

Summary
Generation of hematopoietic stem cells (HSCs) from pluripotent stem cells (PSCs) could potentially provide unlimited HSCs for clinical transplantation, a curative treatment for numerous blood diseases. However, to date, bona fide HSC generation has been largely unsuccessful in vitro. We have previously described proof of concept for in vivo HSC generation from PSCs via teratoma formation. However, our first-generation system was complex and the output low. Here, we further optimize this technology and demonstrate the following: (1) simplified HSC generation using transcription factor overexpression; (2) improved HSC output using c-Kit-deficient host mice, and (3) that teratomas can be transplanted and cryopreserved. We demonstrate that overexpression of Gfi1b, c-Fos, and Gata2, previously reported to transdifferentiate fibroblasts into hematopoietic progenitors in vitro, can induce long-term HSC formation in vivo. Our in vivo system provides a useful platform to investigate new strategies and re-evaluate existing strategies to generate HSCs and study HSC development.
Keywords: hematopoietic stem cell, induced pluripotent stem cell, teratomas, hemogenic endothelium, Gfi1b, cFos, Gata2
Graphical Abstract
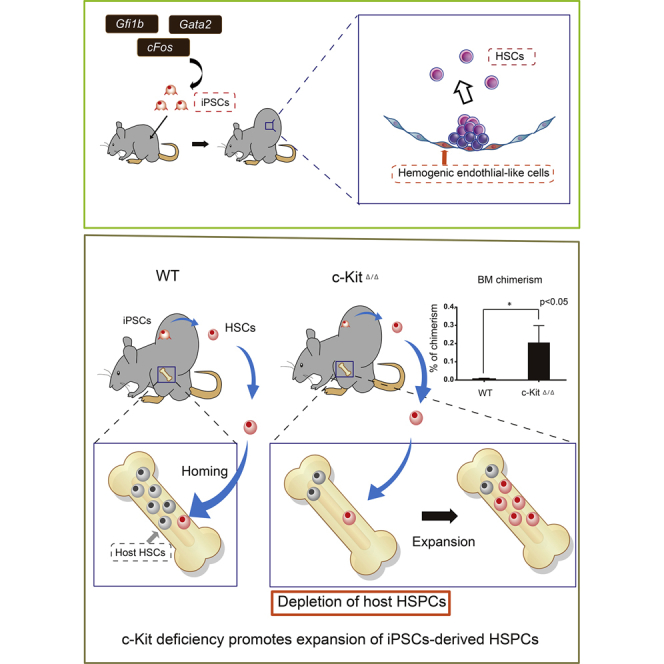
Highlights
-
•
iPSC overexpressing Gfi1b, c-Fos, and Gata2 (GFG) form HSC-producing teratomas
-
•
Hemogenic endothelial-like and peripheral blood cells emerge in GFG teratomas
-
•
Depletion of host HSCs promotes expansion of long-term, transplantable HSCs
-
•
Teratomas can be cryopreserved and transplanted
Tsukada et al. demonstrate that teratomas formed in vivo by Gfi1b-, c-Fos-, and Gata2-overexpressing iPSCs give rise to functional, long-term HSCs. Teratoma-derived HSCs and hematopoietic cells were detected in peripheral blood, expanded to the bone marrow after depletion of host HSCs, and showed successful engraftment in serial transplantation assays. This work underscores the importance of evaluating differentiation strategies in vivo.
Introduction
Pluripotent stem cells (PSCs) can differentiate into any bodily cell type. Somatic cells can also be reprogrammed to pluripotency by overexpression of “Yamanaka factors,” generating inducible PSCs (iPSCs) (Takahashi and Yamanaka, 2006). Optimization of these technologies over the past 10 years has enabled the safe generation of patient-specific iPSCs for prospective clinical applications. Now the major hurdle in the translational application of iPSC technologies is developing strategies to differentiate PSCs into clinically relevant functional cell types.
