

Abstract
Stroke, characterized by a disruption of blood supply to the brain, is a major cause of morbidity and mortality worldwide. Although humanin, a 24-amino acid polypeptide, has been identified to have multiple neuroprotective functions, the level of humanin in plasma has been demonstrated to decrease with age, which likely limits the effects against stroke injury. A potent humanin analogue, S14G-humanin (HNG), generated by replacement of Ser14 with glycine, has been demonstrated to have 1,000-fold stronger biological activity than humanin. The present study established an in vitro oxygen glucose deprivation/reoxygenation (OGD/R) model using SH-SY5Y neuroblastoma cells to mimic the in vivo ischemia/reperfusion injury in stroke. Adding HNG (0–10 µg/l) to SH-SY5Y cells to different extents blocked OGD/R-induced reduction of cell viability and antioxidative capacity, as well as decreased the elevated apoptosis rate induced by OGD/R, with the most evident effects at 1 µg/l HNG. Janus kinase 2 (Jak2)/signal transducer and activator of transcription 3 (Stat3) signaling was attenuated in OGD/R processes, yet reactivated with HNG treatment. FLLL32 (5 µM), a specific inhibitor of the signal, abolished effects of HNG on anti-apoptosis and antioxidation in OGD/R processes. Co-treatment with HNG and FLLL32 failed to interrupt upregulation of cytochrome c, B-cell lymphoma 2-associated X protein and cleaved caspase-3 provoked by OGD/R. Similar to FLLL32, Jak2/Stat3 signaling activated by HNG was also repressed by inhibitor of phosphoinositide 3-kinase (PI3K; 10 µM LY294002) or protein kinase B (AKT; 5 µM MK-2206 2HCl). These data collectively indicated that HNG has neuroprotective effects against OGD/R by reactivating Jak2/Stat3 signaling through the PI3K/AKT pathway, suggesting that HNG may be a promising agent in the management of stroke.
Keywords: humanin analogue, oxygen glucose deprivation/reoxygenation, Janus kinase 2/signal transducer and activator of transcription 3, phosphoinositide 3-kinase/protein kinase B
Introduction
Stroke is a devastating disease that results from abnormal blood supply to the brain. It represents one of the leading cause of mortality and disability worldwide (1). According to the World Health Organization, 15 million individuals suffer from stroke worldwide each year. Of these, 5 million succumb and a further 5 million are permanently disabled (1). However, no effective approach has been established to prevent or treat this brain damage (2). Stroke may be classified into ischemic stroke and hemorrhagic stroke. The former represents the majority of cases in clinical data and occurs when blood flow is obstructed in brain arteries by blood clots (3). The latter is caused by the rupture of blood vessels, for instance in trauma (3). Cerebral ischemia in strokes blocks delivery of oxygen and nutrients, and the brain has a considerably low tolerance to lack of these substances as it does not store oxygen and glycogen, but has a high oxygen consumption rate and rapid metabolism (4,5). Although rapid restoration of blood supply normalizes the demands for physiological processes, this reperfusion gives rise to other serious problems, such as oxidative stress, inflammatory responses, mitochondrial dysfunction and ion imbalances, which paradoxically antagonize the beneficial effect, resulting in more severe pathological state directly related to the intensity of stroke injury (2). Therefore, suppression of a series of disastrous effects induced by ischemia/reperfusion is essential for the treatment of strokes.
