iQ | 10 | −70 | 5.73 ± 0.150 | 0.010 ± 0.0031 | |||
| GluA2iQ | CNIH3 | 6 | −70 | 35.9 ± 2.69 | 0.13 ± 0.021 | ||
| GluA2iQ‐C528L | 7 | −70 | 4.67 ± 0.173 | 0.062 ± 0.049 | |||
| GluA2iQ‐C528L | CNIH3 | 7 | −70 | 11.1 ± 1.15 | 0.037 ± 0.0070 | ||
| GluA2iQ‐L789F | 6 | −70 | 4.70 ± 0.224 | 0.013 ± 0.0030 | |||
| GluA2iQ‐L789F | CNIH3 | 6 | −70 | 4.43 ± 0.211 | 0.025 ± 0.0053 | ||
| GluA2iQ‐A793F | 6 | −70 | 9.13 ± 1.10 | 0.030 ± 0.0066 | |||
| GluA2iQ‐A793F | CNIH3 | 8 | −70 | 34.1 ± 5.10 | 0.44 ± 0.025 | ||
| GluA2iQ‐A793L | 7 | −70 | 5.17 ± 0.403 | 0.036 ± 0.015 | |||
| GluA2iQ‐A793L | CNIH3 | 5 | −70 | 5.05 ± 0.832 | 0.060 ± 0.017 | ||
| GluA2iQ‐A793G | 7 | −70 | 9.00 ± 1.42 | 0.10 ± 0.025 | |||
| GluA2iQ‐A793G | CNIH3 | 5 | −70 | 8.92 ± 0.711 | 0.066 ± 0.019 | ||
| GluA2iQ‐C528L‐A793F | 8 | −70 | 6.23 ± 0.444 | 0.069 ± 0.051 | |||
| GluA2iQ‐C528L‐A793F | CNIH3 | 5 | −70 | 15.2 ± 1.94 | 0.12 ± 0.028 | ||
| Data set 2 (intein trans spliced) | |||||||
| GluA2iQ | 11 | −70 | 4.87 ± 0.312 | 0.012 ± 0.0017 | |||
| GluA2iQ | Stg | 10 | −70 | 14.5 ± 0.707 | 0.20 ± 0.018 | ||
| GluA2iQ(DDD) | Stg | 15 | −70 | 12.5 ± 0.806 | 0.11 ± 0.011 | ||
| GluA2iQ(AGA) | Stg | 11 | −70 | 12.0 ± 0.934 | 0.17 ± 0.029 | ||
| GluA2iQ(EGE) | Stg | 5 | −70 | 8.85 ± 0.661 | 0.10 ± 0.0095 | ||
| Data set 3 (intein trans spliced) | |||||||
| GluA2iQ | 8 | −60 | 5.78 ± 0.520 | 0.014 ± 0.0028 | |||
| GluA2iQ | Stg | 10 | −60 | 15.0 ± 1.18 | 0.19 ± 0.020 | ||
| GluA2iQ‐C528L | 7 | −60 | 5.87 ± 0.219 | 0.056 ± 0.0081 | |||
| GluA2iQ‐C528L | Stg | 10 | −60 | 9.20 ± 0.973 | 0.083 ± 0.016 | ||
| GluA2iQ‐L789F | 11 | −60 | 4.25 ± 0.248 | 0.034 ± 0.0060 | |||
| GluA2iQ‐L789F | Stg | 8 | −60 | 6.71 ± 0.356 | 0.022 ± 0.0019 | ||
| GluA2iQ‐A793F | 13 | −60 | 7.62 ± 0.427 | 0.053 ± 0.011 | |||
| GluA2iQ‐A793F | Stg | 12 | −60 | 10.3 ± 0.834 | 0.084 ± 0.015 | ||
| GluA2iQ‐G804A | 7 | −60 | 5.15 ± 0.254 | 0.040 ± 0.010 | |||
| GluA2iQ‐G804A | Stg | 16 | −60 | 16.4 ± 1.29 | 0.26 ± 0.026 | ||
| GluA2iQ‐M807L | 6 | −60 | 6.05 ± 0.837 | 0.057 ± 0.027 | |||
| GluA2iQ‐M807L | Stg | 10 | −60 | 12.5 ± 1.03 | 0.14 ± 0.019 | ||
| GluA2iQ‐L808F | 8 | −60 | 5.64 ± 0.204 | 0.019 ± 0.0025 | |||
| GluA2iQ‐L808F | Stg | 8 | −60 | 12.3 ± 1.68 | 0.20 ± 0.031 | ||
| Data set 4 (intein trans spliced) | |||||||
| GluA2iQ | Stg | 10 | −60 | 12.9 ± 1.08 | 0.17 ± 0.025 | ||
| GluA2iQ | Stg‐KK52EE | 7 | −60 | 28.6 ± 5.31 | 0.45 ± 0.040 | ||
| GluA2iQ | Stg‐KK53Enull | 7 | −60 | 23.6 ± 2.88 | 0.48 ± 0.036 | ||
| GluA2iQ | Stg‐K53null | 9 | −60 | 14.9 ± 1.14 | 0.21 ± 0.022 | ||
| GluA2iQ | Stg‐KK52AA | 6 | −60 | 15.9 ± 1.50 | 0.31 ± 0.027 | ||
| GluA2iQ | Stg‐E70K | 5 | −60 | 14.4 ± 1.24 | 0.11 ± 0.015 | ||
| GluA2iQ | Stg‐ED84RK | 7 | −60 | 15.5 ± 2.52 | 0.17 ± 0.036 | ||
| GluA2iQ | Stg‐DAD86KAK | 6 | −60 | 15.7 ± 0.581 | 0.22 ± 0.022 | ||
| GluA2iQ | Stg‐EAD90KAK | 6 | −60 | 22.4 ± 3.04 | 0.47 ± 0.077 | ||
| Data set 5 (intein trans spliced) | |||||||
| GluA2iQ | Stg | 20 | −60 | 13.3 ± 0.572 | 0.20 ± 0.014 | ||
| GluA2iQ | Stg‐KK52EE | 19 | −60 | 21.1 ± 1.42 | 0.43 ± 0.013 | ||
| GluA2iQ | Stg‐KK53Enull | 12 | −60 | 20.6 ± 1.75 | 0.42 ± 0.018 | ||
| GluA2iQ‐C528L | Stg | 16 | −60 | 8.82 ± 0.747 | 0.11 ± 0.020 | ||
| GluA2iQ‐C528L | Stg‐KK52EE | 15 | −60 | 13.0 ± 1.32 | 0.23 ± 0.025 | ||
| GluA2iQ‐C528L | Stg‐KK53Enull | 10 | −60 | 15.5 ± 1.90 | 0.29 ± 0.042 | ||
| one‐way ANOVA with Dunnett's post hoc (vs. GluA2iQ+Stg) | |||||||
| Data set 6 (dual expression without using intein) | τ (weighted) | SS/P | |||||
| GluA2iQ | Stg | 9 | −60 | 10.4 ± 0.531 | 0.11 ± 0.010 | ||
| GluA2iQ‐C528L | Stg | 10 | −60 | 6.90 ± 0.409 | 0.059 ± 0.41 | 0.0049 (**) | 0.0571 (n.s.) |
| GluA2iQ‐L789F | Stg | 10 | −60 | 4.72 ± 0.388 | 0.027 ± 0.0038 | 0.0001 (****) | 0.0004 (***) |
| GluA2iQ‐A793F | Stg | 10 | −60 | 6.25 ± 0.526 | 0.099 ± 0.020 | 0.0006 (***) | 0.978 (n.s) |
| GluA2iQ‐G804A | Stg | 8 | −60 | 12.5 ± 0.813 | 0.16 ± 0.016 | 0.24 (n.s.) | 0.2144 (n.s.) |
| GluA2iQ‐M807L | Stg | 10 | −60 | 12.8 ± 1.26 | 0.15 ± 0.021 | 0.111 (n.s.) | 0.35 (n.s.) |
| GluA2iQ‐L808F | Stg | 9 | −60 | 10.7 ± 0.660 | 0.14 ± 0.013 | 0.9997 (n.s.) | 0.7755 (n.s.) |
| Data set 7 (dual expression without using intein) | |||||||
| GluA2iQ | Stg | 9 | −60 | 11.5 ± 0.579 | 0.14 ± 0.015 | ||
| GluA2iQ | Stg‐KK52EE | 8 | −60 | 21.1 ± 1.65 | 0.40 ± 0.022 | 0.0002 (***) | 0.0001 (****) |
| GluA2iQ | Stg‐KK53Enull | 6 | −60 | 15.6 ± 1.02 | 0.34 ± 0.015 | 0.2079 (n.s.) | 0.0001 (****) |
| GluA2iQ | Stg‐K53null | 7 | −60 | 15.3 ± 1.96 | 0.24 ± 0.035 | 0.2164 (n.s.) | 0.0223 (*) |
| GluA2iQ | Stg‐KK52AA | 8 | −60 | 15.1 ± 2.03 | 0.22 ± 0.030 | 0.2713 (n.s.) | 0.0802 (n.s.) |
The experiment data were categorized in seven data sets. Small differences in gating parameters of the same construct (for example GluA2iQ alone) are likely due to uncontrollable small variations in experimental conditions. A holding potential of −60 mV was preferred for experiments using stargazin because the current amplitude was overall greater. Mean ± SEM are shown. Numbers of experimental replicates (outside‐out patches) are indicated by n. No data was re‐used between data sets. Statistical significance was examined within each data set using one‐ or two‐way ANOVA with appropriate post hoc tests for multiple comparisons as descried in the text.