Adult hematopoietic stem cells (HSCs) reside in the bone marrow (BM) and have the unique ability to reform the entire blood system (Eaves, 2015). HSC function is defined by a cell-intrinsic transcriptional program as well as cell-extrinsic signaling emanating from the BM microenvironment. Long-term HSC function provides the basis of BM transplantation, a curative therapy for a range of hematological disorders. Derivation of functional HSCs from PSCs has therefore been a long-sought goal of regenerative medicine (Wahlster and Daley, 2016). However, we are currently unable to generate truly functional HSCs from PSCs in vitro. This is likely due to our inability to fully recapitulate the complex and temporally dynamic in vivo microenvironmental signaling program that specifies HSCs during development (Medvinsky et al., 2011). In mice, transplantable HSCs emerge from hemogenic endothelial (HE) precursors, notably in the dorsal aorta (Zovein et al., 2008). Expression of the transcription factor (TF) Runx1 can be used to track this process (North et al., 1999); live-cell imaging has captured hematopoietic stem/progenitor cell (HSPC) formation at sites of Runx1+ HE cells (Bertrand et al., 2010, Boisset et al., 2010).
Certain attempts to generate HSCs thus far have used transgene overexpression to either direct the differentiation of PSCs, or alternatively, to transdifferentiate other somatic cell types. Several approaches have been reported to generate hematopoietic cells in vitro (Doulatov et al., 2013, Riddell et al., 2014, Sandler et al., 2014). In two very recent reports, conversion of mouse and human endothelial cells to engraftable HSCs was achieved by overexpression of several TFs (Sugimura et al., 2017, Lis et al., 2017).
In another study, Pereira et al. (2013) reported that overexpression of three TFs (Gfi1b, c-Fos, and Gata2) could transdifferentiate mouse fibroblasts into HE-like intermediates that could further mature into hematopoietic progenitor cells (HPCs). However, fully functional HSCs were not generated by this method.
As an alternative, we (and others) have previously demonstrated proof of concept for the in vivo generation of fully functional HSCs from PSCs via teratoma formation (Suzuki et al., 2013, Amabile et al., 2013). However, our first-generation in vivo differentiation system (Suzuki et al., 2013) had several limitations: (1) PSCs needed to be co-injected with OP9 stromal cells and hematopoietic cytokines (SCF and TPO) administered via micropump; (2) we could not identify the site of HSC emergence; and; (3) HSC formation was slow, taking 2–3 months. Here, we systemically overcome these limitations and provide an optimized in vivo HSC formation protocol. Furthermore, we demonstrate that in vivo overexpression of Gfi1b, c-Fos, and Gata2 during teratoma formation is sufficient to generate functional long-term HSCs.
Results
Gfi1b, c-Fos, and Gata2 Overexpression Induces Hematopoietic Cell Formation in Teratomas
Teratomas contain tissues from all three germ layers, and we previously demonstrated that teratomas can generate HSCs (Suzuki et al., 2013). However, this required co-injection of OP9 stromal cells and continuous administration of cytokines (Suzuki et al., 2013). We hypothesized that induction of TFs related to HSCs and/or the HSC microenvironment could improve HSC generation in teratomas. To this end, we investigated three distinct TF combinations: (1) Gfi1b, c-Fos, and Gata2 (GFG), which have been reported to generate HE-like cells and HPCs from fibroblasts (Pereira et al., 2013); (2) Erg, HoxA9, and Rora (EAR), which have been reported to generate HPCs during PSC embryoid body formation (Doulatov et al., 2013), and (3) Foxc1, reported to be highly expressed in CXCL12-abundant reticular (CAR) cells, an important component of the HSC BM niche (Omatsu et al., 2014).
To enable the identification of iPSC-derived HSCs, we first established iPSCs from a C57BL/6-Ly5.1 mouse to allow immunophenotypic discrimination from C57BL/6-Ly5.2 host mouse blood cells. Next, we cloned the above TFs into a doxycycline (Dox)-inducible (Tet-On) lentivirus expression vector (Figure 1A). Stable iPSC lines were generated and Dox-inducible transgene expression was confirmed using qPCR (Figure S1A). Next, we injected C57BL/6-Ly5.1 iPSCs (2 × 106) subcutaneously into host C57BL/6-Ly5.2 mice, and administered Dox for 4 weeks once teratomas reached >2 cm in diameter (6–8 weeks post-injection) to induce TF expression (Figure 1B). Importantly, this was undertaken without co-injection of stromal cells or cytokines.
Figure 1.

