

Abstract
Recent studies have provided evidence for a regulatory role of GLI-similar (GLIS) transcription factors in reprogramming, maintenance and differentiation of several stem and progenitor cell populations. GLIS1, in conjunction with several other reprogramming factors, was shown to markedly increase the efficiency of generating induced pluripotent stem cells (iPSC) from somatic cells. GLIS2 has been reported to contribute to the maintenance of the pluripotent state in hPSCs. In addition, GLIS2 has a function in regulating self-renewal of hematopoietic progenitors and megakaryocytic differentiation. GLIS3 plays a critical role during the development of several tissues. GLIS3 is able to promote reprogramming of human fibroblasts into retinal pigmented epithelial (RPE) cells. Moreover, GLIS3 is essential for spermatogonial stem cell renewal and spermatogonial progenitor cell differentiation. During pancreas development, GLIS3 protein is first detectable in bipotent pancreatic progenitors and pro-endocrine progenitors and plays a critical role in the generation of pancreatic beta cells. Here, we review the current status of the roles of GLIS proteins in the maintenance and differentiation of these different stem and progenitor cells.
Keywords: GLI-similar (GLIS), pluripotent stem cells, reprogramming, spermatogonial stem cells (SSCs), hematopoietic stem cells (HSCs), pancreatic beta cells, Krüppel-like zinc finger protein, pancreas, transcription, diabetes, acute megakaryoblastic leukemia (AMKL)
Introduction
The GLI Similar 1-3 (GLIS) proteins form a subfamily of Krüppel-like zinc finger transcription factors that are closely-related to the GLI and ZIC subfamilies (1-8). Members of these three subfamilies share a highly homologous DNA binding domain (DBD) consisting of five Cys2His2-type zinc finger motifs. However, these proteins exhibit little homology outside their DBD region.
The DBD mediates the interaction of GLIS1-3 to GLIS binding sequences, referred to as GLISBS, in the regulatory regions of target genes (7,9,10). The GLISBS consists of the G-rich consensus sequence, (A/G)GG(G/A)GG (Figure 1). GLIS1-3 can function as activators as well as repressors of gene transcription. The transcriptional activation by GLIS proteins is mediated through the recruitment of coactivators, such as CREB binding protein (CREBBP, also referred to as CBP) (11), that interact with a transactivation domain (TAD) at their C-terminus. Transcriptional repression by GLIS2 has been reported to involves recruitment of corepressors, such as the C-terminal binding protein 1 (CtBP1) (12). GLIS proteins can undergo a number of posttranslational modifications, including ubiquitination, methylation, phosphorylation, and sumoylation, that can influence their subcellular localization, protein stability, protein-protein interaction, and/or transcriptional activity (13-16). However, rather little is known about the upstream signaling pathways (e.g., protein kinases) that regulate GLIS1-3 activity.
Figure 1.

Schematic of GLIS1-3 proteins and their roles in stem and progenitor cells. The DBD consisting of five zinc finger motifs (ZF1-5) and the TAD are indicated. The DBD recognizes a G-rich GLISBS in the regulatory region of target genes. Regulation of transcriptional activation or repression by GLIS proteins is mediated through their recruitment of co-activator or co-repressor complexes, respectively. GLIS functions in various stem and progenitor cells are indicated. DBD, DNA binding domain; TAD, transactivation domain; GLISBS, GLIS binding sequence.
GLIS1-3 have been reported to play a critical role in the regulation of many physiological processes and are implicated in various pathologies (6,7). Deficiency in GLIS3 causes abnormalities in multiple tissues, including development of neonatal diabetes, congenital hypothyroidism, polycystic kidney disease, infertility, and several neurological disorders (7,8,17-23). GWAS studies have linked single nucleotide polymorphisms (SNPs) in GLIS3 to increased risk for type 1 as well as type 2 diabetes (24,25). GLIS3 is a critical regulator of insulin gene expression and essential for pancreatic β-cell generation, thyroid hormone biosynthesis, the maintenance of normal kidney functions and normal spermatogenesis (8,23). Deficiency in GLIS2 leads to the development of nephronophthisis, a cystic renal disease characterized by renal atrophy, fibrosis, and inflammation (5,18). The fibrosis appears to involve epithelial-mesenchymal transition (EMT) of renal epithelial cells. A translocation involving GLIS2 has been implicated in acute myeloid leukemia (26-28). Beyond its role in reprogramming, relatively little is known about the biological functions of GLIS1 (29). GWAS studies reported an association between SNPs in GLIS1 and increased risk of autism spectrum disorder and Alzheimer’s disease (30,31).
Recent studies demonstrated that GLIS1-3 are expressed in a number of stem/progenitor cell populations, suggesting a possible role for these proteins in the regulation of maintenance, differentiation, or self-renewal of these cells. In this report, we present a short overview of the function of GLIS1 in reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) and the emerging roles of GLIS proteins in several stem/progenitor cell populations.
GLIS1 as pro-reprogramming factor
It has been now well-established that iPSCs can be generated from multiple somatic cell types (32,33). This, together with the establishment of protocols that enable PSCs and iPSCs to differentiate into a variety of differentiated cell types of all three germ layers, including pancreatic β cells, cardiomyocytes, and various immune and neuronal cell types, has greatly enhanced the interest in the potential of stem cell therapies and regenerative medicine. Although many safety concerns remain, including tumor formation and immune rejection, the generation of progenitor and differentiated cell types from patient-histocompatible (autologous or HLA-matched) iPSCs should reduce complications by host immune responses.