Immunofluorescence microscopy
Cells were prepared on a poly‐d‐lysine coated coverglass, transfected, and induced with DOX as described above. Cells were fixed with warm fixative (4% formaldehyde in 0.1m D‐PBS (pH 7.5), at 37°C) for 8 min at 24–36 h post‐induction. After three D‐PBS washes, the total Stg–1D4 and GluA2 were labelled with anti‐Rho1D4 antibody and anti‐GluA2‐CT, respectively, diluted in 1× GDB (0.2% gelatin, 0.6% Triton X‐100, 33 mm phosphate buffer, pH 7.4, and 0.9 m NaCl). Anti‐rabbit IgG antibody conjugated with Alexa 488 and anti‐mouse IgG conjugated with Alexa 568 were used as the secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA). Cells were imaged using a fluorescence microscope (Olympus) with a ×40 objective lens. Images were recorded on a CCD camera (Hamamatsu Orca).
Results
Mutating residues in the transmembrane helices of GluA2 that are not conserved in kainate receptors
CNIH3 and Stg interact with AMPARs but not with kainate receptors (KARs) (Shanks et al. 2010, 2014). We therefore reasoned that critical residues for complex formation and functional modulation are unique to AMPARs. Using the X‐ray crystal structures (Sobolevsky et al. 2009; Yelshanskaya et al. 2014; PDB: 3KG2 and 4U4G) as guides we identified residues that are exposed to the membrane in the TMD (M1–4) of GluA2 and are not conserved in GluK2. Each residue was mutated to another hydrophobic amino acid or to the complementary KAR residue to prevent gross alteration of topology. Bulky aromatic residues, such as W, F and Y, at the surface were mutated whether or not they were conserved in KARs. Residues in M4 critical for tetrameric assembly were excluded (Salussolia et al. 2013). This identified a total of 42 residues (14 in M1, 2 in M1–2 linker, 11 in M2–3, 15 in M4; Fig. 1, Table 1) to screen for their contribution to GluA2–auxiliary subunit complex stability in detergent. All TMD mutants of GluA2 expressed at similar levels when expressed alone in HEK cells.
Figure 1. Selection of TMD mutants of GluA2.
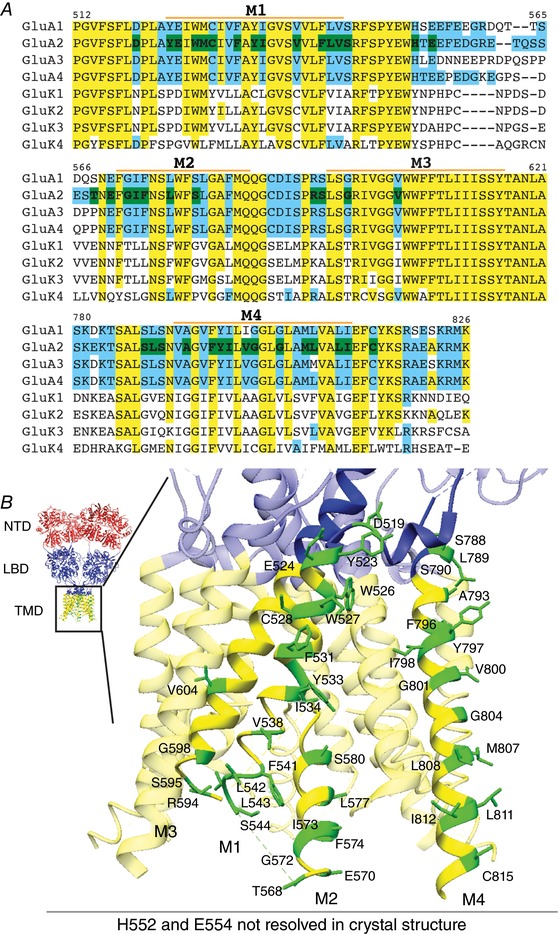
A, CLUSTALW alignment of M1–M4 of AMPARs and KARs. Yellow: conserved residues between GluA2 and GluK2; blue: residues not conserved between GluA2 and GluK2; green: residues of GluA2 mutated in this study. Residues different between GluA2 and GluK2 in the TMD regions, as well as bulky aromatic residues in the TMD were mutated. M1–4 labels indicate the extent of each transmembrane helix. B, GluA2 TMD residues that were studied in this report are indicated on the crystal structure (PDB: 4U4G). The domains are coloured red for the N‐terminal domain (NTD), blue for the ligand‐binding domain (LBD) and LBD–TMD linkers, and yellow for the transmembrane domain (TMD). All mutated residues are coloured green and labelled in the zoomed view of one GluA2 subunit. H522 and E554 are unresolved in the crystal structure and not shown, but occur on the intracellular linker between M1 and M2.
Table 1.
Summary of TMD mutants
| GluA2 mutants | GluK2 mutants | ||
|---|---|---|---|
| M1 | M1–2 linker | M4 | M1 |
| D519L | H552G | S788L | Y566C* |
| Y523L | E554K | L789F# | |
| E524L | S790L | M4 | |
| E524D | M2 | A793F∧ | V817L# |
| W526L | T568L | A793G∧ | G821A∧ |
| M527L | E570L | A793L∧ | |
| C528L* | E570N | F796L | |
| C528F* | G572A | Y797L | |
| C528Y* | I573A | I798A | |
| F531L | F574L | V800A | |
| F531A | L577F | G801A | |
| Y533L | S580L | G804A | |
| I534L | M807L | ||
| I534A | L808F | ||
| V538C | M3 | L811A | |
| F541L | R594L | I812A | |
| L542A | S595A | C815L | |
| V543A | G598A | ||
| S544K | V604A |
First three columns show the mutations to GluA2 in the format of original residue followed by residue number followed by the mutated residue. The fourth column shows the mutations to GluK2 residues that align to significantly affected residues in GluA2. The highlighted residues are complementary between AMPAR and KAR.
Mutation in TMD residues affects GluA2–CNIH3 stability
We rationalize the use of dodecyl maltoside (DDM) detergent in the biochemical studies of the GluA2–CNIH3 complex based on the observations that (1) co‐IP assay in 0.1∼0.3% DDM was able to detect robust interaction between GluA2 and CNIH3, and (2) the GluA2–CNIH3 complex was stable enough in DDM for the complex to be detected using negative stain single particle analysis (Shanks et al. 2014). Although we cannot equate the stability of the complexes in detergent to that in the membrane, the differences in the co‐IP efficiency between mutant and wild‐type complexes in detergent suggest that the chemical properties of the interaction have been changed by the mutation. Seven residues (D519L, E524L, W526L, C528L, L789F, A793F and F796L, red in Fig. 2 A) reduced the CNIH3 co‐IP efficiency below 20% of wild‐type, as determined by one‐way ANOVA with Dunnett's post hoc test. Four of these (D519L, E524L, W526L and F796L) had negligible or no recordable ion channel activity, and thus were removed from further analysis. The remaining three residues, C528, L789 and A793, form a patch of surface spanning M1 and M4 helices of adjacent subunits located near the extracellular side of the membrane in the X‐ray structure of GluA2 (Fig. 2 C, green). These residues were part of the protein interaction interface in the detergent micelle embedded complex formed between GluA2 and Stg (Twomey et al. 2016, 2017; Zhao et al. 2016b). Members of the CNIH family use a shared mechanism for complex formation, as these mutations in M1 (C528L) and M4 (L789F and A793F) also destabilized interactions with CNIH1 and 2 (Fig. 3 A). Two residues (G804L and L808F) in M4 significantly stabilized the GluA2–CNIH3 complex, as judged by more than 2‐fold increase in co‐IP efficiency (Fig. 2 A). These results suggest that the cytoplasmic side of M4, where G804 and L808 are located, also contribute to complex stability in detergent.
Figure 3. GluA2 TMD residues in M1 (C528) and M4 (L789 and A793) are critical for complex formation with CNIH1–3.
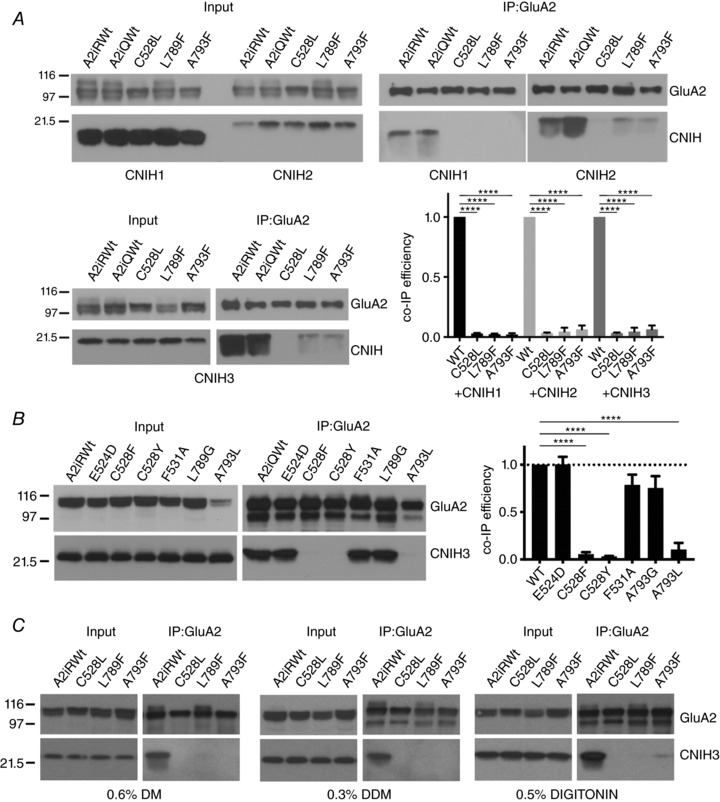
A, representative western blots of co‐IP experiments. HEK cell lysates containing GluA2 mutants and CNIH1–3 (Input) were subject to IP using the FLAG epitope tag in GluA2. Co‐IPs of CNIH1–3 are shown. Wild‐type GluA2iQ and GluA2iR were used as positive controls. Mutants are derived from GluA2iR. A bar graph summary of quantification is shown bottom right. Statistical significance was determined by one‐way ANOVA with post hoc Dunnett's multiple comparison test against the GluA2iR WT (**** P < 0.0001; n = 3; mean ± SEM). B, representative western blots of co‐IP experiments. HEK cell lysates containing GluA2iQ mutants and CNIH3 (Input) were subject to IP using the FLAG epitope tag in GluA2. Co‐IPs of CNIH3 are shown. GluA2iQ WT was used as positive control. A bar graph summary of quantification is on the right. Statistical significance was determined by one‐way ANOVA with post hoc Dunnett's multiple comparison test against the GluA2iQ WT (**** P < 0.0001; n = 3; mean ± SEM). C, western blot of a representative co‐IP of CNIH3 when GluA2iR variants were immunoprecipitated after dissolving the identical initial materials in three different detergents of varying strength (DM, DDM and digitonin). Higher level of co‐IP of A793F was observed in 0.5% digitonin relative to DM and DDM.