Humanin, a 24-amino acid polypeptide, was discovered by Hashimoto et al (6) in 2001 through a cDNA library survey in surviving occipital lobe neurons of a human patient with Alzheimer's disease. Humanin is encoded from the 16S ribosomal RNA (rRNA) region of the mitochondrial genome, thus it is one of the few mitochondrial derived peptides (7). Human mitochondrial DNA contains 37 genes encoding 13 proteins, 22 transfer RNAs, and 2 rRNAs (7). Previous results have revealed that humanin exerts various protective actions in multiple cell types and animal models. A study by Sreekumar et al (8) demonstrated that humanin functions to protect human retinal pigment epithelial cells from oxidative stress, senescence and mitochondrial dysfunction partly by Janus kinase 2 (Jak2)/signal transducer and activator of transcription 3 (Stat3) signal activation and caspase-3 inhibition. Similarly, humanin rescues Stat3 from amyloid β (Aβ)-induced disruption and causes the downregulation of caspase-3, thereby exerting neuroprotective actions against Aβ toxicity in rats (9). Cardiac injury induced by ischemia/reperfusion is also attenuated by humanin analogue pretreatment, as evidenced by decreased arrhythmia incidence, cardiac dysfunction and infarct size, as well as improved cardiac mitochondrial function (10). These data raised the hypothesis that humanin may have neuroprotective functions counteracting adverse effects induced by ischemia/reperfusion. However, humanin, which is secreted from cells, decreases with age in human plasma levels, while stroke incidence notably increases with age and culminates in elderly individuals (11). This may lead to limited protection by humanin at physiological levels from stroke in the elderly, even though humanin may have the ability to counteract stroke impairment.
A potent humanin analogue, S14G-humanin (HNG), is generated by replacement of Ser14 with glycine, and this substitution potently increases humanin activity (12). The present study used HNG to evaluate its potential neuroprotective effects against ischemia/reperfusion in vitro and to identify the molecular mechanism underlying this protection. The present study demonstrated that HNG may be a promising agent for neuroprotection from ischemia/reperfusion injury via humanin-analogous function.
Materials and methods
Cell culture and treatments
SH-SY5Y is a human-derived neuroblastoma cell line widely used for in vitro models that mimic neurons in physiological and pathological states. Recently, a study claimed that human SH-SY5Y cells may have been mixed with SH-SY5Y cells from mice, resulting in the potential contamination of human SH-SY5Y cells (13). However, the present study did not involve differences between human and murine species; thus, the present study is not likely influenced by the use of human SH-SY5Y cells in case they were contaminated with murine SH-SY5Y cells.
SH-SY5Y cells used in the present study were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were initially maintained in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and streptomycin in a 5% CO2 incubator at 37°C, as a routine cell culture method. After 70–80% confluence was reached, the cells were subjected to oxygen glucose deprivation/reoxygenation (OGD/R) processes in the presence or absence of HNG (0.01–10 µg/l, H6161; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 37°C. For OGD/R, cells were cultured for 16 h in culture medium with deprivation of glucose and serum in a sealed chamber that created a hypoxic condition (0.5% O2) at 37°C. Subsequently, the cells were maintained under normoxic conditions (reoxygenation, at 37°C) in normal medium for an additional 9 h. FLLL32 (S7259), LY294002 (S1105) and MK-2206 2HCl (S1078; all Selleck Chemicals, Houston, TX, USA) are specific inhibitors of Stat3, PI3K and AKT, respectively. These inhibitors (5 µM FLLL32, 10 µM LY294002 or 5 µM MK-2206 2HCl) were added to the cells during OGD/R processes at 37°C.
Cell viability assay
SH-SY5Y cell viability was evaluated with a cell counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Briefly, the cells were seeded at 1×104 cells/well in 96-well plates and cultured for 48 h at 37°C before the cells were subjected to the aforementioned treatments. Subsequently, 10 µl CCK-8 solution was added to each well and the plates were incubated for an additional 5–8 h at 37°C. The optical density of each well was measured using a Synergy HT microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) at 450 nm.
Analysis of apoptosis rate
Cells were collected by centrifugation (800 × g) at room temperature for 10 min. The apoptosis rate of SH-SY5Y cells was evaluated by staining with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) using an Annexin V-FITC Apoptosis Detection kit I (BD Biosciences, San Jose, CA, USA). In brief, 5 µl Annexin V-FITC and 5 µl propidium iodide were added to the cells, followed by incubation for 15 min at room temperature in the dark. The extent of apoptosis was analyzed with a dual laser flow cytometer (BD Biosciences) and estimated using ModFit LT software version 2.0 (Verity Software House, Inc., Topsham, ME, USA).