Initial overexpression of OCT3/4 (POU5F1), SOX2, and KLF4 (OSK) are widely used for the reprogramming of somatic cells into iPSCs (32). However, the efficiency of generating iPSCs is very low, which has been attributed to difficulties in overcoming epigenetics barriers in the starting cell (33). Co-expression of C-MYC increases the efficiency, but also enhances the potential tumorigenicity of iPSC-derived differentiated cells. Recently, using a screen analyzing 1,437 transcription factors for their ability to promote reprogramming efficiency, GLIS1 was found to greatly enhance the number of iPSC colonies generated when co-expressed with OSK (referred to as OSKG) in either human or mouse dermal fibroblasts (29,34). Inversely, down-regulation of GLIS1 expression by shRNAs reduced the OSK-induced generation of iPSC colonies in mouse fibroblasts suggesting that endogenous GLIS1 is able to promote OSK-mediated reprogramming. The iPSCs derived from OSKG reprogramming exhibited a similar morphology and expressed many of the PSC marker genes, including NANOG, ZFP42 (REX1), SOX2, and OCT3/4. Moreover, subcutaneous transplantation of OSKG iPSCs formed teratomas containing differentiated cells from all three germ layers. These data demonstrated that GLIS1 functions as a strong promoter of somatic cell reprogramming. It was further shown that GLIS1 co-purified with OCT3/4, SOX2, and KLF4 suggesting that it is part of a larger complex of these proteins (29). Deletion analysis indicated that the zinc finger domain and N-terminus of GLIS1 were required for its interaction with KLF4. Whether these transcriptional regulators bind to their own enhancer sequences and interact with each other via co-activator complexes and whether this involves looping of chromatin in order to bring DNA-bound transcriptional complexes closer together needs further study.
The increase in reprogramming efficiency by GLIS1 was also demonstrated by Yoshioka et al. employing a different strategy to generate iPSCs using a modified Venezuelan equine encephalitis (VEE) RNA virus expressing OCT4, SOX2, KLF4 and GLIS1 (OSKG) (35). This virus has the advantage that it does not use a DNA intermediate for replication, thereby eliminating the potential for genomic integration and instability. Transfection with VEE-OSKG enhanced the generation of iPSC clones. The VEE-OSKG-induced iPSCs exhibited many hallmarks of embryonic stem cells and in vivo generated tissues from all three germ layers.
The p53 pathway has been reported to suppress OSK-mediated reprogramming in mouse and human fibroblasts (36); however, the increase in reprogramming efficiency by GLIS1 was found to be independent of the p53 pathway (29). Gene profiling analysis demonstrated that GLIS1 significantly increased the expression of several genes that were previously reported to enhance reprogramming, including the estrogen-related receptor β (ESRRB1), lin-28 homologue (LIN28A), v-Myc avian myelocytomatosis viral oncogene neuroblastoma and lung carcinoma derived homologs (MYCN and MYCL, respectively), tetraspanin 18 (TSPAN18), neurogranin (NRGN), several Wnt genes (WNT3, WNT6, WNT8A, WNT10A), and forkhead box A2 (FOXA2) (29). During the generation of iPSCs from fibroblasts by OSK, mesenchymal-epithelial transition (MET) is required for reprogramming (37). Several genes related to MET were found to be regulated by GLIS1, including increased expression of FOXA2, an inhibitor of epithelial-mesenchymal transition (EMT). ChIP analysis indicated that MYCN and MYCL were regulated directly by GLIS1, whereas FOXA2, as well as ESRRB1 and LIN28A transcription were regulated by an indirect mechanism. Together, these findings suggest that GLIS1 is able to enhance the generation of iPSCs by stimulating several pro-reprogramming pathways, including WNT signaling and MET related genes, some of which are regulated directly by GLIS1.
The proto-oncogene C-JUN has been shown to be induced during the differentiation of PSCs into endoderm and down-regulated during reprogramming of fibroblasts into iPSCs, indicating that its expression inversely correlates with pluripotency (38). This is further supported by data showing that ectopic expression of C-JUN in mouse pluripotent stem cells (mPSCs) suppresses the expression of pluripotent marker genes and induces the expression of mesenchymal-lineage associated genes and differentiation along the endoderm lineage. Inversely, inhibition of C-JUN expression by shRNAs or inhibition of C-JUN function by expressing a dominant-negative C-JUN enhances iPSC reprogramming. Similarly, expression of Jun dimerization protein 2 (JDP2), a repressor of C-JUN mediated transactivation, promotes OSKM-mediated reprogramming and can substitute for OCT3/4. Moreover, JDP2 together with lysine demethylase KDM2B (JHDM1b), ID1/3, the nuclear receptor LRH1 (NR5A2), Spalt Like Transcription Factor 4 (SALL4), and GLIS1 are sufficient to reprogram fibroblasts, thereby providing an alternative approach to generate iPSCs (38). As observed for OSK-mediated reprogramming, GLIS1 is not a strict requirement, but greatly promotes reprogramming.
GLIS2 and pluripotent stem cells
Cytosine-phosphate-Guanine (CpG) islands (CGIs), regions enriched in CpG sites, are often associated with the promoter of many mammalian genes (39). Methylation of cytosines in CpGs by DNA methyltransferases plays a critical role in the epigenetic regulation of gene expression and in stable gene silencing. Comparison of genome-wide CGI methylation analysis of hPSC lines and somatic tissues identified a CpG methylation signal specific for hPSCs (40). Many of the PSC-specific methylation sites were associated with transcriptional repressors and activators. GLIS2 was among the genes in which a PSC-associated CGI was consistently unmethylated in hPSCs and methylated in somatic tissues. However, no correlation was found between this CGI and the expression pattern of GLIS2. Knockdown of GLIS2 expression by siRNA in hPSCs was accompanied by a down-regulation of the pluripotent marker genes, OCT4, SOX2, and NANOG, and increased expression of endoderm-related genes, including HNF4α, GATA6, and α-fetoprotein (AFP), as well as several trophoblast-associated genes. Although GLIS2 binding sites are present in OCT4 and NANOG, whether these genes are directly regulated by GLIS2 requires further study. Together, these findings indicate that down-regulation of GLIS2 expression induces loss of the stem cell phenotype and suggest that GLIS2 might be playing a role in maintaining the pluripotent state of PSCs. However, in contrast to GLIS1, GLIS2 had no significant effect on the generation of iPSC colonies when expressed together with the programming factors OCT4, SOX2, KLF4, L-MYC, a p53 shRNA, and LIN28 in human dermal fibroblasts (40). Knockdown of OCT4 in hPSCs reduces GLIS2 expression suggesting that GLIS2 transcription might be regulated by OCT4. This was supported by GLIS2 promoter sequence analysis showing the presence of OCT4 binding sites and ChIP-qPCR data demonstrating OCT4 occupancy at the GLIS2 promoter region. Thus, epigenetic and transcriptional regulation of GLIS2 might contribute to the maintenance of the pluripotent state in hPSCs.