We examined if C528, L789 and A793 are sufficient to induce binding with CNIH3. The C528, L789 and A793 are unique in AMPARs and correspond to residues Y566, V817 and G821, respectively, in GluK2 (see Fig. 1 A, Table 1). When converted to the amino acids of GluA2, the resulting GluK2 mutants did not acquire the ability to interact with CNIH3. Specifically, GluK2 mutants that have single amino acid substitution, Y566C and G821A, as well as double and triple substitutions, Y566C/V817L, V817L/G821A, Y566C/G821A and Y566C/V817L/G821A, all failed to convert GluK2 into a CNIH3‐interacting subunit (data not shown). We interpreted this to indicate that the sufficiency condition was not satisfied because other GluA2‐specific residues could contribute to the interaction at a subcritical level.
The effects on AMPAR gating of mutant GluA2–CNIH3 complexes with reduced stability in detergent
CNIH3 slows AMPAR desensitization (Schwenk et al. 2009). Using this property as a readout, we examined the effects of the aforementioned mutations on AMPAR gating modulation by recording glutamate‐evoked currents from outside‐out patches pulled from TetON HEK cells co‐expressing the GluA2iQ mutants with CNIH3 using a fast ligand application. To ensure efficient co‐expression of GluA2 and CNIH3 in each transfected cell and to eliminate variability in expression levels of two proteins, both proteins were co‐expressed by using a DOX‐inducible expression system from a single plasmid. A submaximal dose of DOX (5 μg ml−1) was used to induce expression while preventing cytotoxicity caused by enhanced gating activity due to CNIH3. The transfected cells were identified by fluorescence signals of mVenus derived from another plasmid that was co‐transfected at 1/15 of the quantity of the total transfected DNA.
The mutations C528L in M1, and L789F in M4, significantly reduced the 1 mm glutamate‐evoked gating modulation by CNIH3 (Fig. 4). The rate of desensitization was measured using two parameters, the weighted exponential decay time constant (τw,des) derived from fitting the data to a bi‐exponential decay model and the steady state to peak (SS/P) current amplitude ratio. In the absence of CNIH3, both C528L and L789F had no effect on desensitization compared to wild‐type (GluA2iQ‐C528L: τw,des = 4.67 ± 0.173 ms, P > 0.99, SS/P = 0.062 ± 0.049, P > 0.99, n = 7; GluA2iQ‐L789F: τw,des = 4.70 ± 0.224 ms, P > 0.99, SS/P = 0.013 ± 0.0030, P > 0.99, n = 6; GluA2iQwt: τw,des = 5.73 ± 0.150 ms, SS/P = 0.010 ± 0.0031, n = 10, holding potential −70 mV; the entire data set 1 in Table 2 was analysed using two‐way ANOVA with Tukey's post hoc test). In the presence of CNIH3, desensitization measured by τw,des was accelerated in C528L and L789F compared to the wild‐type (Fig. 4 A–C and Ga and b), consistent with the observed decline in complex stability (GluA2iQ‐C528L+CNIH3: τw,des = 11.1 ± 1.15 ms, P < 0.0001, SS/P = 0.037 ± 0.0070, P = 0.51, n = 7; GluA2iQ‐L789F+CNIH3: τw,des = 4.43 ± 0.211 ms, P < 0.0001, SS/P = 0.025 ± 0.0053, P = 0.36, n = 6; GluA2wt+CNIH3: τw,des = 35.9 ± 2.69 ms, SS/P = 0.13 ± 0.021, n = 6, holding potential −70 mV; Table 2). In particular, the L789F mutation was insensitive to CNIH3.
Figure 4. Effect of GluA2 mutants on gating modulation by CNIH3.
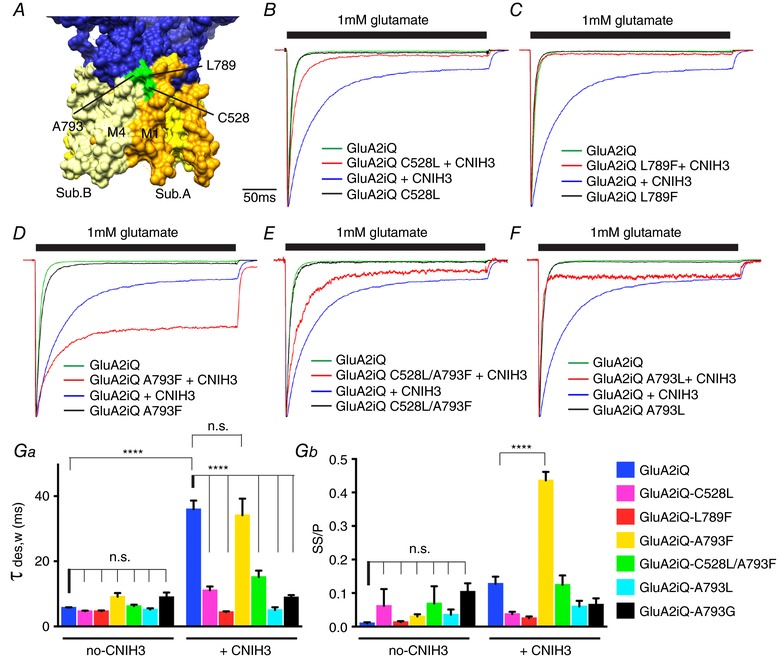
A, the locations of C528 (M1), L789 (M4) and A793 (M4) are indicated. B–F, representative recordings obtained from outside‐out patches in response to 1 mm glutamate application for 300 ms. The traces were normalized to the peak amplitudes to facilitate comparison of the gating kinetics. The constructs transfected into TetON HEK cells are indicated. Ga and b, summary of τw,des and steady state/peak (SS/P) amplitude ratios. Statistical significance against GluA2iQ and GluA2iQ+CNIH3, respectively, was determined by two‐way ANOVA with post hoc Tukey's multiple comparison test (**** P < 0.001; mean ± SEM). Also see, data set 1 in Table 2.
In contrast, the A793F mutation promoted slowing of desensitization by CNIH3 (Fig. 4 D, Ga, and b). Without CNIH3, the GluA2iQ‐A793F and GluA2iQwt had similar gating kinetics (GluA2iQwt: τw,des = 5.73 ± 0.150 ms, SS/P = 0.010 ± 0.0031, n = 10; GluA2iQ‐A793F: τw,des = 9.13 ± 1.10 ms, P > 0.99, SS/P = 0.030 ± 0.0066, P > 0.99, n = 6). A prominent difference in gating was revealed when CNIH3 was present (GluA2wt+CNIH3: τw,des = 35.93 ± 2.69 ms, SS/P = 0.13 ± 0.021, n = 6; GluA2iQ‐A793F+CNIH3: τw,des = 34.1 ± 5.10 ms, P > 0.99, SS/P = 0.44 ± 0.025, P < 0.001, n = 8). Specifically, the SS/P ratio but not τw,des was significantly increased by the A793F mutation compared to wild‐type when CNIH3 was co‐expressed. Interestingly, the initial phase of desensitization was affected less compared to the later phase, which contributes to establishing the steady state current. This observation may imply that the mutation increased the probability to re‐open the desensitized receptors. The enhanced slowing of desensitization caused by the combination of A793F and CNIH3 indicates that this GluA2 mutant forms a functional complex with CNIH3 in the membrane. Despite a strong gain of function of GluA2‐A793F with CNIH3, the complex was unstable in DDM detergent compared to the wild‐type assembly (Fig. 2 A and Bb). We have explored the effect of DM, DDM and digitonin on complex stability. In particular, digitonin stabilizes AMPAR–Stg complex (Zhao et al. 2016a). Neither of these detergents produced significant improvements in complex stability of C528L, L789F or A793F (Fig. 3 C).
The functional interaction between the C528L and A793F mutations was examined by recording currents from a mutant GluA2 that carried both mutations (Fig. 4 E and Ga and b). Without CNIH3, the GluA2‐C528L‐A793F double mutant desensitized at an intermediate rate between those of the individual mutations (GluA2iQ‐C528L‐A793F: τw = 6.23 ± 0.444 ms, SS/P = 0.069 ± 0.051, n = 8). In the presence of CNIH3, the rate of desensitization was also at an intermediate level between those of C528L and A793F single mutants (GluA2iQ‐C528L‐A793F+CNIH3: τw = 15.2 ± 1.94 ms, SS/P = 0.12 ± 0.028, n = 5), and thus the effects on gating introduced by the individual TMD mutations appear to cancel each other in the double mutant.
We investigated additional mutations at the functionally sensitive residues C528 and A793 (C528F and C528Y, and A793G and A793L, respectively), in order to further evaluate their effects on complex stability and gating modulation. The C528 plays a significant role in complex stability since modifying this residue to hydrophobic L, F, or Y reduced co‐IP efficiency of CNIH3 with GluA2 in both DDM (Fig. 3 B) and digitonin (data not shown). However, glutamate application produced no currents in the C528F and C528Y. Changing the A793 produced clearer results. The A793G and A793L destabilized the GluA2–CNIH3 complex in both DDM (Fig. 3 B) and digitonin (data not shown). Consistently, both mutations significantly neutralized the effect of CNIH3 by speeding desensitization (GluA2iQ‐A793G+CNIH3: τw = 8.92 ± 0.711 ms, SS/P = 0.066 ± 0.019, n = 5; GluA2iQ‐A793G: τw = 9.00 ± 1.42 ms, SS/P = 0.10 ± 0.025, n = 7; GluA2iQ‐A793L+CNIH3: τw = 5.05 ± 0.832 ms, SS/P = 0.060 ± 0.017, n = 5; GluA2iQ‐A793L: τw = 5.17 ± 0.403 ms, SS/P = 0.036 ± 0.015, n = 7) (Fig. 4 F and Ga and b). Collectively, these results identify the neighbourhood containing C528, L789 and A793 as a potential mediator for gating modulation by CNIH3 (Fig. 4 A). Importantly these residues are exposed to the surface of the TMD, accessible to CNIH3.