Screening Transcription Factor Expression for Hematopoiesis within Teratomas
(A) Schematic diagram of the doxycycline (Dox)-inducible system for the expression of transcription factor (TF) combinations.
(B) Strategy to induce hematopoietic cells from lentivirally transduced iPSCs through teratoma formation. Dox was administrated for 4 weeks after teratoma size reached 2 cm in diameter, after which teratomas were collected for histological analysis.
(C) Representative H&E-stained teratoma sections, with morphological features labeled. Trabecular bone-like structure, TB (and black arrows); bone marrow cells, BMCs (and red arrows); endothelial cells, ECs (and white arrows); epithelial, EPCs (and black arrows).
(D) Representative flow cytometric plots of CD45.1 and CD45.2 expression in peripheral blood (PB) from teratoma-bearing Ly5.2 host mice at 14–16 weeks after Ly5.1 iPSC injection.
(E) Average percentage of CD45.1+ PB cells in host mice. Data are the means ± SD from two independent experiments (n = 6). ∗p < 0.05.
(F) Average percentages of hematopoietic cell lineages within the CD45.1+ population in (E). Gr-1+, granulocytes; Mac-1+, macrophages; B220+, B cells; CD4+, CD4+ T cells; and CD8+, CD8+ T cells. Data are means ± SD from two independent experiments (n = 3).
After the 4-week Dox exposure, we analyzed the peripheral blood (PB) of engrafted mice by flow cytometry and the teratoma by histology (Figure 1B). We observed distinct differences between teratomas generated in the different experimental regimens (Figures 1C and 1D). For example, the GFG iPSC-derived teratomas contained a large number of endothelial and epithelial-like cells by H&E staining (Figure 1C). By contrast, Foxc1-derived teratomas formed bone cortices, cartilage, and BM-like cells (Figure 1C). While CD45.1+ donor-derived blood cells were not detected in the host PB following injected with wild-type iPSCs or Foxc1 iPSCs, both EAR iPSCs and GFG iPSCs reconstituted multi-lineage hematopoiesis 14–18 weeks post-injection (Figures 1D and 1E), with GFG iPSCs generating approximately 2-fold more hematopoietic cells (Figures 1E and 1F). These data demonstrate that GFG iPSC-derived teratomas differentiate into hematopoietic cells more efficiently compared with the other groups.
To evaluate the potential consequences of the GFG cassette on HSCs, we generated GFG transgenic mice from GFG embryonic stem cells. Leaky expression could not be detected (Figure S1B), and no difference in colony-forming ability was seen (Figure S1C). Reactivation of the reprogramming factors could also not be detected in iPSC-derived CD45+ cells (Figure S1D).
Identification of Hemogenic Endothelium within GFG iPSC-Derived Teratomas
Given that GFG expression directly induces HE-like cells from mouse fibroblast in vitro (Pereira et al., 2013), we hypothesized that endothelial cells (ECs) within the GFG iPSC-derived teratomas (Figure 2A) might in fact resemble HE cells. By co-staining with Cytokeratin and CD31, we could identify CD31+ endothelial-lined cystic structures, which were also CD144/VE-cadherin+ (Figures S2A and S2B). We further confirmed the presence of Runx1-expressing ECs in GFG teratoma sections by immunostaining for Runx1 and CD31 (Figure 2B). Moreover, we could even identify hematopoietic cell clusters budding from these ECs (Figure 2B). CD45+ hematopoietic cells could also be identified within the endothelial structures, suggesting teratoma vasculature was perfused with blood (Figure S2C).
Figure 2.

Identification of Hematopoietic-Generating Tissue in Teratomas
(A) Representative images from GFG teratoma sections stained with H&E. These tissues contain a large number of endothelial-like cells (ECs), annotated with black arrows. Hematopoietic cells (HCs) appearing to directly bud from ECs are annotated with white arrows.
(B) Representative images from GFG iPSC-derived teratoma sections stained with Runx1 and CD31. Runx1+ cells were detected by DAB (brown color) and CD31+ were detected by Bajoran purple (Purple color), with sites of double staining identified as putative sites of hematopoietic cell emergence, as marked by white arrows.
(C) Representative images of teratoma tissue clearing in chemical cocktails and computational analysis (CUBIC); non-cleared on the left, cleared on the right.