GLIS2 and hematopoietic stem cells (HSCs)
HSCs constitute a functionally heterogeneous cell population with respect to their self-renewal, life span, and differentiation capabilities (41). Transplanted HSCs can reconstitute the entire hematopoietic system and are being used in curative therapy for several blood and immune diseases, including leukemia and myeloma. Since the number of HSCs for transplantation can be limited, many studies have been focusing on improving the efficiency of HSC transplantation. In a strategy to identify genes that improve in vivo repopulation ability of HSCs, GLIS2 was identified as one of several genes required for optimal repopulation (42). GLIS2 was found to be expressed in the Lineage-SCA-1+C-KIT+ (LSK) subpopulation of hematopoietic precursors, but not in Lineage- populations that were either SCA-1−C-KIT+ or SCA-1−C-KIT−. Down-regulation of GLIS2 expression by shRNA in murine LSK cells caused a reduction in their repopulation potential when transplanted into lethally irradiated mice. Down-regulation of GLIS2 was associated with increased apoptosis, inhibition of HSC differentiation, and decreased cell survival downstream of HSCs. These observations suggested that GLIS2 contributes positively to HSC repopulation activity.
Recently, translocations involving the GLIS2 and CBFA2/RUNX1 Translocation Partner 3 (CBFA2T3 or ETO2) genes has been linked to a subtype of pediatric acute megakaryoblastic leukemia (AMKL) (26-28,43). These translocations result in high expression of a CBFA2T3-GLIS2 fusion protein. Ectopic expression of CBFA2T3-GLIS2 or GLIS2 in murine hematopoietic progenitors causes a significant increase in the number of both immature CD41+C-KIT+ and maturing CD41+CD42+ megakaryocytes; however, the number of CD41+CD42+ cells was several-fold higher in GLIS2 expressing cells than in CBFA2T3-GLIS2 expressing cells (44). The latter suggests that GLIS2 promotes megakaryocytic differentiation, whereas CBFA2T3-GLIS2 favors maintaining the immature stage. A different study showed that both CBFA2T3-GLIS2 and GLIS2 expression can increase self-renewal capacity in hematopoietic progenitors (26). Together, these findings show that CBFA2T3-GLIS2 enhances self-renewal and inhibits differentiation, whereas ectopic GLIS2 expression has a variable effect on self-renewal and promotes megakaryocytic differentiation. Based on these observations one might conclude that the development of AMKL in patients with CBFA2T3-GLIS2 translocations is related to increased self-renewal of hematopoietic progenitors and an inhibition of normal hematopoietic differentiation.
Emerging roles for GLIS3 in stem and progenitor cells
Several studies have shown that during mammalian development, as well as in the adult, GLIS3 is expressed in various stem and progenitor cells, including bipotent pancreatic progenitors and spermatogonial stem cells (SSCs), suggesting that GLIS3 may have a regulatory function in these cells (6,7,23,45-47). Recently, using a computational approach to identify core transcription factors that control cell identity, GLIS3 was identified as one of the factors predicted to promote reprogramming of human fibroblasts into retinal pigmented epithelial (RPE) cells (47). The investigators then demonstrated that lentiviral expression of GLIS3, in combination with that of the transcription factors PAX6, OTX2, MITF, SIX3, and FOXD1, was able to promote reprogramming of human foreskin fibroblasts into RPE-like cells consistent with a regulatory role for GLIS3 in cell lineage determination.
Role of GLIS3 in SSCs
GLIS3 was recently identified as a key regulator of early stages of spermatogenesis and shown to have a critical function in SSCs (23). In mammalian testes, SSCs arise from non-mitotic gonocytes after they migrate to the basement membrane compartment of seminiferous tubules shortly after birth. Vitamin A and its derivatives, anti-Mullerian hormone (AMH) and FGF signaling are involved in the regulation of the gonocyte to SSC transition (48-50). The differentiation of gonocytes into SSCs is marked by the translocation of the transcription factor FOXO1 from the cytoplasm to the nucleus. FOXO1 remains expressed in undifferentiated spermatogonia, but is not present in C-KIT+ differentiated spermatogonia (51).
SSCs are defined by the expression of inhibitor of DNA binding 4 (ID4) and paired box 7 (PAX7) (48,52,53). SSCs have an extensive self-renewal capacity and provide a source for the production of spermatozoa throughout life (54-58). Thus, preserving the delicate balance between self-renewal and differentiation is necessary for maintaining the pool of SSCs as well as for the continuous generation of mature spermatogonia. Fibroblast growth factor 2 (FGF2), produced by several testicular cell types, and glial cell line-derived neurotrophic factor (GDNF), produced by Sertoli cells, are critical for SSC self-renewal (48,54,55,59). GDNF through its interaction with the GDNF receptor complex, consisting of C-RET and the GDNF family receptor α1 (GFRα1), activates the PI3K/AKT pathway and induces the expression of several genes, including the RNA-binding protein NANOS2, LHX1, ETV5 and POU3F1, all of which support self-renewal. In addition to these factors, the Krüppel-type transcription factor, zinc finger and BTB domain containing 16 (ZBTB16, also named PLZF) also plays a role in the control of SSC self-renewal (48,60). The SSCs can convert into spermatogonial progenitor cells (SPCs), which together are also referred to as undifferentiated spermatogonia. Inhibin beta A subunit (INHBA, also referred to as Activin A), bone morphogenetic protein 4 (BMP4), and neuregulin 1 (NRG1) have been implicated in regulating this differentiation (48,54,55,59). Based on their morphological characteristics SPCs are classified into A-single cell (As), A-paired (Apr) and A-aligned (Aal4, Aal8, Aal16) spermatogonia. A subpopulation of As cells is believed to function as SSCs (48,54,55,61). The Apr and Aal4-al16 transient amplifying cells are irreversibly committed to differentiation. This is associated with a gradual loss in self-renewal capacity and reduced expression of GFRα1 and NANOS2. The loss of the expression of these genes and that of FOXO1, PLZF, and E-cadherin (CDH1), together with the induction of C-KIT mark the differentiation of SPCs into differentiated spermatogonia, which subsequently undergo meiosis leading ultimately to the formation of mature spermatozoa.