Effect of single amino acid substitutions in the TMD on GluA2–Stg stability
In contrast to CNIH3, Stg co‐IP with all GluA2 mutants in DDM at efficiencies greater than 40% with the exception of I798A and R594L mutations in M4 and M3, respectively (Fig. 5 A and Ba–c). Here, we rationalize the use of DDM because it has been proven to partially maintain the structural integrity of the GluA2–Stg complex (Twomey et al. 2016). We identified several mutations (C528L, F531L, G804L, L808F and I812A) that significantly increased the stability of the GluA2–Stg complex by greater than twofold, as determined by one‐way ANOVA with Dunnett's post hoc test. The residues C528 and F531 were mapped in the extracellular‐middle part of M1, whereas G804, M807, L808 and I812A were mapped near the middle cytoplasmic side on M4 helix of GluA2 (Fig. 5 C). These regions of M1 and M4 were found to form an interaction interface with Stg (Twomey et al. 2016; Zhao et al. 2016b), consistent with the idea that modification of this interface influences complex stability.
Intein‐mediated trans‐splicing produces functional GluA2–Stg fusion
We used two different strategies to study the effect of six GluA2 TMD mutants described above (C528L, L789F, A793F, and G804A, M807L and L808F) on Stg‐dependent gating modulation in HEK cells. In the first approach, we expressed each GluA2 mutant with Stg so that the two proteins would be covalently conjugated. The rationale for taking this approach is that AMPAR–Stg fusion constructs were previously exploited in studies investigating the molecular mechanisms of Stg‐dependent gating modulation of AMPARs, as well as in a cryoEM structural study of the complex (Shi et al. 2009; Shelley et al. 2012; Dawe et al. 2016; Twomey et al. 2016). Here, instead of generating a single polypeptide fusion construct, we applied intein‐mediated trans‐splicing (Iwai et al. 2006) to covalently link the two proteins after each protein is translated separately, as described below. The advantage of using an intein approach, as opposed to a single polypeptide construct expression, is that the two entities are expressed separately and fold at much higher efficiencies than the GluA2–Stg fused complex (data not shown).
The second approach we adopted, which would be more physiological, is to co‐express GluA2 and Stg as individual entities from a single plasmid using the identical configuration described above to study the effect of CNIH3 on gating.
To induce the intein trans‐splicing event, we fused the N‐ and C‐termini fragments of the human codon optimized NpuDnaE split intein to the C‐terminus of GluA2iQ and N‐terminus of Stg, respectively, generating GluA2iQ–IntN and IntC–Stg (Fig. 6 A). When the two fusion proteins were co‐expressed in HEK cells a spontaneous intein trans‐splicing reaction generated GluA2iQ–Stg and reconstituted NpuDnaE split intein, the latter resulting from fusion of N‐ and C‐terminal fragments of the split inteins. The trans‐splicing event was efficient and the majority of the GluA2iQ fused with Stg within 24–36 h of co‐expression (Fig. 6 B). When examined by immunofluorescence microscopy, strong GluA2iQ immunoreactivity was localized at the cell surface when GluA2iQ–IntN and IntC–Stg were co‐expressed (Fig. 6 C). The 1 mm glutamate‐evoked currents recorded from patches derived from HEK cells co‐expressing GluA2iQ–IntN and IntC–Stg exhibit Stg‐mediated slowing of desensitization and increase of steady state current (GluA2iQ alone: τw,des = 4.87 ± 0.312 ms, SS/P = 0.012 ± 0.0017, n = 11; GluA2iQwt–Stg: τw,des = 14.5 ± 0.707 ms, SS/P = 0.20 ± 0.018, n = 10, holding potential = −70 mV, τw,des and SS/P: P < 0.0001) (Fig. 6 D, Table 2 data set 2). The amplitudes of current measured from intein‐mediated fusion were greater than the conventional fusion construct based on a previous design (Shi et al. 2009) (data not shown). The basic residues ‘KGK’ (residues 718–720) at the D2 lobe of LBD of GluA2 subunit were proposed to play critical roles in gating modulation by interacting with the acidic residues in the extracellular loop of Stg (Fig. 6 E, left) (Twomey et al. 2016; Zhao et al. 2016b). The GluA2iQ(KGK→D) mutant reduced Stg modulation, when examined in the context of the GluA2–Stg tethered fusion construct (Dawe et al. 2016). We tested the identical mutant using the intein‐mediated tethering protocol and confirmed similar phenotypes (data not shown), validating the intein method as an alternative to the direct fusion constructs. We extended this observation and generated additional mutations in this region that convert KGK to DDD, EGE and AGA (Fig. 6 E–H, Table 2 data set 2). The results indicate that converting the residues from basic to acidic is critical for the loss‐of‐function phenotype.
Figure 6. Recording currents from GluA2 and Stg complexes tethered by intein trans‐splicing.
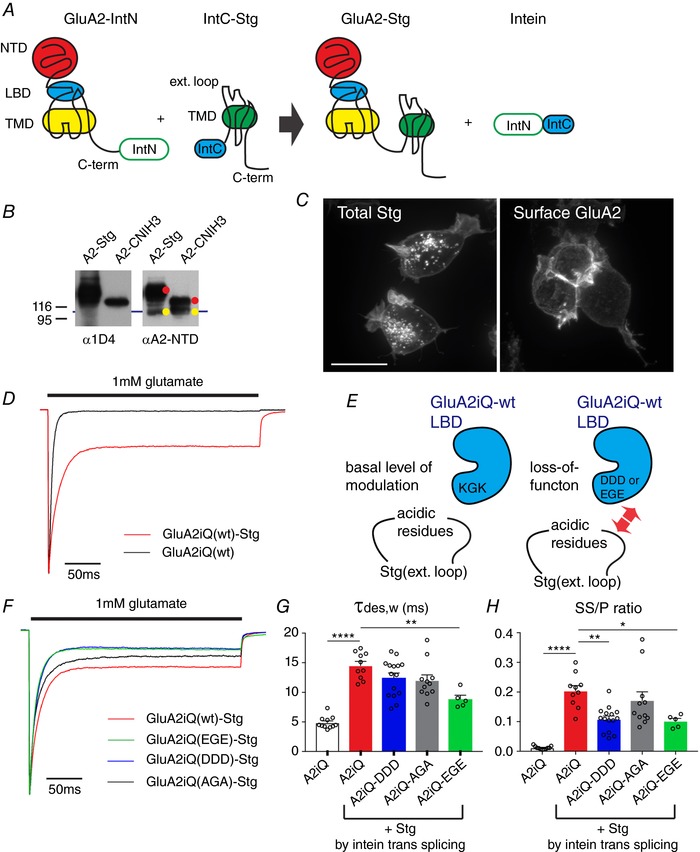
A, schematic diagram of the construct design and intein trans‐splicing reaction. B, western blot of lysates obtained from TetON HEK cells co‐expressing GluA2–IntN and IntC–Stg or IntC–CNIH3. Auxiliary subunits are tagged at the C‐terminus with 1D4 epitope. Yellow indicates unspliced GluA2–IntN and red indicates intein trans‐splicing products that are GluA2 tethered with Stg or CNIH3. C, confocal images of TetON HEK cells expressing GluA2–IntN and IntC–Stg. Left, total staining by anti‐1D4 in order to detect Stg. Right, surface labelling by anti‐GluA2‐NTD. Scale bar 10 μm. D, representative peak‐normalized glutamate‐evoked recording from patches containing GluA2iQ alone (black) or GluA2iQ–IntN+IntC–Stg (Red). E, a schematic model introducing the ‘KGK’ sequence in the D2 lobe of LBD and its role in Stg‐dependent gating modulation of AMPAR. F, representative recordings from Stg+GluA2 mutants with ‘KGK’ sequence replaced by ‘DDD’, ‘EGE’ and ‘AGA’, using intein trans‐splicing tethering scheme. Stg+GluA2iQ wild‐type (wt) is shown as a reference. Peak normalized glutamate‐evoked recording from patches are shown. G and H, summary of τw,des and SS/P amplitude ratios. Statistical significance of each pair indicated by both ends of horizontal bars was determined by one‐way ANOVA with post hoc Tukey's multiple comparison test (* P < 0.05; ** P < 0.01; **** P < 0.0001; mean ± SEM). Individual data points are shown as open circles.
The A793F mutation in M4 has opposing effect on gating modulation by CNIH3 and Stg
The A793F mutation in M4 expressed a gain‐of‐function phenotype by enhancing the slowing of desensitization induced by CNIH3. Surprisingly, the identical mutation expressed a loss‐of‐function phenotype with respect to gating modulation by Stg. The rate of desensitization was at a value between wild‐type GluA2 with and without Stg (GluA2iQ‐A793F+Stg: τw,des = 10.3 ± 0.834 ms, SS/P = 0.084 ± 0.015, n = 12; GluA2iQ‐A793F: τw,des = 7.62 ± 0.427 ms, SS/P = 0.053 ± 0.011, n = 13; GluA2iQwt+Stg: τw,des = 15.0 ± 1.18 ms, SS/P = 0.19 ± 0.020, n = 10, at holding potential −60 mV. Stg tethered by intein trans‐splicing; Fig. 7 A and D–F, left; Table 2 data set 3; two‐way ANOVA with Tukey's post hoc test). Similar results were obtained when GluA2 and Stg were co‐expressed as individual entities (Fig. 7 E and F, right; Table 2 data set 6; two‐way ANOVA with Tukey's post hoc test). Importantly, the A793F mutation weakens Stg‐mediated slowing of AMPAR desensitization while the complex remains intact (Fig. 5). Because A793F creates opposing effects on CNIH3 and stargazin, A793 would be a pivotal target for drugs that act specifically on AMPARs bound to certain auxiliary subunits. Furthermore, the opposing effects of CNIH3 and stargazin on the same mutant A793F suggests that the mechanisms recruited by each auxiliary subunit in order to slow desensitization are different.