(D) Localization of eR1+ cells (green) and DAPI (blue) in 2D reconstructed image between eR1-derived and eR1-GFG-derived teratoma.
(E) Localization of eR1+ cells (green) and DAPI (blue) in 3D reconstructed images between eR1-derived and eR1-GFG-derived teratoma.
(F) Representative flow cytometric plots for CD45 and GFP expression in the teratoma from eR1-EGFP iPSC-derived teratomas.
(G) Percentage GFP+CD45+ cells in teratomas, using gating displayed in (F) (n = 3). ∗p < 0.05.
Runx1 enhancer activity can be used to identify HE cells (Swiers et al., 2013). To study this in the teratoma, we established iPSCs from a eR1-EGFP transgenic reporter mouse (Ng et al., 2010). The activity of an enhancer for Runx1 (eR1), formerly called Runx1+24 or +23, is known to specifically mark HE cells within CD31+ endothelium in the embryo as well as committed CD45+ HSPCs (Nottingham et al., 2007). To comprehensively visualize all GFP+ cells within the teratoma, we applied tissue clearing and 3D volumetric imaging or CUBIC (Susaki et al., 2014) (Figure 2C). Using this method, we were further able to identify GFP+ endothelial structures within GFG-derived teratomas (Figures 2D and 2E). Notably, GFP+ cells were much more frequent within the GFG-derived teratomas than the controls (Figure 2E).
We further confirmed that eR1-EGFP-iPSCs were giving rise to GFP+CD45+ HSPCs by flow cytometric analysis of the teratomas. We detected a distinct GFP+CD45+ cell population, which was nearly 10-fold higher in GFG-derived teratomas compared with the controls (average value: control, 0.06% ± 0.02%; GFG, 0.8% ± 0.3%) (Figures 2F and 2G). Therefore, GFG expression induces eR1-EGFP expression within teratomas and generates GFP+CD45+ hematopoietic cells, similar to that described in vitro (Pereira et al., 2013). However, the low frequency of teratoma-derived blood cells in this system precluded our further analysis of HSC formation and function. It is worth noting that GFG induction in iPSCs could not induce eR1-GFP expression, or surface markers associated with HE or hematopoietic cell commitment (Figure S2D), suggesting hematopoietic cell induction following GFG expression occurred within the context of teratoma development/differentiation.
Conditional c-Kit Deficiency Promotes Expansion of iPSC-Derived HSPCs In Vivo
Adult HSCs take up lifelong residence in the BM. To improve teratoma-derived hematopoietic cell frequencies, we hypothesized that HSC-deficient hosts would promote expansion of iPSC-derived HSCs. HSCs can engraft in c-Kit-deficient mice without irradiation (Cosgun et al., 2014). We therefore generated host mice with inducible, hematopoietic-specific c-Kit deletion by breeding C57BL/6-Ly5.2 c-Kitflox/flox Mx1-Cre transgenic mice where c-Kit deletion was achieved by poly(I:C) administration (Figure S3A). Importantly, white blood cell, hemoglobin, and platelet counts were lower in c-Kit-deficient mice (c-KitΔ/Δ) than in the controls (Figure S3B), and the BM was hypocellular (Figure S3C). We further confirmed long-term multi-lineage donor reconstitution in Ly5.2 c-KitΔ/Δ hosts without irradiation following transplantation of 1 × 107 Ly5.1 BM donor cells (Figures S3D and S3E).
Having validated a mouse model that was permissive to HSC engraftment, we next wanted to determine whether the c-KitΔ/Δ model could improve teratoma-derived HSC formation. We injected GFG iPSCs subcutaneously into Mx1-Cre;c-Kitflox/flox host mice and administered poly(I:C) following teratoma formation (and Dox treatment) to deplete host HPSCs (Figure 3A). After 4–6 weeks, we observed a 10-fold increase in the frequency of iPSC-derived hematopoietic cells within the PB of the c-KitΔ/Δ hosts compared with control mice (control group, 0.02% ± 0.02%; c-Kit Δ/Δ, 0.3% ± 0.2%) (Figure 3B). We also found similar increases in iPSC-derived cell frequencies within the BM (control group, 0.01% ± 0.001%; c-KitΔ/Δ, 0.2% ± 0.1%) (Figure 3C). Notably, poly(I:C) did not influence the generation of teratoma-derived PB cells in wild-type mice (Figure S4A).