In the developing testis, GLIS3 exhibits a specific spatiotemporal pattern of expression (23). GLIS3 mRNA expression was detected in the testis cord at E15.5 of mouse development and in early postnatal testis. GLIS3 protein is present at early stages of spermatogenesis, but undetectable in adult testis. At postnatal day 1 (PND1), GLIS3 protein is restricted to germ cells and only expressed in gonocytes, but not in somatic cells, including Sertoli and Leydig cells (23). At PND4 and 7, GLIS3 remains expressed in SSCs and PSCs; however, its level gradually decreases in Aal4-al16 SPCs and GLIS3 is no longer expressed in C-KIT+ differentiated spermatogonia. During SPC differentiation the decrease in GLIS3 protein expression occurs after the repression of GFRα1 and before the down-regulation of PLZF (Figure 2). Its expression pattern is consistent with the hypothesis that GLIS3 has regulatory functions in gonocytes, SSCs, and SPCs.
Figure 2.
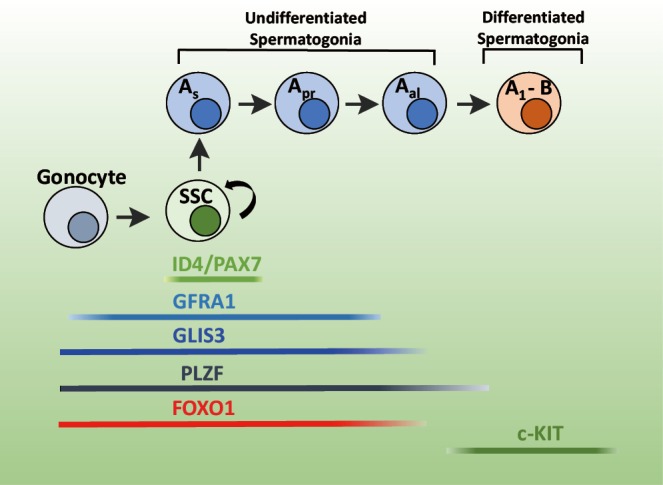
Schematic model of GLIS3 expression during neonatal spermatogenesis. After SSCs develop from gonocytes shortly after birth, they give rise to a series of SPCs (As, Apr, and Aal4-16), which then give rise to A and B type differentiated spermatogonia. GLIS3 is expressed in gonocytes, SSCs, and most SPCs. Its expression is gradually decreased during the Aal stage and absent in C-KIT+ spermatogonial cells. GLIS3 expression is compared to that of several other transcription factors. ID4 and PAX7 are enriched in SSCs. Loss of GLIS3 expression greatly inhibits the generation of SSCs and SPCs. SSCs, spermatogonial stem cells; SPCs, spermatogonial progenitor cells.
Study of the effect of GLIS3-deficiency on neonatal testis development showed that at birth the number of gonocytes was only slightly reduced in testes of Glis3KO mice compared to WT mice, whereas the number of GFRα1+ and PLZF+ SSCs and SPCs was significantly decreased in testes from PND7 Glis3KO mice (23). The loss of SSCs and SPCs appeared not to be due to increased apoptosis, but related to reduced proliferation and differentiation. As a consequence, this leads to a dramatic reduction in the generation of C-KIT+ differentiated spermatogonia as well as the subsequent development of spermatids and spermatozoa. Analysis of the subcellular localization of FOXO1 protein, which marks the gonocyte-SSC transition, showed that GLIS3-deficiency greatly impairs the translocation of FOXO1 to the nucleus suggesting that the reduced number of SSCs and SPCs in Glis3KO testes might in part be related to an inhibition of the differentiation gonocytes into SSCs. These observations suggest that GLIS3 might play a role in the regulation of the gonocyte-SSC transition.
Gene profiling analysis showed that at PND7 a large number of genes were dramatically decreased in Glis3KO testis compared to WT testis (23). These included several genes associated with SSC self-renewal and SPC differentiation, such as ETV5, RET, LHX1, POU5F1, BRACHYURY, SALL4, BCL6B, PIWIL4, GFRα1, and PLZF (23). Among them, ETV5, BCL6B, LHX1, and BRACHYURY are known downstream targets of the GFRΑ1/RET pathway. These results support a role for GLIS3 in SSC self-renewal and SPC differentiation. This might in part be related to the reduced expression of GFRα1, which is important in self-renewal and the generation of SPCs. These observations indicate that GLIS3 plays a critical role in the generation of SSCs and SPCs and is essential for spermatogenesis and male fertility. Identification of genes that are directly regulated by GLIS3, may provide greater insights into the molecular mechanism by which GLIS3 regulates the early stages of neonatal spermatogenesis.
GLIS3 function during pancreas development
A role for GLIS3 in pancreatic β-cells became first apparent from a study of human patients, in whom deletions in and around the GLIS3 gene were linked to a syndrome referred to as neonatal diabetes and congenital hypothyroidism (NDH), in 3 consanguineous families (17). More recently, additional deletions and point mutations in GLIS3 have been identified in individuals with NDH (22,62). In addition, a number of GWAS studies have linked SNPs in GLIS3 to Type 2 diabetes (63-66), Type 1 diabetes (24,67), and with abnormal β-cell function (25,68-70). Interestingly, GLIS3 is one of only a handful of genes that has been linked to both Type 1 and Type 2 Diabetes (71), highlighting its possible critical role in the control of both β-cell function and survival. This was further supported by the study of GLIS3-deficient mice, which develop hyperglycemia and hypoinsulinemia due to aberrant β cell generation and insulin gene expression (6,20,45,72-75).