Measurement of superoxide dismutase (SOD) activity and methane dicarboxylic aldehyde (MDA) concentration in SH-SY5Y cells
SOD activity in the SH-SY5Y cells was measured using SOD activity detection kits (cat. no. KGT00150-2, Nanjing KeyGen Biotech Co., Ltd., Nanjing, China), according to the manufacturer's protocol. The detection method was based on the reaction principle that SOD existed in our samples and inhibited the process of superoxide transforming WST-8, a 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt, to a stable water-soluble WST-8 formazan. The presence of WST-8 formazan may be evaluated through testing the optical density at 450 nm. SOD activity was determined by calculating the inhibition rate of generation of WST-8 formazan.
For the MDA assay, 1 ml 0.6% thiobarbituric acid was added to 1 ml supernatant (centrifugation at 3,200 × g, at 4°C for 20 min) of the SH-SY5Y cell lysate in a 2.5-ml tube. The tube was subsequently placed in boiling water for 15 min and then placed on ice to cool down (~20 min), before the optical density was measured at 532 nm. The results were expressed in nmol MDA/g protein.
Western blot analysis
Total protein (20 µg/lane), which was extracted from the SH-SY5Y cells with radioimmunoprecipitation assay lysis reagent (Sigma-Aldrich; Merck KGaA), was separated on 10–12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Merck KGaA). The membrane was blocked with 5% non-fat milk overnight at 4°C. Following this, the proteins were probed with the following primary antibodies: Jak2 (1:1,000; ab108596; Abcam, Cambridge, UK), phosphorylated (p)-Stat3 Y705 site (1:1,000; 9131; CST Biological Reagents Co., Ltd., Shanghai, China), p-Stat3 S727 site (1:500; ab30647; Abcam), cytochrome c (1:200; ab53056; Abcam), B-cell lymphoma 2-associated X protein (Bax; 1:300; sc-493; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), caspase-3 (1:1,000; 19677–1-AP; ProteinTech Group, Inc., Chicago, IL, USA) and β-actin (1:800; sc-47778; Santa Cruz Biotechnology, Inc.) at 37°C for 2 h. The membranes were washed with TBS-Tween-20 three times (10 min each) at room temperature. Then, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies that target mouse immunoglobulin (Ig)G (ab97040; Abcam) or rabbit IgG (A0545; Sigma) at a dilution of 1:2,000 for 2 h at room temperature. Reactive proteins were detected using Pierce Enhanced Chemiluminescent and SuperSignal™ Chemiluminescent substrates (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The blots on the membranes were scanned to quantify the proteins (Odyssey; LI-COR Biosciences, Lincoln, NE, USA and AlphaView 3.0 system; ProteinSimple; Bio-Techne, Minneapolis, MN, USA).
Statistical analysis
All values were expressed as mean ± standard error of the mean. Differences between groups were analyzed by one-way analysis of variance with SPSS 13.0 (SPSS, Inc., Chicago, IL, USA), followed by Bonferroni post hoc analyses, as appropriate. P<0.05 was considered to indicate a statistically significant difference.
Results
HNG has neuroprotective functions against OGD/R
To determine the neuroprotective functions of HNG against OGD/R, SH-SY5Y cells were subjected to OGD/R in the presence or absence of HNG. Cell viability tests demonstrated that OGD/R induced significant reduction of SH-SY5Y cell viability compared with the control (P<0.05; Fig. 1A), while adding doses of HNG (0.01–10 µg/l) to the cells to some extent inhibited OGD/R-induced reduction in the cell viability. SH-SY5Y cells incubated with 1, 5 or 10 µg/l HNG had significantly higher cell viability than the cells without HNG treatment in the OGD/R process (P<0.05). OGD/R also resulted in a significant increase in apoptosis of SH-SY5Y cells compared with the control (P<0.05; Fig. 1B and C), and incubation with doses of HNG decreased cell apoptosis. Notably, 1 µg/l HNG significantly decreased apoptosis by ~60% in OGD/R, compared with the group without HNG addition (P<0.05). These data indicated that HNG has cytoprotective and anti-apoptotic functions in neural cells under OGD/R status.