Figure 7. Effects of key residues in M1 and M4 of GluA2 on Stg‐dependent gating modulation.
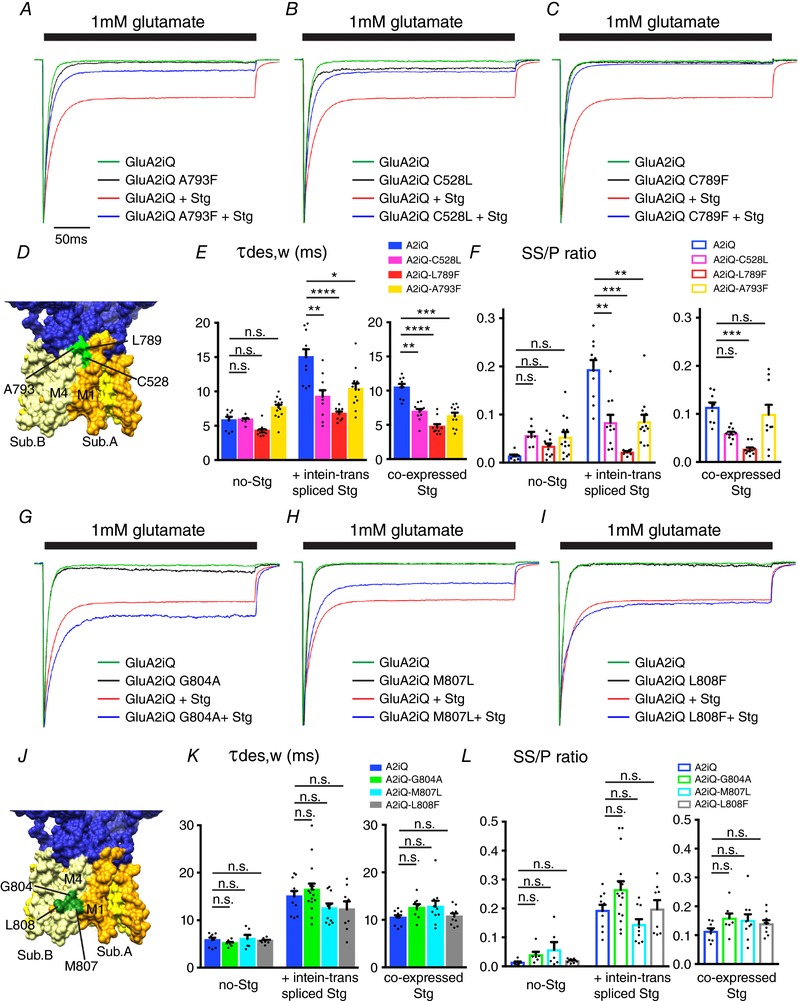
A–C and G–I, representative recordings obtained from outside‐out patches in response to 1 mm glutamate application for 300 ms. The traces were normalized to the peak amplitudes to facilitate comparison of the gating kinetics. The constructs transfected into TetON HEK cells are indicated. D and J, locations of the mutants studied in this figure (PDB: 4U4G). E, F, K and L, summary of τw,des and SS/P amplitude ratios. GluA2 variants and Stg were tethered by intein trans‐splicing or co‐expressed as non‐tethered individual entities, as indicated. Individual data points are shown as dots. Statistical significance was determined by one‐way ANOVA with post hoc Tukey's test (* P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001; mean ± SEM) from data sets 3 (inteine trans‐spliced) and 6 (co‐expressed), respectively.
The C528L and L789F mutations reduce gating modulation by both CNIH3 and Stg
The C528L mutation in M1 destabilized the GluA2–CNIH3 complex but stabilized the GluA2–Stg complex by twofold. Interestingly, the magnitude of gating modulation of AMPAR by Stg was significantly reduced when C528L mutation was present (GluA2iQ‐C528L+Stg: τw,des = 9.20 ± 0.973 ms, P = 0.0024, SS/P = 0.083 ± 0.016, P = 0.0047, n = 10; GluA2iQwt+Stg: τw,des = 15.0 ± 1.18 ms, SS/P = 0.19 ± 0.020, n = 10, at holding potential −60 mV; Stg tethered by intein trans‐splicing; Fig. 7 B, E and F, left). Similar to the case of A793F mutation, the loss of modulation by Stg is unlikely a consequence of complex dissociation. Stg remained bound to the GluA2‐C528L yet gating modulation was diminished. The L789F mutation in M4, which weakens CNIH3 interaction but not Stg, had very similar effect on gating modulation as the C528L (GluA2iQ‐L789F+Stg: τw,des = 6.71 ± 0.356 ms, P < 0.0001, SS/P = 0.022 ± 0.0019, P < 0.0001, n = 8, in comparison with GluA2iQwt+Stg; Stg tethered by intein trans‐splicing; Fig. 7 C, E and F). These effects were not influenced by whether Stg was tethered to or co‐expressed with GluA2 mutants (Fig. 7 E and F, right; Table 2 data set 6). Interestingly, the increased complex stability in DDM did not correlate with stronger gating modulation by Stg, highlighting that these mutant AMPAR–Stg complexes are structurally intact but gating modulation deficient.
The effects on gating of AMPAR–Stg complex stabilizing mutants in M4
The mutations in M4 (G804A, M807L and L808F) that increased the stability of the GluA2–Stg complex could potentially alter gating modulation by modifying a protein interaction interface. As such, the effect of these mutations on Stg‐mediated slowing of desensitization was examined (Fig. 7 G–L, Table 2 data sets 3 and 6). In the absence of Stg, all three mutants had gating kinetics similar to wild‐type GluA2 (GluA2iQ‐G804A: τw,des = 5.15 ± 0.254 ms, SS/P = 0.040 ± 0.010, n = 7, P > 0.99; GluA2iQ‐M807L: τw,des = 6.05 ± 0.837 ms, SS/P = 0.057 ± 0.027, n = 6, P > 0.99; GluA2iQ‐L808F: τw,des = 5.64 ± 0.204 ms, SS/P = 0.019 ± 0.0025, n = 8, P > 0.99; GluA2iQ: τw,des = 5.78 ± 0.520 ms, SS/P = 0.014 ± 0.0028, n = 8, at holding potential −60 mV; Stg tethered by intein trans‐splicing). Contrary to our expectation, G804A, M807L and L808F had negligible effect on Stg‐mediated slowing of desensitization (GluA2iQ‐G804A+Stg: τw = 16.4 ± 1.29 ms, SS/P = 0.26 ± 0.026, n = 16, P > 0.99; GluA2iQ‐M807L+Stg: τw,des = 12.5 ± 1.03 ms, SS/P = 0.14 ± 0.019, n = 10, P > 0.99; GluA2iQ‐L808F+Stg: τw,des = 12.3 ± 1.68 ms, SS/P = 0.20 ± 0.031, n = 8, P > 0.99; GluA2iQ–Stg: τw,des = 15.0 ± 1.18, SS/P = 0.19 ± 0.020, n = 10, at holding potential −60 mV; Stg tethered by intein trans‐splicing), which was not statistically significant. Again, these effects were not influenced by whether Stg was tethered to or co‐expressed with GluA2 mutants (Fig. 7 K and L, right). These results point out that simply increasing the stability of the GluA2–Stg complex is not necessarily accompanied by a parallel increase in gating modulation.
Testing cooperative function between the extracellular and intra‐membrane interaction in gating modulation
Various mechanisms appear to control TARP‐mediated slowing of desensitization. Swapping the extracellular loops of different TARPs is sufficient to convert the gating characteristics from one TARP to another (Milstein et al. 2007). Consistent with this observation, mutations in the extracellular LBD have been isolated that reduce the effect of Stg on gating (MacLean et al. 2014; Dawe et al. 2016). In contrast, the residues critical for the action of the γ‐8‐dependent AMPAR antagonist LY3130481 were mapped to TM3 and TM4 of γ‐8 (Kato et al. 2016), indicating that the residues inside the membrane and potentially those that interface GluA subunits support allosteric modulation of gating. The latter observation is in agreement with the critical roles of TMD in Stg function (Ben‐Yaacov et al. 2017).
How do the functional residues at the TMD surface of GluA2 identified in our study relate to the roles of extracellular domains? Dependency between the two mutations introduced at these domains is testable by examining if the phenotype of a double mutant is additive or dominant of that expressed by a single mutation. We, therefore, studied the gating phenotypes generated by introducing two mutations in the GluA2–Stg complex, one in the extracellular domain of Stg and another on the surface of the TMD of GluA2.
Mutation in Lp1 of Stg that influences gating modulation
To do this we first searched for gain‐of‐function mutations in Lp1 of Stg that slow desensitization and/or increase SS/P ratio significantly greater than wild‐type. The electrostatic attraction between structural elements of GluA2 and Stg has emerged as a candidate mechanism for gating modulation (Dawe et al. 2016; Twomey et al. 2016; Zhao et al. 2016b). We therefore searched for charged residues in Lp1 of Stg that could potentially make contacts with the basic ‘KGK’ motif (residues 718–720) in the D2 lobe of the LBD and, when mutated into opposite charges, would result in a gain or loss of function of Stg‐mediated gating modulation. The candidate basic residues, hypothesized to create gain‐of‐function Stg mutants with charge reversal, are limited in Lp1 of Stg and highlighted in red in Fig. 8 A and B. The residues 85–91 were hypothesized to be important from their proximity to the LBD in the cryoEM structure of the complexes (Twomey et al. 2016; Zhao et al. 2016b).