Development of the mouse pancreas begins with a dorsal and ventral outgrowth from the pancreatic progenitor cells in dorsal foregut endoderm [reviewed in (76-78)]. Both outgrowths follow a similar lineage commitment, with branching morphogenesis leading by E12.5 to distinct cell populations in the “tip” and “trunk” compartments. The tip region contains multipotential pancreatic cells (MPCs), defined by their co-expression of PDX-1, PTF1A, NKX6.1, SOX9, and HNF1β, which can generate all pancreatic cell types and control overall pancreas size (79). By E14.5, these cells largely switch to a pro-acinar program and subsequently differentiate to mature acinar cells. The trunk domain, in which PDX1+SOX9+HNF1β+NKX6.1+PTF1A− bipotent progenitor cells reside, will give rise to both preductal cells, which subsequently differentiate into mature pancreatic ductal cells, and pro-endocrine cells. The latter will give rise to all 5 different lineages of hormone-producing endocrine cells (Figure 3). GLIS3 expression is first observed at the protein level in this trunk domain, but is never observed in the PTF1A+ tip domain (45). Although little is known about the mechanism that induces GLIS3 expression, the transcription factors HNF1β has been implicated in the regulation of GLIS3 transcription (80). Subsequently, during a period known as the “secondary transition”, cells within this trunk domain will make a lineage fate decision between ductal cells and endocrine cells, a process that is not totally understood. Induction of neurogenin 3 (NGN3) is linked to the commitment of bipotent progenitors to the pro-endocrine lineage (76-78). Notch signaling is thought to play an important role in this decision (81,82), possibly through upregulation of HES1, which by repressing NGN3 expression promotes ductal cell differentiation (76,83-86). Unlike several transcription factors that are restricted to either the ductal cells (such as HES1) or endocrine cells (such as NGN3), GLIS3 remains expressed in both endocrine and ductal cells (45,84,87). The latter indicates that GLIS3 may not play a major role in the duct/proendocrine lineage fate decision.
Figure 3.
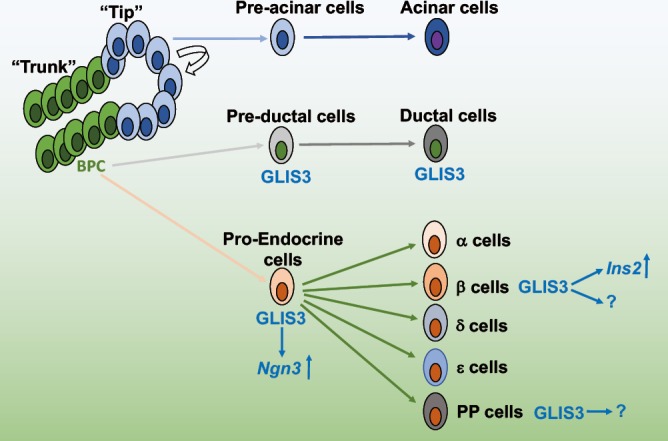
Schematic model of GLIS3 protein expression during pancreas development. GLIS3 is expressed early in the bipotent “trunk” domain of the developing pancreas, and maintains its expression in both pre-ductal and pro-endocrine cell types. In endocrine cells, GLIS3 becomes restricted to β-cells and PP-cells. BPC, bipotent progenitor cells.
GLIS3 knockout mice appear grossly normal, but develop severe hyperglycemia soon after birth and die within 10 days (7,20,21,74). Interestingly, overall pancreas size does not appear to be affected in knockout mice, supporting the observation that GLIS3 is not expressed in the MPCs that determine pancreas organ size (20,79). However, these mice do show a reduction in the number of all endocrine cell types as well as islets, suggesting that GLIS3 could play important roles in early pro-endocrine cell development. This is supported by findings showing that GLIS3 is expressed during this stage of lineage decision and participates in the direct regulation of NGN3 expression, which is essential for pro-endocrine cell development (20,45,88). Mice lacking NGN3 fail to develop all endocrine lineages (89), therefore, the reduced expression of NGN3 in GLIS3 knockout mice might at least in part explain the reduction in the number of islets and endocrine cells in these mice. Pro-endocrine cells delaminate from the trunk domain and form islet clusters through a poorly understood mechanism involving an EMT (90,91). During endocrine lineage determination, GLIS3 remains expressed in the insulin-producing β-cells and pancreatic polypeptide (PPY)-producing (PP) cells and becomes repressed in the other endocrine cell types. Whether GLIS3 plays any role in endocrine lineage determination requires further study. In pancreatic β and PP-cells, GLIS3 expression is maintained into adulthood, suggesting that GLIS3 has additional roles in regulating certain functions in these cells postnatally. This is supported by studies showing that GLIS3 is able to induce INS2 and PPY expression in cell lines and regulates INS2 directly by binding GLISBS sites in the INS2 proximal promoter (11,45,72,73). Collectively, these data indicate that GLIS3 has two important functions in pancreatic islets. An early role in promoting the bipotent progenitor-pro-endocrine cell transition, which involves direct regulation of NGN3 (and possibly other genes), and a role in mature β-cells where it is involved in maintaining β-cell identity and function as well as the regulation of INS2 expression (Figure 3). In addition, GLIS3 might also have a role in regulating the proliferation and survival of endocrine progenitors and or immature β cells.