Figure 1.

HNG recovers SH-SY5Y cell viability and inhibits apoptosis in OGD/R. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. In OGD/R processes, the cells were incubated with doses of HNG (0.01–10 µg/l). (A) Cell viability and (B and C) apoptosis assays were performed following OGD/R treatments. *P<0.05 vs. Con; #P<0.05 vs. 0 µg/l HNG OGD/R group. OGD/R, oxygen glucose deprivation/reoxygenation; HNG, S14G-humanin; FITC, fluorescein isothiocyanate; Con, control group.
HNG inhibits oxidative stress induced by OGD/R
SOD is an important enzyme that eliminates oxygen free radicals in neurons (14), thus SOD activity is widely used as a marker to evaluate anti-oxidative ability of cells. SOD activity in SH-SY5Y cells was significantly reduced following OGD/R compared with control cells (P<0.01; Fig. 2A). Adding 1, 5 or 10 µg/l HNG to the cells significantly increased SOD activity in cells compared with cells not treated with HNG (P<0.05), with SOD activity peaking at 1 µg/l.
Figure 2.

HNG improves the anti-oxidative capacity of SH-SY5Y cells in OGD/R. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. In OGD/R processes, the cells were incubated with doses of HNG (0.01–10 µg/l). (A) SOD activity and (B) MDA concentration in the SH-SY5Y cells were detected after the OGD/R treatments. **P<0.01 vs. Con; #P<0.05 vs. 0 µg/l HNG OGD/R group. OGD/R, oxygen glucose deprivation/reoxygenation; HNG, S14G-humanin; SOD, superoxide dismutase; MDA, methane dicarboxylic aldehyde; Con, control group.
MDA is the final oxidation product of unsaturated fatty acids in cells (14). As unsaturated fatty acids are more vulnerable to oxidative species than proteins and nucleic acids, the generated MDA becomes a sensitive indicator of oxidative status in cells (14). MDA concentration in SH-SY5Y cells significantly increased in the OGD/R process compared with that in cells of the control treatment (P<0.01; Fig. 2B), while supplement with 1, 5 or 10 µg/l HNG significantly inhibited the increase in MDA concentration compared with cells not treated with HNG (P<0.05). These results indicated that HNG inhibited the reduction in anti-oxidative ability triggered by OGD/R and the generation of oxidative species.
HNG reactivates the Jak2/Stat3 signaling pathway that is inhibited in OGD/R processes
It was reported that the Jak2/Stat3 signaling pathway underlies the multiple protective mechanisms of humanin (8,9). Thus, the present study detected the expression levels of Jak2 and p-Stat3 following treatment with HNG in OGD/R conditions. As demonstrated in Fig. 3, OGD/R significantly lowered protein levels of Jak2 (P<0.05) and p-Stat3 at the Y705 site (P<0.05) compared with the control. Adding 0.1 µg/l HNG to the cells significantly increased protein levels of Jak2 (P<0.05) and p-Stat3 at the Y705 site (P<0.05) in OGD/R conditions compared with cells not treated with HNG. Furthermore, protein levels of Jak2 (P<0.01), p-Stat 3 at the Y705 site (P<0.05) and p-Stat3 at the S727 site (P<0.05) demonstrated significant increases with treatment with 1 and 5 µg/l HNG in OGD/R conditions compared with cells not treated with HNG. Although 10 µg/l HNG still significantly increased Jak2 protein levels (P<0.05) compared with cells not treated with HNG, it failed to significantly elevate p-Stat3 levels at the Y705 and S727 sites.
Figure 3.