Figure 3.

c-Kit Deficiency Promotes Expansion of Hematopoietic Stem Cells from GFG-Derived iPSCs
(A) Strategy to induce HSCs from iPSCs using c-Kit deficient host mice. iPSCs were injected subcutaneously and Dox administered for 4 weeks to induce GFG expression (as in Figure 1B). Host mice were then administered polyI:C at weeks 14–16. At 20 weeks, WBMCs from teratoma-bearing mice were transplanted into irradiated mice, and PB/BM chimerism tracked. Secondary transplantations were performed at 16 weeks after the primary transplantation.
(B) Representative flow cytometric plots for CD45.1 and CD45.2 expression in the PB from teratoma-bearing C57BL/6 and c-Kit-deficient mice after 4–6 weeks poly(I:C) administration.
(C) Percentage CD45.1+ chimerism in the PB (left) and BM (right). Data are the means ± SD from two independent experiments (n = 6). ∗∗p < 0.005, ∗p < 0.05.
(D) Percentage of (GFG iPSC-derived) CD45.1+ PB chimerism of in primary and secondary recipient mice (n = 5).
(E and F) Colony potential of 2.0 × 104 host CD45.2+ and GFG-derived CD45.1+ BM cells of secondary recipients, after 11-day culture in Methocult. Total number of colonies displayed in (E) and colony type displayed in (F). Data are means ± SD from two independent experiments (n = 3). Colony cells were morphologically identified as neutrophils (n), macrophages (m), erythroblasts (E) and megakaryocytes (M). ∗∗p < 0.005.
(G) Representative flow cytometric plots and gating for the Kit+Sca1+Lineage− fraction within the BM of secondary recipient mice transplanted with GFG teratoma-derived blood cells 24 weeks post-transplantation. Lin−, lineage negative cells; KSL, Lin−Kit+Sca1+ cells.
To assess whether these hematopoietic cells contain engraftable HSCs, we transplanted 1 × 107 whole bone marrow cells (WBMCs) from teratoma-host mice into irradiated C57BL/6-Ly5.2 recipient mice (Figure 3A). We detected CD45.1+ PB cells in multiple lineages over 16 weeks (Figure 3D). To functionally confirm long-term HSC capacity within the original CD45.1+ cells, 1 × 107 WBMCs from the primary recipients were transplanted into irradiated secondary recipient mice. Multi-lineage CD45.1+ PB cells could be detected long-term in these secondary recipients (and even in tertiary recipients), demonstrating long-term HSC activity (Figures 3D, S4B, and S4C). In addition, we detected immunophenotypic CD45.1+C-KIT+SCA-1+Lineage− HPSCs in the BM of secondary recipients (Figure 3G). However, we noted that a 30% reduction in colony-forming units within the CD45.1+ WBMC population compared with CD45.2+ host BM cells, suggesting teratoma-derived BM cells are not identical to normally produced BM cells (Figures 3E, 3F, S4D, and S4E). However, these data demonstrate that GFG expression within teratomas generates functional HSCs that expand within c-Kit-deficient mice.
Transplantable Teratomas Provided Stable and Continuous Source of HSPCs
Finally, to improve on the time-consuming nature of these experiments, we investigated the following: (1) serial transplantation of freshly isolated teratomas and (2) use of cryopreservation of teratoma blocks. We performed transplantation of freshly isolated or cryopreserved teratoma blocks into host recipient mice (Figure 4A). In both cases, the transplanted teratomas survived and expanded (Figure 4B). Moreover, transplantable teratoma blocks generated hematopoietic cells that seeded the BM of c-KitΔ/Δ host mice (Figure 4C). Using this transplantation approach, we are able to shorten hematopoietic cell generation time by 5 weeks, from 10 to 5 weeks (Figure 4D). The frequency of teratomas that generated CD45.1+ blood cells was also higher in the transplanted hosts, compared with injection of iPSCs (Figure 4E). We also confirmed CD45.1+ WBMCs from these hosts could transplant into recipients (Figure S4F). Combined, these data demonstrate that transplantable teratomas constitute stable and continuous sources of hematopoietic cells.
Figure 4.