Conclusions or future perspectives
As we have reviewed here, GLIS proteins have a variety of functions, both in stem cell maintenance and differentiation, and in the development of various tissues. GLIS1 greatly enhances reprogramming efficiency, whereas GLIS2 seems to have a role in regulating stem cell maintenance and differentiation in PSCs and hematopoietic precursors. GLIS3 plays a critical role in the development and maintenance of several different tissues. GLIS3 is essential for SSC renewal and differentiation, and seems to have a still largely undefined function in pancreatic bipotent progenitors. Moreover, GLIS3 plays key role in the generation of pancreatic pro-endocrine and endocrine cells, particularly insulin-producing β cells. A better understanding of the regulation of gene expression/transcription by GLIS proteins will provide greater insights into the mechanisms by which they control various biological processes and their roles in disease. GLIS proteins undergo a number of posttranslational modifications that might influence their interactions with other proteins, protein stability, and their transcriptional activity. The identification of the upstream signaling pathways that regulate GLIS activity will provide not only greater insights into the mechanism of action of GLIS proteins, but may also lead to new therapeutic strategies in the management of various diseases, in which GLIS proteins are implicated, such as diabetes.
Acknowledgements
Funding: This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, the National Institutes of Health (Z01-ES-100485 to AMJ).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Zhang F, Jetten AM. Genomic structure of the gene encoding the human GLI-related, Krüppel-like zinc finger protein GLIS2. Gene 2001;280:49-57. 10.1016/S0378-1119(01)00764-8 [DOI] [PubMed] [Google Scholar]
- 2.Lamar E, Kintner C, Goulding M. Identification of NKL, a novel Gli-Kruppel zinc-finger protein that promotes neuronal differentiation. Development 2001;128:1335-46. [DOI] [PubMed] [Google Scholar]
- 3.Zhang F, Nakanishi G, Kurebayashi S, et al. Characterization of Glis2, a novel gene encoding a Gli-related, Krüppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J Biol Chem 2002;277:10139-49. 10.1074/jbc.M108062200 [DOI] [PubMed] [Google Scholar]
- 4.Nakashima M, Tanese N, Ito M, et al. A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech Dev 2002;119:21-34. 10.1016/S0925-4773(02)00291-5 [DOI] [PubMed] [Google Scholar]
- 5.Kim YS, Kang HS, Herbert R, et al. Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol Cell Biol 2008;28:2358-67. 10.1128/MCB.01722-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang HS, ZeRuth G, Lichti-Kaiser K, et al. Gli-similar (Glis) Krüppel-like zinc finger proteins: insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol Histopathol 2010;25:1481-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lichti-Kaiser K, ZeRuth G, Kang HS, et al. Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease. Vitam Horm 2012;88:141-71. 10.1016/B978-0-12-394622-5.00007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lichti-Kaiser K, ZeRuth G, Jetten AM. Transcription factor GLI-similar 3 (GLIS3): Implications for the development of congenital hypothyroidism. J Endocrinol Diabetes Obes 2014;2:1024. [PMC free article] [PubMed] [Google Scholar]
- 9.Beak JY, Kang HS, Kim Y-S, et al. Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1). Nucleic Acids Res 2008;36:1690-702. 10.1093/nar/gkn009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasanth S, ZeRuth G, Kang HS, et al. Identification of nuclear localization, DNA binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2 (Glis2). J Biol Chem 2011;286:4749-59. 10.1074/jbc.M110.165951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ZeRuth GT , Takeda Y, Jetten AM. The Krüppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol Endocrinol 2013;27:1692-705. 10.1210/me.2013-1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SC, Kim YS, Jetten AM. Krüppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1). Nucleic Acids Res 2005;33:6805-15. 10.1093/nar/gki985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ZeRuth GT , Yang XP, Jetten AM. Modulation of the transactivation function and stability of Krüppel-like zinc finger protein Gli-similar 3 (Glis3) by Suppressor of Fused. J Biol Chem 2011;286:22077-89. 10.1074/jbc.M111.224964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramachandran H, Schäfer T, Kim Y, et al. Interaction with the Bardet-Biedl gene product TRIM32/BBS11 modifies the half-life and localization of Glis2/NPHP7. J Biol Chem 2014;289:8390-401. 10.1074/jbc.M113.534024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran H, Herfurth K, Grosschedl R, et al. SUMOylation Blocks the Ubiquitin-Mediated Degradation of the Nephronophthisis Gene Product Glis2/NPHP7. PloS One 2015;10:e0130275. 10.1371/journal.pone.0130275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ZeRuth GT , Williams JG, Cole YC, et al. HECT E3 Ubiquitin Ligase Itch Functions as a Novel Negative Regulator of Gli-Similar 3 (Glis3) Transcriptional Activity. PloS One 2015;10:e0131303. 10.1371/journal.pone.0131303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006;38:682-7. 10.1038/ng1802 [DOI] [PubMed] [Google Scholar]
- 18.Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet 2007;39:1018-24. 10.1038/ng2072 [DOI] [PubMed] [Google Scholar]
- 19.Kang HS, Beak JY, Kim YS, et al. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol 2009;29:2556-69. 10.1128/MCB.01620-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang HS, Kim YS, ZeRuth G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol 2009;29:6366-79. 10.1128/MCB.01259-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Chang BH, Yechoor V, et al. The Krüppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia 2011;54:2595-605. 10.1007/s00125-011-2255-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimitri P, Habeb AM, Gurbuz F, et al. Expanding the Clinical Spectrum Associated With GLIS3 Mutations. J Clin Endocrinol Metab 2015;100:E1362-9. 10.1210/jc.2015-1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HS, Chen LY, Lichti-Kaiser K, et al. Transcription Factor GLIS3: A New and Critical Regulator of Postnatal Stages of Mouse Spermatogenesis. Stem Cells 2016;34:2772-83. 10.1002/stem.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703-7. 10.1038/ng.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105-16. 10.1038/ng.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruber TA, Larson Gedman A, Zhang J, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 2012;22:683-97. 10.1016/j.ccr.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiollier C, Lopez CK, Gerby B, et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 2012;209:2017-31. 10.1084/jem.20121343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masetti R, Pigazzi M, Togni M, et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 2013;121:3469-72. 10.1182/blood-2012-11-469825 [DOI] [PubMed] [Google Scholar]