HNG reactivates Jak2/Stat3 signaling that is inhibited by OGD/R. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. In OGD/R processes, the cells were incubated with doses of HNG (0.01–10 µg/l). Protein expression levels of Jak2, p-Stat3 (Y705) and p-Stat3 (S727) were detected using western blotting following the OGD/R treatments. *P<0.05 vs. Con; #P<0.05 and ##P<0.01 vs. 0 µg/l HNG OGD/R group. Jak2, Janus kinase 2; Stat3, signal transducer and activator of transcription 3; OGD/R, oxygen glucose deprivation/reoxygenation; HNG, S14G-humanin; p, phosphorylated; Con, control group.
Jak2/Stat3 signaling mediates neuroprotective functions of HNG against OGD/R
To determine whether Jak2/Stat3 signaling mediated neuroprotective functions of HNG against OGD/R, a specific inhibitor (FLLL32) of this signal was added to the cells together with HNG. Cell viability assay demonstrated that 1 µg/l HNG significantly reversed the decrease in cell viability (P<0.05) induced by OGD/R; however, co-treatment with 1 µg/l HNG and 5 µM FLLL32 was unable to significantly restore the cell viability (Fig. 4A). The increased apoptosis rate by OGD/R was significantly suppressed by treatment with 1 µg/l HNG (P<0.05), while adding 5 µM FLLL32 to the cells significantly abrogated the apoptosis-inhibitory effect of HNG (P<0.05; Fig. 4B).
Figure 4.

Protection by HNG from neuron apoptosis in OGD/R is related to Jak2/Stat3 signaling. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. In OGD/R processes, the cells were treated with 1 µg/l HNG alone or 1 µg/l HNG and 5 µM FLLL32 in combination. FLLL32 is a specific inhibitor of Jak2/Stat3 signaling. (A) Cell viability, (B) apoptosis rate and (C) western blot assays were undertaken following OGD/R treatments. *P<0.05 vs. control; #P<0.05 vs. 0 µg/l HNG OGD/R group; &P<0.05 vs. OGD/R + 1 µg/l HNG group. HNG, S14G-humanin; OGD/R, oxygen glucose deprivation/reoxygenation; Jak2, Janus kinase 2; Stat3, signal transducer and activator of transcription 3; FITC, fluorescein isothiocyanate; Bax, B-cell lymphoma 2-associated X protein.
OGD/R-triggered apoptosis is related to the induction of mitochondrial dysfunction and caspase-3 activation. Data from western blot analyses demonstrated that OGD/R significantly upregulated protein levels of cytochrome c, Bax and cleaved caspase-3 (P<0.05) compared with control cells; however, HNG significantly inhibited the upregulation triggered by OGD/R (P<0.05; Fig. 4C). The inhibitory effect was not observed with co-treatment with 1 µg/l HNG and 5 µM FLLL32. These data collectively indicated that HNG effectively inhibits OGD/R-induced apoptosis via activation of Jak2/Stat3 signaling.
Results also demonstrated that FLLL32 interfered with HNG functions in anti-oxidation. FLLL32 significantly abolished HNG-triggered increase in SOD activity and reduction in MDA concentration, when compared with the OGD/R group (P<0.05; Fig. 5).
Figure 5.