Transplantation and Freeze Preservation of Teratoma Blocks
(A) Strategy transplant and freeze-preserved teratomas for hematopoietic cell formation. Teratomas (26–30 weeks old) were divided into (1.5 × 1.5 × 1.5 cm) blocks and were transplanted into c-Kit-deficient mice with 4 weeks of Dox administration (with or without initial freeze preservation), followed by poly(I:C) administration.
(B) Average diameter of the transplanted teratoma blocks after transplantation (n = 3).
(C) Representative flow cytometric plots for CD45.1 and CD45.2 from the PB of host mice 5–7 weeks after transplantation with GFG teratoma blocks (n = 5).
(D) Average percentage CD45.1+ PB cells in host mice following transplantation with teratoma blocks, frozen blocks, or iPSCs. Data are the means ± SD from two independent experiments (n = 6 iPSCs, n = 16 teratoma blocks, n = 3 frozen blocks).
(E) Frequency of teratomas giving rise to CD45.1 PB cells from transplanted iPSCs and teratoma blocks.
Discussion
Here, we report an optimized system to generate HSC in vivo via teratoma formation for faster, higher-efficiency HSC production from iPSCs without co-injection of stromal cells or continuous provision of cytokines. While Pereira et al. (2013) were able to generate HPCs from fibroblasts in vitro by overexpressing GFG, transplantable HSCs could not be detected. Here, we have demonstrated that within an in vivo environment (iPSC-derived teratomas), GFG expression generated functional long-term HSCs. Our data therefore highlight the importance of in vivo evaluation of directed and transdifferentiation approaches. We envision in vivo HSC generation via teratoma formation as an important assay to assess new (and existing) approaches to generate engraftable HSCs. We also highlight the potential of our system to study mechanisms of HSC generation using an eR1-EGFP reporter system (Ng et al., 2010). In summary, we demonstrate that GFG overexpression in vivo within mouse teratomas can generate fully functional HSCs.
Experimental Procedures
Mice
C57BL/6-Ly5.2 (Ly5.2) and C57BL/6-Ly5.1 (Ly5.1) mice were purchased from Japan SLC (Shizuoka, Japan) and Sankyo Lab Service (Tsukuba, Japan), respectively. For generation of Mx1-Cre c-Kitflox/flox mice, c-Kitlox66-71/lox66-71 mice (Kimura et al., 2011) were crossed with Mx1-Cre mice transgenic mice (Kuhn et al., 1995). Mice received 250 μg of poly(I:C) intraperitoneally seven times every other day to induce Cre expression. Animal experiments were approved by the Animal Care and Use Committee, Institute of Medical Science, University of Tokyo.
Cell Culture
Mouse iPSCs were obtained by reprogramming tail-tip fibroblasts of C57BL/6-Ly5.1 mice with Oct4, Sox2, and Klf4, and then maintained as previously described (Nakagawa et al., 2008).
Lentivirus
Inducible lentivirus vectors were derived from the self-inactivating lentiviral vector CS-TRE-PRE-Ubc-tTA-I2G (Yamaguchi et al., 2012). Lentiviral production was performed as previously described (Shibuya et al., 2003).
Reverse Transcription PCR
Total RNA was extracted using TRIzol (Invitrogen) and reverse transcribed (High Capacity cDNA RT Kit; Applied Biosystems). qPCR was performed using the Universal Probe Library System (Roche Diagnostics; see Table S1). Samples were normalized to Gadph expression, using TaqMan probes (Applied Biosystems).
Teratoma Formation
Two million murine iPSCs were administered subcutaneously into C57BL/6(Ly5.2) or Mx1-Cre c-Kitflox/flox mice (6–10 weeks old) with Matrigel. We measured teratoma growth every month and analyzed the ratio of CD45.1+ cells in PB every month. Where indicated, TF expression was induced after teratoma formation by Dox (2 μg/mL) administration via the water supply for 4 weeks. After 4 weeks of Dox administration, mice received poly(I:C) intraperitoneally.
BM Transplantation Assay
BM cells (1 × 107) from teratoma-bearing mice were transplanted into lethally irradiated (9.5 Gy) wild-type C57BL/6-Ly5.2 recipient mice. BM cells (1 × 107) from the primary recipient mice were transplanted 16 weeks after primary BM transplantation into lethally irradiated secondary recipients.