- 29.Maekawa M, Yamaguchi K, Nakamura T, et al. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011;474:225-9. 10.1038/nature10106 [DOI] [PubMed] [Google Scholar]
- 30.Kuo PH, Chuang LC, Su MH, et al. Genome-Wide Association Study for Autism Spectrum Disorder in Taiwanese Han Population. PloS One 2015;10:e0138695. 10.1371/journal.pone.0138695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deming Y, Li Z, Kapoor M, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 2017;133:839-56. 10.1007/s00401-017-1685-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-76. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 33.Plath K, Lowry WE. Progress in understanding reprogramming to the induced pluripotent state. Nat Rev Genet 2011;12:253-65. 10.1038/nrg2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maekawa M, Yamanaka S. Glis1, a unique pro-reprogramming factor, may facilitate clinical applications of iPSC technology. Cell Cycle 2011;10:3613-4. 10.4161/cc.10.21.17834 [DOI] [PubMed] [Google Scholar]
- 35.Yoshioka N, Gros E, Li H-R, et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 2013;13:246-54. 10.1016/j.stem.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009;460:1132-5. 10.1038/nature08235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Liang J, Ni S, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010;7:51-63. 10.1016/j.stem.2010.04.014 [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Han Q, Peng T, et al. The oncogene c-Jun impedes somatic cell reprogramming. Nat Cell Biol 2015;17:856-67. 10.1038/ncb3193 [DOI] [PubMed] [Google Scholar]
- 39.Baubec T, Schübeler D. Genomic patterns and context specific interpretation of DNA methylation. Curr Opin Genet Dev 2014;25:85-92. 10.1016/j.gde.2013.11.015 [DOI] [PubMed] [Google Scholar]
- 40.Pells S, Koutsouraki E, Morfopoulou S, et al. Novel Human Embryonic Stem Cell Regulators Identified by Conserved and Distinct CpG Island Methylation State. PloS One 2015;10:e0131102. 10.1371/journal.pone.0131102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci 2016;1370:82-96. 10.1111/nyas.13016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmfeldt P, Ganuza M, Marathe H, et al. Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J Exp Med 2016;213:433-49. 10.1084/jem.20150806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Rooij JDE, Hollink IHIM, Arentsen-Peters STCJM, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013;27:2280-8. 10.1038/leu.2013.87 [DOI] [PubMed] [Google Scholar]
- 44.Thirant C, Ignacimouttou C, Lopez CK, et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell 2017;31:452-65. 10.1016/j.ccell.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 45.Kang HS, Takeda Y, Jeon K, et al. The Spatiotemporal Pattern of Glis3 Expression Indicates a Regulatory Function in Bipotent and Endocrine Progenitors during Early Pancreatic Development and in Beta, PP and Ductal Cells. PloS One 2016;11:e0157138. 10.1371/journal.pone.0157138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim YS, Nakanishi G, Lewandoski M, et al. GLIS3, a novel member of the GLIS subfamily of Krüppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res 2003;31:5513-25. 10.1093/nar/gkg776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Alessio AC, Fan ZP, Wert KJ, et al. A Systematic Approach to Identify Candidate Transcription Factors that Control Cell Identity. Stem Cell Reports 2015;5:763-75. 10.1016/j.stemcr.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manku G, Wang Y, Merkbaoui V, et al. Role of retinoic acid and platelet-derived growth factor receptor cross talk in the regulation of neonatal gonocyte and embryonal carcinoma cell differentiation. Endocrinology 2015;156:346-59. 10.1210/en.2014-1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teletin M, Vernet N, Ghyselinck NB, et al. Roles of Retinoic Acid in Germ Cell Differentiation. Curr Top Dev Biol 2017;125:191-225. 10.1016/bs.ctdb.2016.11.013 [DOI] [PubMed] [Google Scholar]
- 50.Pui HP, Saga Y. Gonocytes-to-spermatogonia transition initiates prior to birth in murine testes and it requires FGF signaling. Mech Dev 2017;144:125-39. 10.1016/j.mod.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 51.Goertz MJ, Wu Z, Gallardo TD, et al. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest 2011;121:3456-66. 10.1172/JCI57984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aloisio GM, Nakada Y, Saatcioglu HD, et al. PAX7 expression defines germline stem cells in the adult testis. J Clin Invest 2014;124:3929-44. 10.1172/JCI75943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helsel AR, Yang QE, Oatley MJ, et al. ID4 levels dictate the stem cell state in mouse spermatogonia. Development 2017;144:624-34. 10.1242/dev.146928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oatley JM, Brinster RL. The germline stem cell niche unit in mammalian testes. Physiol Rev 2012;92:577-95. 10.1152/physrev.00025.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol 2013;29:163-87. 10.1146/annurev-cellbio-101512-122353 [DOI] [PubMed] [Google Scholar]
- 56.Nagano MC, Yeh JR. The identity and fate decision control of spermatogonial stem cells: where is the point of no return? Curr Top Dev Biol 2013;102:61-95. 10.1016/B978-0-12-416024-8.00003-9 [DOI] [PubMed] [Google Scholar]
- 57.Song HW, Wilkinson MF. Transcriptional control of spermatogonial maintenance and differentiation. Semin Cell Dev Biol 2014;30:14-26. 10.1016/j.semcdb.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griswold MD. Spermatogenesis: The Commitment to Meiosis. Physiol Rev 2016;96:1-17. 10.1152/physrev.00013.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hogarth CA. Transcriptional/translational regulation of mammalian spermatogenic stem cells. Adv Exp Med Biol 2013;786:105-28. 10.1007/978-94-007-6621-1_7 [DOI] [PubMed] [Google Scholar]