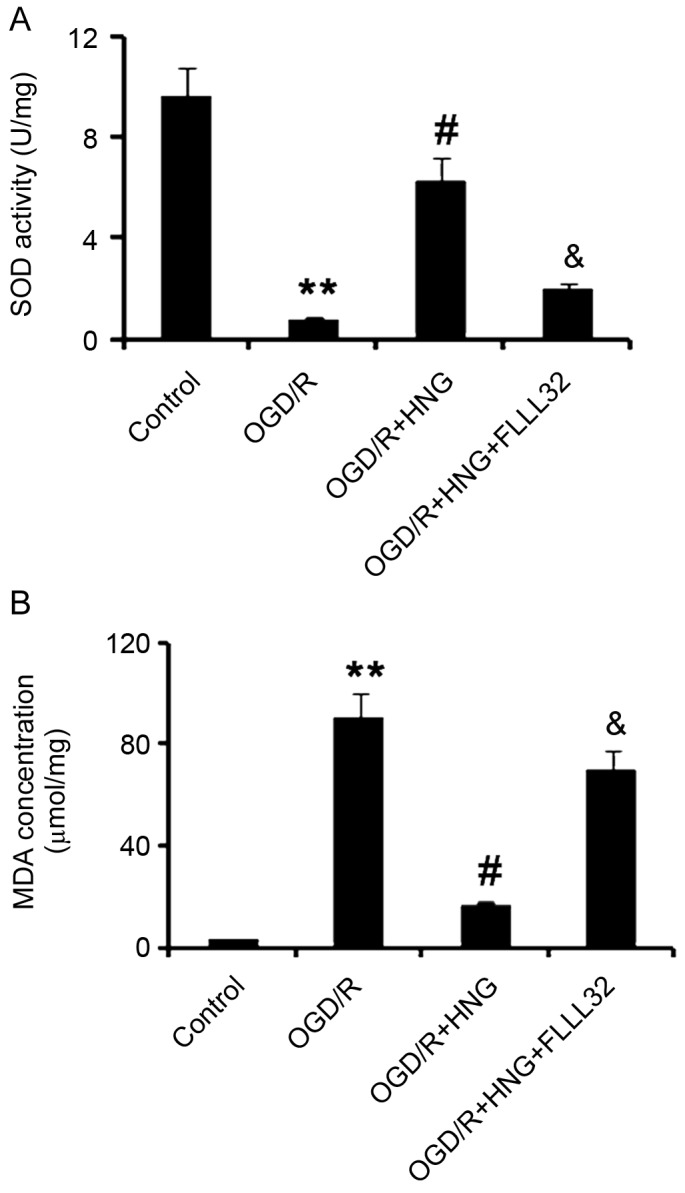
Jak2/Stat3 signaling mediates HNG-promoting anti-oxidative capacity of neurons in OGD/R. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. In OGD/R processes, the cells were treated with 1 µg/l HNG alone or 1 µg/l HNG and 5 µM FLLL32 in combination. FLLL32 is a specific inhibitor of Jak2/Stat3 signaling. (A) SOD activity and (B) MDA concentration in the SH-SY5Y cells were detected following OGD/R treatments. **P<0.01 vs. control group; #P<0.05 vs. OGD/R group; &P<0.05 vs. OGD/R + 1 µg/l HNG group. Jak2, Janus kinase 2; Stat3, signal transducer and activator of transcription 3; HNG, S14G-humanin; OGD/R, oxygen glucose deprivation/reoxygenation; SOD, superoxide dismutase; MDA, methane dicarboxylic aldehyde.
HNG activates Jak2/Stat3 signaling through the PI3K/AKT pathway
Jak2/Stat3 signaling is well documented to be regulated by the PI3K/AKT pathway (15–17). To understand the influence of the PI3 K/AKT pathway on Jak2/Stat3 signaling when HNG is used to counteract OGD/R of neurons, HNG was used in co-treatment with specific inhibitors of PI3K and AKT in OGD/R processes. Similar to the inhibitor of Jak2/Stat3 signaling, FLLL32, inhibitors of PI3K (10 µM LY294002) and AKT (5 µM MK-2206 2HCl) significantly downregulated (P<0.05) Jak2 protein expression levels that were promoted by HNG in OGD/R processes (Fig. 6). Upregulated p-Stat3 at the Y705 site by HNG demonstrated significant reduction when treated with 5 µM FLLL32 (P<0.01). Inhibitors of PI3K and AKT also significantly lowered p-Stat3 levels at the Y705 site (P<0.05) compared with controls, although they were not as effective as FLLL32. p-Stat3 level at the S727 site was significantly decreased by FLLL32 (P<0.05), LY294002 (P<0.05) and MK-2206 2HCl (P<0.01) treatments compared with control treatments. These data indicated that the activation of Jak2/Stat3 signaling by HNG is, at least partly, through the PI3K/AKT pathway.
Figure 6.