Figure 8. Gain‐of‐function mutations in Stg Lp1 and Lp2.
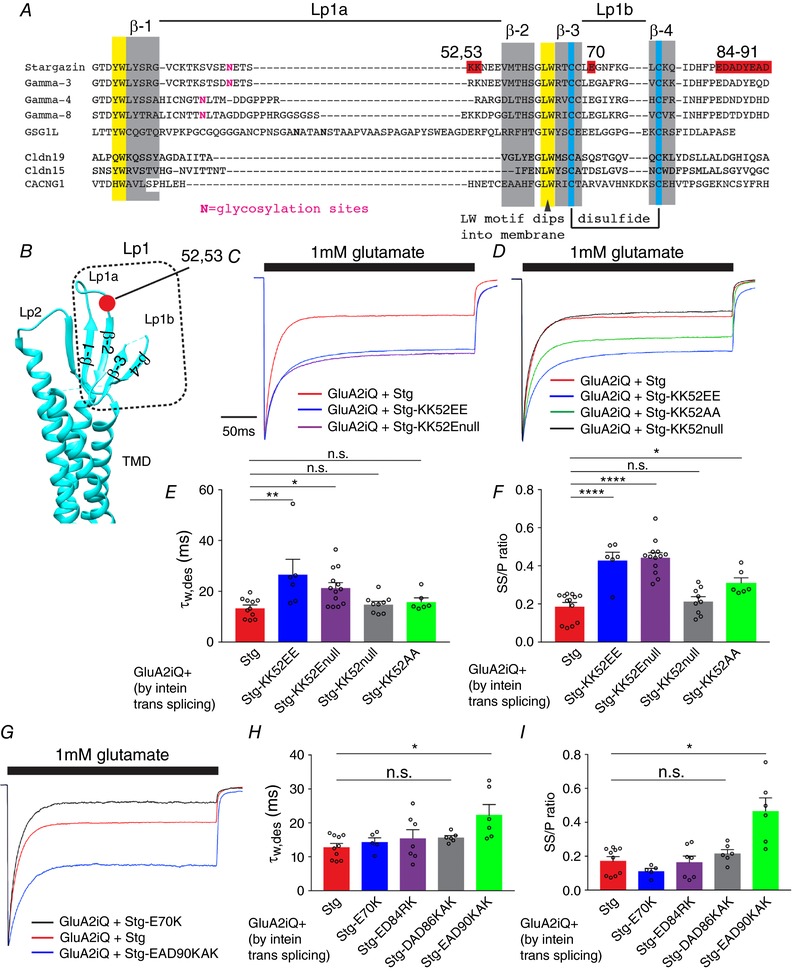
A, alignment of Stg, TARP γ‐3, 4, 8 and related members of the claudin family. The red residues were interrogated by mutation. The extracellular loop 1 (Lp1) is divided into Lp1a and Lp1b. Secondary structure elements and post‐translational modifications are indicated. β‐Strands (β‐1–4) and disulfide bonds are highlighted with grey and blue, respectively. B, locations that correspond to the secondary structure elements in A are mapped onto the crystal structure of claudin 19, a homologue of Stg. Lp2, extracellular loop2. The position of residues 52–53 is in red. C, D and G, representative recordings obtained from outside‐out patches in response to 1 mm glutamate application for 300 ms. The amplitude of each trace was normalized to the peak to facilitate comparison of the gating kinetics. The constructs transfected into TetON HEK cells are indicated. E, F, H and I, summary of τw,des and SS/P amplitude ratios. Individual data points are shown as open circles. Statistical significance against GluA2iQ+Stg (in red) was determined by one‐way ANOVA with post hoc Dunnett's comparison test (* P < 0.05; ** P < 0.01; **** P < 0.0001; mean ± SEM).
The Stg(KK52EE), converting two lysines to two glutamates at positions 52–53, is a gain‐of‐function mutant that slows desensitization greater than the wild‐type Stg (GluA2iQwt–Stg(KK52EE): τw,des = 28.6 ± 5.31 ms, P < 0.01, SS/P = 0.45 ± 0.040, P < 0.0001, n = 7, compared to GluA2iQwt–Stg; Fig. 8 C, E and F, Table 2 data set 4). This effect was observed regardless of whether Stg was tethered via intein trans‐splicing or co‐expressed (Table 2 data set 7). This is consistent with the idea that a more acidic Lp1 of Stg is favourable for stronger gating modulation. To further establish this concept, we introduced variations to the original Stg(KK52EE) mutation and evaluated the outcomes. The Stg(KK52Enull) that replaces two lysines with a single glutamate was sufficient to produce a gain of function (Fig. 8 C, E and F). Conversely, removing the two lysines in Stg(KK52null) produced no effects (Fig. 8 D, E and F). Importantly, converting two lysines to alanines in Stg(KK52AA) had a weak gain of function (Fig. 8 D, E and F), indicating a hydrophobic residue could partially mimic the effect of glutamate. A similar hypothesis is that Stg(E70K) mutation should make the Stg Lp1 more basic and result in a loss of function. However, Stg(E70K) had virtually no effect on modulation of desensitization by Stg (P = 0.79, Fig. 8 G–I, Table 2 data set 4). More surprisingly, contrary to the proposed models, a trend for gain of function was observed for Stg(EAD90KAK) mutation (τw,des = 22.4 ± 3.04 ms, SS/P = 0.47 ± 0.077, n = 6, P = 0.046), while Stg(ED84RK) and Stg(DAD86KAK) had no effects (P > 0.99 and P > 0.95, respectively). These results were unexpected; one would predict that more basic Stg Lp1 should lead to a loss of function, thereby emphasizing the incompleteness of the prevailing mechanistic models based on electrostatic interactions between Stg and AMPAR (Dawe et al. 2016; Twomey et al. 2016; Zhao et al. 2016a). Collectively, these results identify novel gain‐of‐function Stg mutants.
Finally, we generated GluA2iQ(C528L)–Stg(KK52EE), which contains both gain‐ and loss‐of‐function mutations in the extracellular domain of Stg and the TMD (M1) of GluA2, respectively. If this mutant GluA2–Stg complex expresses a phenotype similar to GluA2iQ(C528L)–Stg(wt), then C528L is required for the action of the extracellular events. On the other hand if the mutant expresses a hybrid phenotype between GluA2iQ(C528L)–Stg(wt) and GluA2iQ(wt)–Stg(KK52EE), then the two mechanisms are independent and additive. Our results indicate that the latter is the case (GluA2iQ(C528L)–Stg(KK52EE): τw,des = 13.0 ± 1.32 ms, P > 0.99, SS/P = 0.23 ± 0.025, P > 0.99, n = 15, compared to GluA2+Stg), suggesting a non‐hierarchical mechanism contributed to by both elements (Fig. 9 A and Ba and b, Table 2 data set 5). In support of this idea similar results were obtained from an analogous double mutant GluA2iQ(C528L)–Stg(KK52Enull) that carries gain‐ and loss‐of‐function mutations in the extracellular domain of Stg and the TMD (M1) of GluA2, respectively (Fig. 9, Table 2 data set 5).
Figure 9. Functional interaction between gain‐of‐function Stg Lp1 mutations and loss‐of‐function TMD mutation of GluA2.
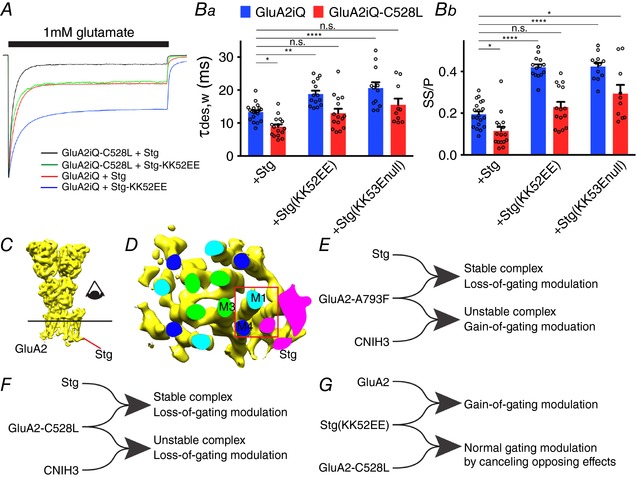
A, representative recordings obtained from outside‐out patches in response to 1 mm glutamate application for 300 ms. The traces were normalized to the peak amplitudes to facilitate comparison of the gating kinetics. The constructs transfected into TetON HEK cells are indicated. Ba and b, summary of τw,des and SS/P amplitude ratios. Individual data points are shown as open circles. Statistical significance against GluA2iQ+Stg was determined by two‐way ANOVA with post hoc Tukey's test (* P < 0.05; ** P < 0.01; **** P < 0.0001; mean ± SEM). C, GluA2–Stg complex (EMD‐8230) was displayed using Chimera and the detergent micelle removed. The line and eye indicates the cross section and the viewing direction, respectively, in D. D, cross section of the transmembrane helices in the GluA2‐Stg complex. M1, 3 and 4 of GluA2 are shown as light blue, green and dark blue, respectively. The cross section of Stg is shown in magenta. The red box is the location where the surface residues C528, L789 and A793 could potentially contact the TMD of Stg. E and F, functional bifurcations of A793L and C528L mutations. Rightward merging of the lines indicates complex formation. Functional consequence of each complex is summarized. G, a diagram summarizing the antagonistic effects of gain‐of‐function Stg(KK52EE) and loss‐of‐function GluA2iQ‐C528C.
Discussion
Surface residues in the TMD of GluA2 play key functional roles
The mechanism used by different auxiliary subunits to slow AMPAR desensitization is likely to have shared components, because a gating deficiency caused by a single GluA2 mutant was partially rescued by both Stg and CNIH2/3 (Dawe et al. 2016). In contrast, the difference in mechanisms among AMPAR auxiliary subunits is less explored. Using extensive mutational analyses, at a single amino acid resolution, we identified several structural elements of the AMPAR TMD that are exposed to the membrane as novel determinants. Among these elements, we found mutations that behave very differently depending on whether they are in complex with CNIH3 or Stg. For example, gating modulations of the GluA2‐A793F mutant induced by CNIH3 and Stg are qualitatively in opposing directions (Fig. 9 E). This divergence suggests that the two auxiliary subunit types recruit identical structural element but utilize different mechanisms to slow channel desensitization.