- 60.Buaas FW, Kirsh AL, Sharma M, et al. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet 2004;36:647-52. 10.1038/ng1366 [DOI] [PubMed] [Google Scholar]
- 61.Brinster RL, Avarbock MR. Germline transmission of donor haplotype following spermatogonial transplantation. Proc Natl Acad Sci USA 1994;91:11303-7. 10.1073/pnas.91.24.11303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alghamdi KA, Alsaedi AB, Aljasser A, et al. Extended clinical features associated with novel Glis3 mutation: a case report. BMC Endocr Disord 2017;17:14. 10.1186/s12902-017-0160-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 2011;44:67-72. 10.1038/ng.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakai K, Imamura M, Tanaka Y, et al. Replication study for the association of 9 East Asian GWAS-derived loci with susceptibility to type 2 diabetes in a Japanese population. PloS One 2013;8:e76317. 10.1371/journal.pone.0076317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muller YL, Piaggi P, Chen P, et al. Assessing variation across 8 established East Asian loci for type 2 diabetes mellitus in American Indians: Suggestive evidence for new sex-specific diabetes signals in GLIS3 and ZFAND3. Diabetes Metab Res Rev 2017;33. [DOI] [PubMed] [Google Scholar]
- 66.Rees SD, Hydrie MZI, O’Hare JP, et al. Effects of 16 genetic variants on fasting glucose and type 2 diabetes in South Asians: ADCY5 and GLIS3 variants may predispose to type 2 diabetes. PloS One 2011;6:e24710. 10.1371/journal.pone.0024710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Awata T, Yamashita H, Kurihara S, et al. A low-frequency GLIS3 variant associated with resistance to Japanese type 1 diabetes. Biochem Biophys Res Commun 2013;437:521-5. 10.1016/j.bbrc.2013.06.102 [DOI] [PubMed] [Google Scholar]
- 68.Boesgaard TW, Grarup N, Jørgensen T, et al. Variants at DGKB/TMEM195, ADRA2A, GLIS3 and C2CD4B loci are associated with reduced glucose-stimulated beta cell function in middle-aged Danish people. Diabetologia 2010;53:1647-55. 10.1007/s00125-010-1753-5 [DOI] [PubMed] [Google Scholar]
- 69.Hu C, Zhang R, Wang C, et al. Variants from GIPR, TCF7L2, DGKB, MADD, CRY2, GLIS3, PROX1, SLC30A8 and IGF1 are associated with glucose metabolism in the Chinese. PloS One 2010;5:e15542. 10.1371/journal.pone.0015542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong K-W, Chung M, Cho SB. Meta-analysis of genome-wide association study of homeostasis model assessment β cell function and insulin resistance in an East Asian population and the European results. Mol Genet Genomics 2014;289:1247-55. 10.1007/s00438-014-0885-6 [DOI] [PubMed] [Google Scholar]
- 71.Wen X, Yang Y. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J Mol Endocrinol 2017;58:R73-85. 10.1530/JME-16-0232 [DOI] [PubMed] [Google Scholar]
- 72.Yang Y, Chang BH, Samson SL, et al. The Krüppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res 2009;37:2529-38. 10.1093/nar/gkp122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Y, Chang BH, Chan L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol Med 2013;5:92-104. 10.1002/emmm.201201398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watanabe N, Hiramatsu K, Miyamoto R, et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett 2009;583:2108-13. 10.1016/j.febslet.2009.05.039 [DOI] [PubMed] [Google Scholar]
- 75.Yang Y, Bush SP, Wen X, et al. Differential Gene Dosage Effects of Diabetes-Associated Gene GLIS3 in Pancreatic β Cell Differentiation and Function. Endocrinology 2017;158:9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol 2013;29:81-105. 10.1146/annurev-cellbio-101512-122405 [DOI] [PubMed] [Google Scholar]
- 77.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 2011;240:530-65. 10.1002/dvdy.22584 [DOI] [PubMed] [Google Scholar]
- 78.Guney MA, Gannon M. Pancreas cell fate. Birth Defects Res C Embryo Today 2009;87:232-48. 10.1002/bdrc.20156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 2007;445:886-91. 10.1038/nature05537 [DOI] [PubMed] [Google Scholar]
- 80.De Vas MG, Kopp JL, Heliot C, et al. Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 2015;142:871-82. 10.1242/dev.110759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murtaugh LC, Stanger BZ, Kwan KM, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA 2003;100:14920-5. 10.1073/pnas.2436557100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greenwood AL, Li S, Jones K, et al. Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech Dev 2007;124:97-107. 10.1016/j.mod.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 83.Apelqvist A, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature 1999;400:877-81. 10.1038/23716 [DOI] [PubMed] [Google Scholar]
- 84.Jensen J, Pedersen EE, Galante P, et al. Control of endodermal endocrine development by Hes-1. Nat Genet 2000;24:36-44. 10.1038/71657 [DOI] [PubMed] [Google Scholar]
- 85.Lee JC, Smith SB, Watada H, et al. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 2001;50:928-36. 10.2337/diabetes.50.5.928 [DOI] [PubMed] [Google Scholar]
- 86.Ahnfelt-Rønne J, Jørgensen MC, Klinck R, et al. Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development 2012;139:33-45. 10.1242/dev.071761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002;129:2447-57. [DOI] [PubMed] [Google Scholar]
- 88.Kim YS, Kang HS, Takeda Y, et al. Glis3 regulates neurogenin 3 expression in pancreatic β-cells and interacts with its activator, Hnf6. Mol Cells 2012;34:193-200. 10.1007/s10059-012-0109-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gradwohl G, Dierich A, LeMeur M, et al. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000;97:1607-11. 10.1073/pnas.97.4.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rukstalis JM, Habener JF. Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expr Patterns 2007;7:471-9. 10.1016/j.modgep.2006.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cole L, Anderson M, Antin PB, et al. One process for pancreatic beta-cell coalescence into islets involves an epithelial-mesenchymal transition. J Endocrinol 2009;203:19-31. 10.1677/JOE-09-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]