Jak2/Stat3 signal activation by HNG in OGD/R is through the PI3K/AKT pathway. SH-SY5Y cells were subjected to OGD/R processes following the procedures of culture in medium with deprivation of glucose and serum in a hypoxic condition (0.5% O2) for 16 h, and then incubation in normoxic conditions in normal medium for an additional 9 h. FLLL32 (5 µM), LY294002 (10 µM) and MK-2206 2HCl (5 µM), which are specific inhibitors of Stat3, PI3K and AKT, respectively, were applied in co-treatment with HNG (1 µg/l) in the OGD/R processes. Protein levels of Jak2, p-Stat3 (Y705) and p-Stat3 (S727) were detected by western blotting following the OGD/R treatments. *P<0.05 and **P<0.01 vs. control (only HNG treatment in OGD/R). Jak2, Janus kinase 2; Stat3, signal transducer and activator of transcription 3; OGD/R, oxygen glucose deprivation/reoxygenation; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; HNG, S14G-humanin; p, phosphorylated.
Discussion
Stroke, which is caused by a disruption of blood supply to the brain, poses a serious threat to the life and health of humans and considerably impairs the quality of life of the patients (18). Brain nerve cells are characterized by high sensitivity to deficiency of oxygen and glucose in cerebral ischemia (5). Recovering the blood supply, termed reperfusion, is a key step to rescue nerve cells from ischemia-induced death, however, it also raises serious problems linked to oxidative stress, inflammation disorder and ion overloading, thus the onset of reperfusion usually produces more severe damage to nerve cells (2). Apoptosis of nerve cells is a common event in stroke, according to a previous report (2). Apoptotic alterations of neurons, including the condensation of nuclei and the activation of caspases, are observed in the ischemic core in the early stage of focal cerebral ischemia (19,20). In rim of the ischemic core, neurons undergo a delayed neuronal death dependent or independent of caspases (19,20). The present study established an in vitro model of OGD/R that mimics the in vivo ischemia/reperfusion injury, and demonstrated that the apoptosis rate of SH-SY5Y cells was notably increased following OGD/R treatment with reduction of cell viability. As neurons are the core components of the central nervous system, forming the basis for neurological functions, neuronal death, particularly in the hippocampus and brain cortex, is a major cause of morbidity and mortality in stroke (2,3). Therefore, one important strategy in treating acute cerebral ischemia and reperfusion injury is to dampen the death of neurons in clinical settings.
Humanin has been extensively documented to have anti-apoptotic effects in pathological and physiological processes, such as ischemia-induced death of various cell types, oxidized low-density lipoprotein-related death of vascular endothelial cells, neuronal death in Alzheimer's disease, as well as Leydig cell death in spermatogenesis (20–24). However, physiological plasma level of humanin markedly decreases with age, which likely results in the limited role in anti-apoptosis in diseases that frequently occur in elderly people, such as stroke (11). HNG, that may be synthesized artificially, has 1,000-fold stronger biological activity than humanin, thus it is referred to as a potent humanin analogue (24). However, it is unclear whether HNG is able to protect neuron cells from death in OGD/R. This study presented new evidence that HNG is an effective inhibitor of apoptosis in OGD/R, which suggests that HNG may be a promising agent to treat stroke by prevention of neuronal apoptosis.
Multiple studies have evidenced that oxidative stress is closely associated with stroke damage (2,18). Reasons behind the occurrence of oxidative stress in stroke are basically elucidated. Ischemia-induced deficiency of oxygen and glucose disrupt mitochondrial homeostasis and metabolism in brain neurons, resulting in the conversion of O2 to O2.− (superoxide radical) and other reactive oxygen species (ROS) rather than utilization of O2 for oxidative phosphorylation and adenosine triphosphate generation (25–27). Furthermore, blood reperfusion following ischemia leads to the activation of nicotinamide adenine dinucleotide phosphate oxidases, lipoxygenase and xanthine oxidase, all of which may produce ROS (25–27). Overproduction of ROS challenges the antioxidative system in neurons and poses oxidative damage to substances, such as lipids, that are rich in neurons but vulnerable to free radicals (25–27). SOD represents an important antioxidant enzyme responsible for the elimination of O2.− with catalytic action (28). As SOD is a protein that is also the target (reactant) of ROS, SOD would lose the catalytic activity due to protein structure damage when exposed to excessive free radicals (28). The present study demonstrated that SOD activity was significantly reduced following OGD/R, which denoted an attenuated anti-oxidative ability in cells. MDA, one typical oxidative product of lipids, increased with OGD/R treatment in the present study, implying that lipids are devastated by free radicals. Adding HNG to SH-SY5Y cells reversed SOD activity and prevented MDA production, thereby attenuating oxidative stress induced by OGD/R.