We used quantitative co‐IP to evaluate complex stability in detergent as the first step to identify these mutants. Disrupting interaction or modifying the interaction interface between GluA2 and auxiliary subunits would in theory interfere with the gating modulation. Studies have demonstrated that weakened interaction parallels a reduced gating modulation (Cais et al. 2014; Shanks et al. 2014). However, when embedded in the membrane, complexes that are otherwise unstable could remain stable and functional. Such dissociation between stability in detergent and function in the membrane was clearly observed in our results. For example, the complex made of GluA2‐A793F and CNIH3 was unstable in DDM, while a robust functional interaction was maintained in the membrane as determined by recording glutamate‐evoked currents. The inconsistency between biochemical interaction in vitro and electrophysiological recordings points out an important example where detergent sensitive interactions could still have a significant functional consequence in the membrane, where embedded complexes are protected by the lipids. In another case, the stability of the GluA2–Stg complex was enhanced by mutations in M4 (G804A, M807L and L808F), but only G804A expressed a modest modulation increase. Conversely, the C528L mutation in M1 increased the complex stability but failed to modulate desensitization. Accordingly, the membrane complexes whose structures were determined with a choice of detergents that maintains the maximal stability may not necessarily represent the most physiological states. The gain‐of‐function Stg mutants identified in this study, such as Stg(KK52EE), offer a new opportunity to investigate the general mechanism of modulation. Structural differences in the mode of binding between gain‐ and/or loss‐of‐function mutant Stg and GluA2 reported here are likely to provide insights into the mechanism of action of AMPAR modulation by auxiliary subunits.
Previously we identified residues in CNIH3 critical for interaction with GluA2 (Shanks et al. 2014). These residues were located at three sites, at the boundary between the TM1 and the extracellular domain, in the extracellular loop and at the cytoplasmic side of TM2. In retrospect, even though these mutations destabilized the complex in detergent, the data clearly showed residual gating modulation by these mutant CNIH3, indicating that their interactions with GluA2 must be partially maintained in the membrane. In the current study we identified residues at the lipid‐facing surface of the GluA2 TMD that are critical for CNIH3 to modulate AMPAR gating. Collectively, these findings raise an interesting new question as to whether any pair of the residues identified by these studies match up to form a molecular interface. Future structural information is likely to provide answers to this question.
Intein trans‐splicing as an effective approach to tether GluA2 and Stg
A high occupancy of auxiliary subunits in the complex is guaranteed by introducing a covalent tether between GluA2 and Stg, and thus expression schemes using GluA2–Stg fusion constructs have been extensively exploited in studying the mechanism of Stg‐mediated gating modulation of AMPARs (Shi et al. 2009; Shelley et al. 2012; Dawe et al. 2016). The mode of interaction between tethered and untethered GluA2–Stg complexes was virtually identical in the cryoEM structures (Twomey et al. 2016; Zhao et al. 2016a). The intein approach reported here has further advantages over the traditional GluA2–Stg fusion constructs because the two entities are expressed separately and fold at much higher efficiencies than the GluA2–Stg fusing (data not shown). A significant improvement in the expression level was achieved and the recording experiments were conducted very efficiently. The intein‐mediated trans‐splicing is an extensively characterized reaction whose mechanistic basis is established with a wealth of structural biology data of the individual intein fragments (Liu, 2000; Iwai et al. 2006). This reaction could in theory be blocked by a steric hindrance if the split‐inteins are fused in a sub‐optimal design. Technically, the resulting GluA2–Stg conjugate is equivalent to a tandem tethered construct that was expressed as a single polypeptide. As our results indicate, recording experiments using the intein‐mediated trans‐spliced complex produced similar results to those obtained by co‐expressing the two proteins as individual entities, indicating that both approaches are equally valid. The intein trans‐splicing is likely to be applicable to the members of tetraspanins, as identical tethering scheme also worked for fusing GluA2 and GSG1L, a distant homologue of TARPs (data not shown).
We also examined if the intein‐mediated trans‐splicing could be applied to CNIH3. Despite efficient trans‐splicing of co‐expressed GluA2iQ–IntN and IntC–CNIH3 in HEK cells, as determined by western blot analysis, the complex accumulated in the intracellular compartment and was never delivered to the cell surface (data not shown). Consistently, glutamate‐evoked currents were not measurable from the transfected cells. The reason why the intein system did not work is unclear, but we have noticed that several GluA2–CNIH3 fusion constructs also did not work, indicating the possibility that placing a fusion protein at the N‐terminus of CNIH3 may interfere with its function. Further investigation would be necessary to address these issues.
Generality of TMD surface residues influencing gating modulation
The action of GluN2C/D‐specific allosteric modulator CIQ requires residues in the pre‐M1, M1 and M4 segments (Ogden & Traynelis, 2013). Two residues in M4 of GluN2C/D (M813 and F817) accelerate positive allosteric action of CIQ. The residues in M1 and M4 that modify CIQ action are in the immediate neighbourhood on the surface of the membrane‐embedded domain of the NMDAR (Karakas & Furukawa, 2014; Lee et al. 2014), where lipids would be in contact. Similarly, the M523W mutation in M4 of the GluN2A subunit influences desensitization of the NMDAR (Ren et al. 2003) and is located at the surface in the crystal structure (Karakas & Furukawa, 2014). Interestingly, we demonstrate the analogous region in AMPARs plays a critical function during gating modulation by auxiliary subunits. Specifically, in GluA2, C528 of M1 is immediately next to L789 and A793 of M4 in the adjacent subunit. These three residues are exposed to the surface of the receptors and are located between the centre of the lipid bilayer and the extracellular space that topologically corresponds to the regions in the GluN2C/D critical for CIQ efficacy and M523 of GluN2A. The M1 and M4 helices in adjacent subunits were critical for Stg‐dependent modulation, as modulation was lost when they were swapped with the corresponding segments in GluK2 (Ben‐Yaacov et al. 2017). More surprisingly, this region, spanning M1 and M4, is part of the interface between GluA2 and Stg in the cryoEM structure (PDB: EMD‐8230) (Fig. 9 C and D). Our results demonstrate that C528 not only influences gating modulation by CNIH3 but also by Stg. Collectively, these observations converge on a hypothesis that a small neighbourhood at the surface of M1 and M4 in adjacent subunits in the membrane plays a critical role in ion channel gating modulation in iGluRs. In NMDARs, this region is required for the action of CIQ; in AMPARs, auxiliary subunits utilize this region to modulate gating.
The mechanism by which membrane exposed residues influence gating is yet to be determined. Lipids and auxiliary subunits that would have direct contact with these residues may introduce molecular networks hovering over the surface of the channel domain and impose restrictions on the physical parameters of the conformational transition associated with gating. A recently discovered TARP γ‐8‐dependent AMPAR inhibitor requires residues near the extracellular side of the TMD of γ‐8 for its action (Kato et al. 2007; Gardinier et al. 2016; Maher et al. 2016). Most importantly, considering the recent cryoEM structures of the GluA2–Stg complex (Twomey et al. 2016; Zhao et al. 2016b), residues we identify here (C528 of M1, and L789 and A793 of M4) are likely to be facing and potentially in contact with the residues critical for the action of this inhibitor. Furthermore, the interaction may involve lipids; however, to substantiate this hypothesis, structural data of the receptor–auxiliary subunit complex embedded in lipid will be essential.
Our results confirm the functional importance of the TMD contacts reported in the recent cryoEM structures. However, the mechanisms for gating modulation of our extracellular domain mutants of Stg remain unexplained. Specifically, the highly acidic residues 84–91 in the extracellular domain of Stg (red in Fig. 8 A) were proposed as a candidate region mediating functionally critical electrostatic interaction with the basic residues in the AMPAR LBD. Based on this hypothesis one would predict that converting residues 84–91 of Stg to basic amino acids should result in a loss of function. On the contrary, we found that replacing these residues with more acidic basic ones had either no effect for Stg(ED84RK) and Stg(DAD86KAK), or produced a small gain‐of‐function phenotype for Stg(EAD90KAK) (Fig. 8 G–I). Furthermore, as discussed in the following, reversing the electrostatic charges in Lp1a and Lp1b produced robust gain‐ and loss‐of gating modulation, respectively, which was not predicted from the structural data.
A central model of AMPAR modulation by TARP is that the extracellular loop 1 (Lp1) of TARPs shapes the gating modulation (Tomita et al. 2005). The role for residues 52–53 of Lp1 in enhancing modulation was consistent with this logic. However, this region was part of an unresolved flexible structure within the molecule and limited mechanistic predictions could be made (Twomey et al. 2016; Zhao et al. 2016b). Among TARPs, γ‐4 and ‐8 have the strongest modulatory effect, conferring on AMPAR the slowest desensitization (Milstein et al. 2007). The Lp1 sequences of γ‐4 and ‐8 contain acidic residues (DD) in the middle compared to Stg and γ‐3 that have weaker action than γ‐4 and ‐8 and have their acidic residues (EE) near the membrane (Fig. 8 A and B). Adding acidic residues to the Lp1 in Stg(KK53EE) induced stronger gating modulation reminiscent of stronger acting γ‐4 and ‐8. Considering that residues in both the Lp1 of Stg and the TMD of GluA2 are essential for gating modulation, this raises a question as to what the relative contribution of each element is. Are the TMD residues at the surface of GluA2 that, when mutated, reduce Stg function essential for the action of the extracellular loops of Stg? Is the event that takes place at the surface of the TMD necessary for the action of Stg or is it an independent molecular pathway that is in place? We find that the contribution of each element is consistent with an independently operated parallel mechanism, in contrast to a hierarchal one, as the phenotype of the double mutant was additive of those of the single mutants (Fig. 9 E–G).