Flow Cytometry
PB and BM were stained with anti-mouse antibodies as detailed in Table S2. Analysis was performed on a FACSCanto using Summit software (Beckman Coulter), and the results were analyzed with FlowJo.
Colony Assays
BM cells (20,000) were added to 1.1 mL of MethoCult GF M3434 (STEMCELL Technologies) and cultured at 37°C with 5% CO2. Colonies were counted after 11 days, cytospun onto slide glasses, and subjected to Hemacolor staining (Merck) for morphological examination.
Histopathology/Immunohistochemistry
Histological findings were assessed by H&E-stained sections of paraffin-embedded teratoma tissue. For immunohistochemical analysis of teratomas, sections were stained with anti-Runx1 monoclonal Ab (Abcam92336) followed by anti-rabbit IgG-HRP. Antibody staining was detected by DAB staining followed by counterstaining with hematoxylin. After heating the microscope slides to 95°C for 5 min, sections were stained with anti-CD31 monoclonal Ab (SPRING BIOSCIENCE SP38) followed by anti-rabbit IgG-HRP. Antibody staining was detected by Bajoran Purple Chromogen Kit (Biocare Medical).
3D Teratoma Imaging
Teratoma clearing protocols were slightly modified from the original CUBIC protocol (Susaki et al., 2014). Teratomas were collected and fixed in 4% paraformaldehyde solution for 2 days. For nuclear staining, teratomas were immersed in DAPI/PBS solution at 37°C for 3 days with gentle shaking. Tissues were embedded in 4 mm diameter glass capillaries with 2% agarose for imaging. Images were acquired using a Zeiss Z1 Lightsheet microscope (Zeiss) and reconstituted into 3D images using Zen software (Zeiss).
Statistics
Calculations of statistically significant differences between samples using the Student's two-tailed t test were performed using Prism 4 software. Error bars displayed are the SD.
Author Contributions
M.T., Y.O., and S.Y. planned and performed the experiments and wrote the manuscript. M.O. helped perform experiments. A.C.W., H.J.B., and H.N. provided scientific discussion and technical support. S.Y., H.B., A.W., and H.N. directed the study and wrote the manuscript.
Acknowledgments
We thank Y. Yamazaki, Y. Ishii, R. Ishida, K. Ito, S. Rafii, M. Otsu, and R. Yamamoto for support and advice. This work was supported by grants from the Japan Society for the Promotion of Science (JSPS) (grant no. 50625580), the Ministry of Education, Culture, Sport, Science, and Technology (Japan), the California Institute of Regenerative Medicine (grant no. LA1-06917), the Siebel Foundation, and the Ludwig Foundation. A.C.W. is a Bloodwise Visiting Fellow.
Published: September 21, 2017
Footnotes
Supplemental Information includes four figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.08.010.
Contributor Information
Hiromitsu Nakauchi, Email: nakauchi@ims.u-tokyo.ac.jp.
Satoshi Yamazaki, Email: y-sato4@ims.u-tokyo.ac.jp.
Supplemental Information
References
- Amabile G., Welner R.S., Nombela-Arrieta C., D’Alise A.M., Di Ruscio A., Ebralidze A.K., Kraytsberg Y., Ye M., Kocher O., Neuberg D.S. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–1264. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand J.Y., Chi N.C., Santoso B., Teng S., Stainier D.Y.R., Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset J.-C., van Cappellen W., Andrieu-Soler C., Galjart N., Dzierzak E., Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464:116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- Cosgun K.N., Rahmig S., Mende N., Reinke S., Hauber I., Schäfer C., Petzold A., Weisbach H., Heidkamp G., Purbojo A. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell. 2014;15:227–238. doi: 10.1016/j.stem.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Doulatov S., Vo L.T., Chou S.S., Kim P.G., Arora N., Li H., Hadland B.K., Bernstein I.D., Collins J.J., Zon L.I. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves C.J. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125:2605–2613. doi: 10.1182/blood-2014-12-570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y., Ding B., Imai N., Nolan D.J., Butler J.M., Rafii S. c-Kit-mediated functional positioning of stem cells to their niches is essential for maintenance and regeneration of adult hematopoiesis. PLoS One. 2011;6:e26918. doi: 10.1371/journal.pone.0026918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R., Schwenk F., Aguet M., Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Lis R., Karrasch C.C., Poulos M.G., Kunar B., Redmond D., Duran J.G.B., Badwe C.R., Schachterle W., Ginsberg M., Xiang J. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. 2017;545:439–445. doi: 10.1038/nature22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A., Rybtsov S., Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. 2011;138:1017–1031. doi: 10.1242/dev.040998. [DOI] [PubMed] [Google Scholar]
- Nakagawa M., Koyanagi M., Tanabe K., Takahashi K., Ichisaka T., Aoi T., Okita K., Mochiduki Y., Takizawa N., Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Ng C.E.L., Yokomizo T., Yamashita N., Cirovic B., Jin H., Wen Z., Ito Y., Osato M. A Runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells. 2010;28:1869–1881. doi: 10.1002/stem.507. [DOI] [PubMed] [Google Scholar]
- North T., Gu T.L., Stacy T., Wang Q., Howard L., Binder M., Marín-Padilla M., Speck N.A. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- Nottingham W.T., Jarratt A., Burgess M., Speck C.L., Cheng J.-F., Prabhakar S., Rubin E.M., Li P.-S., Sloane-Stanley J., Kong-A-San J. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood. 2007;110:4188–4197. doi: 10.1182/blood-2007-07-100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y., Seike M., Sugiyama T., Kume T., Nagasawa T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 2014;508:536–540. doi: 10.1038/nature13071. [DOI] [PubMed] [Google Scholar]
- Pereira C.F., Chang B., Qiu J., Niu X., Papatsenko D., Hendry C.E., Clark N.R., Nomura-Kitabayashi A., Kovacic J.C., Ma’Ayan A. Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell. 2013;13:205–218. doi: 10.1016/j.stem.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J., Gazit R., Garrison B.S., Guo G., Saadatpour A., Mandal P.K., Ebina W., Volchkov P., Yuan G.C., Orkin S.H. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler V.M., Lis R., Liu Y., Kedem A., James D., Elemento O., Butler J.M., Scandura J.M., Rafii S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature. 2014;511:312–318. doi: 10.1038/nature13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya K., Shirakawa J., Kameyama T., Honda S.-I., Tahara-Hanaoka S., Miyamoto A., Onodera M., Sumida T., Nakauchi H., Miyoshi H. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J. Exp. Med. 2003;198:1829–1839. doi: 10.1084/jem.20030958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R., Jha D.K., Han A., Soria-Valles C., da Rocha E.L., Lu Y.-F., Goettel J.A., Serrao E., Rowe R.G., Malleshaiah M. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545:432–438. doi: 10.1038/nature22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susaki E.A., Tainaka K., Perrin D., Kishino F., Tawara T. Resource whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157:726–739. doi: 10.1016/j.cell.2014.03.042. [DOI] [PubMed] [Google Scholar]
- Suzuki N., Yamazaki S., Yamaguchi T., Okabe M., Masaki H., Takaki S., Otsu M., Nakauchi H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 2013;21:1424–1431. doi: 10.1038/mt.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiers G., Baumann C., O’Rourke J., Giannoulatou E., Taylor S., Joshi A., Moignard V., Pina C., Bee T., Kokkaliaris K.D. Early dynamic fate changes in haemogenic endothelium characterized at the single-cell level. Nat. Commun. 2013;4:2924. doi: 10.1038/ncomms3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Wahlster L., Daley G.Q. Progress towards generation of human haematopoietic stem cells. Nat. Cell Biol. 2016;18:1111–1117. doi: 10.1038/ncb3419. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Hamanaka S., Kamiya A., Okabe M., Kawarai M., Wakiyama Y., Umino A., Hayama T., Sato H., Lee Y.S. Development of an all-in-one inducible lentiviral vector for gene specific analysis of reprogramming. PLoS One. 2012;7:e41007. doi: 10.1371/journal.pone.0041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovein A.C., Hofmann J.J., Lynch M., French W.J., Turlo K.A., Yang Y., Becker M.S., Zanetta L., Dejana E., Gasson J.C. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008;3:625–636. doi: 10.1016/j.stem.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.