The elements we identified in this study are only a part of the complete mechanism. The critical roles of the cytoplasmic domains of the GluA2 and stargazin (Tomita et al. 2005; Ben‐Yaacov et al. 2017) remain unexplained. Within the TMD of the AMPAR, M2, which corresponds to the region next to the selectivity filter in potassium channels, is accessible to objects in the membrane and cytoplasm. The M2 could potentially be an effective element to govern gating modulation. Further investigation in this area would be necessary to address these issues.
Additional information
Competing interests
The authors have no conflict of interest.
Author contributions
T.N. conceived and supervised the project. All authors designed the experiments and analysed data. N.H. and T.N. made cDNA constructs of mutants. E.Z. made cell lines. N.H. and E.Z. conducted co‐IP. T.N. conducted electrophysiology. T.N. wrote the manuscript with inputs from the other authors. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH grants (R01HD061543 and R21MH102546) to T.N.
Acknowledgements
We thank Kenny Futai (U Mass) for discussion. We thank members of Nakagawa lab for critically reading the manuscript.
N. Hawken and E. Zaika contributed equally.
References
- Ben‐Yaacov A, Gillor M, Haham T, Parsai A, Qneibi M & Stern‐Bach Y (2017). Molecular mechanism of AMPA receptor modulation by TARP/stargazin. Neuron 93, 1126–1137.e4. [DOI] [PubMed] [Google Scholar]
- Cais O, Herguedas B, Krol K, Cull‐Candy SG, Farrant M & Greger IH (2014). Mapping the interaction sites between AMPA receptors and TARPs reveals a role for the receptor N‐terminal domain in channel gating. Cell Rep 9, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS & Nicoll RA (2000). Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408, 936–943. [DOI] [PubMed] [Google Scholar]
- Dawe GB, Musgaard M, Aurousseau MR, Nayeem N, Green T, Biggin PC & Bowie D (2016). Distinct structural pathways coordinate the activation of AMPA receptor‐auxiliary subunit complexes. Neuron 89, 1264–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y & Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina AN, Blain KY, Maruo T, Kwiatkowski W, Choe S & Nakagawa T (2011). Separation of domain contacts is required for heterotetrameric assembly of functional NMDA receptors. J Neurosci 31, 3565–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardinier KM, Gernert DL, Porter WJ, Reel JK, Ornstein PL, Spinazze P, Stevens FC, Hahn P, Hollinshead SP, Mayhugh D, Schkeryantz J, Khilevich A, De Frutos O, Gleason SD, Kato AS, Luffer‐Atlas D, Desai PV, Swanson S, Burris KD, Ding C, Heinz BA, Need AB, Barth VN, Stephenson GA, Diseroad BA, Woods TA, Yu H, Bredt D & Witkin JM (2016). Discovery of the first α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor antagonist dependent upon transmembrane ampa receptor regulatory protein (TARP) γ‐8. J Med Chem 59, 4753–4768. [DOI] [PubMed] [Google Scholar]
- Gill JK, Savolainen M, Young GT, Zwart R, Sher E & Millar NS (2011). Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA 108, 5867–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Shi Y, Suh YH, Zheng CY, Blankenship SM, Roche KW & Nicoll RA (2013). Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron 77, 1083–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai H, Zuger S, Jin J & Tam PH (2006). Highly efficient protein trans‐splicing by a naturally split DnaE intein from Nostoc punctiforme . FEBS Lett 580, 1853–1858. [DOI] [PubMed] [Google Scholar]
- Jackson AC & Nicoll RA (2011). The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 70, 178–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova E, Lorca RA, Kaur I, Barisone GA, Li B, Ishimaru T, Trimmer JS, Mohapatra DP & Diaz E (2010). SynDIG1: an activity‐regulated, AMPA‐ receptor‐interacting transmembrane protein that regulates excitatory synapse development. Neuron 65, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E & Furukawa H (2014). Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Burris KD, Gardinier KM, Gernert DL, Porter WJ, Reel J, Ding C, Tu Y, Schober DA, Lee MR, Heinz BA, Fitch TE, Gleason SD, Catlow JT, Yu H, Fitzjohn SM, Pasqui F, Wang H, Qian Y, Sher E, Zwart R, Wafford KA, Rasmussen K, Ornstein PL, Isaac JT, Nisenbaum ES, Bredt DS & Witkin JM (2016). Forebrain‐selective AMPA‐receptor antagonism guided by TARP γ‐8 as an antiepileptic mechanism. Nat Med 22, 1496–1501. [DOI] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Ho MT, Yu H, Tu Y, Siuda ER, Wang H, Qian YW, Nisenbaum ES, Tomita S & Bredt DS (2010). Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron 68, 1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Zhou W, Milstein AD, Knierman MD, Siuda ER, Dotzlaf JE, Yu H, Hale JE, Nisenbaum ES, Nicoll RA & Bredt DS (2007). New transmembrane AMPA receptor regulatory protein isoform, γ‐7, differentially regulates AMPA receptors. J Neurosci 27, 4969–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen RV, Stroeder J, Coussen F, Hafner AS, Petersen JD, Renancio C, Schmitz LJ, Normand E, Lodder JC, Rotaru DC, Rao‐Ruiz P, Spijker S, Mansvelder HD, Choquet D & Smit AB (2016). Shisa6 traps AMPA receptors at postsynaptic sites and prevents their desensitization during synaptic activity. Nat Commun 7, 10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X & Gouaux E (2014). NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XQ (2000). Protein‐splicing intein: Genetic mobility, origin, and evolution. Annu Rev Genet 34, 61–76. [DOI] [PubMed] [Google Scholar]
- MacLean DM, Ramaswamy SS, Du M, Howe JR & Jayaraman V (2014). Stargazin promotes closure of the AMPA receptor ligand‐binding domain. J Gen Physiol 144, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher MP, Wu N, Ravula S, Ameriks MK, Savall BM, Liu C, Lord B, Wyatt RM, Matta JA, Dugovic C, Yun S, Ver Donck L, Steckler T, Wickenden AD, Carruthers NI & Lovenberg TW (2016). Discovery and characterization of AMPA receptor modulators selective for TARP‐γ8. J Pharmacol Exp Ther 357, 394–414. [DOI] [PubMed] [Google Scholar]
- Milstein AD, Zhou W, Karimzadegan S, Bredt DS & Nicoll RA (2007). TARP subtypes differentially and dose‐dependently control synaptic AMPA receptor gating. Neuron 55, 905–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK & Traynelis SF (2013). Contribution of the M1 transmembrane helix and pre‐M1 region to positive allosteric modulation and gating of N‐methyl‐D‐aspartate receptors. Mol Pharmacol 83, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Honse Y, Karp BJ, Lipsky RH & Peoples RW (2003). A site in the fourth membrane‐associated domain of the N‐methyl‐D‐aspartate receptor regulates desensitization and ion channel gating. J Biol Chem 278, 276–283. [DOI] [PubMed] [Google Scholar]
- Salussolia CL, Gan Q, Kazi R, Singh P, Allopenna J, Furukawa H & Wollmuth LP (2013). A eukaryotic specific transmembrane segment is required for tetramerization in AMPA receptors. J Neurosci 33, 9840–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, Muller CS, Bildl W, Baehrens D, Huber B, Kulik A, Klöcker N, Schulte U & Fakler B (2012). High‐resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 74, 621–633. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B & Klöcker N. (2009). Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 323, 1313–1319. [DOI] [PubMed] [Google Scholar]
- Shanks NF, Cais O, Maruo T, Savas JN, Zaika EI, Azumaya CM, Yates JR 3rd, Greger I & Nakagawa T (2014). Molecular dissection of the interaction between the AMPA receptor and cornichon homolog‐3. J Neurosci 34, 12104–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks NF, Maruo T, Farina AN, Ellisman MH & Nakagawa T (2010). Contribution of the global subunit structure and stargazin on the maturation of AMPA receptors. J Neurosci 30, 2728–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks NF, Savas JN, Maruo T, Cais O, Hirao A, Oe S, Ghosh A, Noda Y, Greger IH, Yates JR 3rd & Nakagawa T (2012). Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep 1, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley C, Farrant M & Cull‐Candy SG (2012). TARP‐associated AMPA receptors display an increased maximum channel conductance and multiple kinetically distinct open states. J Physiol 590, 5723–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lu W, Milstein AD & Nicoll RA (2009). The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron 62, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP & Gouaux E (2009). X‐ray structure, symmetry and mechanism of an AMPA‐subtype glutamate receptor. Nature 462, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Gratacos‐Batlle E, Farrant M & Cull‐Candy SG (2014). Molecular mechanisms contributing to TARP regulation of channel conductance and polyamine block of calcium‐permeable AMPA receptors. J Neurosci 34, 11673–11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA & Bredt DS (2005). Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA & Bredt DS (2003). Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol 161, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J & Sobolevsky AI (2016). Elucidation of AMPA receptor–stargazin complexes by cryo‐electron microscopy. Science 353, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J & Sobolevsky AI (2017). Channel opening and gating mechanism in AMPA‐subtype glutamate receptors. Nature 549, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Mack V, Sprengel R, Kavenstock N, Li KW, Stern‐Bach Y, Smit AB, Seeburg PH & Monyer H (2010). CKAMP44: a brain‐specific protein attenuating short‐term synaptic plasticity in the dentate gyrus. Science 327, 1518–1522. [DOI] [PubMed] [Google Scholar]
- Yelshanskaya MV, Li M & Sobolevsky AI (2014). Structure of an agonist‐bound ionotropic glutamate receptor. Science 345, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen S, Yoshioka C, Baconguis I & Gouaux E (2016a). Architecture of fully occupied GluA2 AMPA receptor ‐ TARP complex elucidated by single particle cryo‐electron microscopy. biorxiv https://doi.org/10.1101/060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chen S, Yoshioka C, Baconguis I & Gouaux E (2016b). Architecture of fully occupied GluA2 AMPA receptor‐TARP complex elucidated by cryo‐EM. Nature 536, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]