Previous reports have demonstrated that the Jak2/Stat3 signaling axis mediated neuroprotective functions of humanin (8,9). Using a Jak2/Stat3 inhibitor, the present study demonstrated the important role of the Jak2/Stat3 pathway in the effects of HNG against OGD/R injury. Expression levels of Jak2 and p-Stat3 decreased when neurons were exposed to OGD/R, indicating an attenuated Jak2/Stat3 signal. A site-directed mutagenesis experiment revealed that cysteine residues, Cys866 and Cys917, act as a redox-sensitive switch controlling the catalytic activity of Jak2 (29). However, there are inconsistencies regarding changes in Jak2 activity under oxidized conditions in previous reports (30,31). Research has suggested that Jak2 activity in response to oxidative stress may also be dependent on the cell line used, due to difference in structural elements of Jak2 (29). In neurons, Jak2 activity is attenuated by oxidative stress induced by cadmium, with a deficiency of Jak2/Stat3 signaling (32). OGD/R inducing the generation of oxidative stress has been well-established in neurons in vivo and in vitro studies. Thus, inhibition of Jak2/Stat3 signaling in the present study may have been related to oxidative stress induced in OGD/R processes. However, HNG reactivated Jak2/Stat3 signaling in OGD/R processes by elevating their protein or phosphorylation levels. Notably, although regulated by oxidative stress, the Jak2/Stat3 cascade may reversely exert inhibitory effects on oxidative stress, thus providing scope for neuroprotection in OGD/R processes. Research has indicated that blockade of the Jak2/Stat3 cascade via a pharmacological inhibitor (AG490) markedly decreases SOD expression, increases ROS production and aggravates myocardial injury (33).
The present study also revealed that activation of the Jak2/Stat3 cascade by HNG prevented mitochondrial dysfunction-mediated apoptosis, because HNG decreased levels of cytochrome c, Bax and cleaved caspase-3; however, these changes were not observed following blockade of the Jak2/Stat3 cascade. Mitochondrial dysfunction is the most fundamental mechanism of cell damage in cerebral ischemia-reperfusion injury as it has critical roles in the apoptotic pathway in ischemic injury (34). Ischemia-reperfusion impairs mitochondrial function and leads to an increase of mitochondrial membrane permeability, resulting in release of cytochrome c, caspase-3 activation and apoptotic cell death (34). Mitochondria thus become important targets for stroke therapy. It has been documented that humanin may alter mitochondrial oxidative phosphorylation and ATP production; however, the exact molecular mechanisms are not well understood (8). A study by Kumfu et al (35) reported that humanin may prevent brain mitochondrial dysfunction in a model of cardiac ischemia-reperfusion injury. The results of the present study demonstrated that HNG inhibited mitochondrial dysfunction-mediated apoptosis via Jak2/Stat3. HNG may replace humanin to attenuate mitochondrial injury in stroke.
The PI3K/AKT pathway is critically important in multiple physiological and pathological processes by regulating various downstream signaling molecules (36). Activation of the PI3K/AKT pathway has been demonstrated to have neuroprotective effects against ischemia-induced neuron apoptosis (36). The Jak2/Stat3 cascade has been documented to be regulated by the PI3K/AKT pathway, mediating the functions in regulation of cell proliferation, anti-apoptosis and anti-senescence, cancer generation and migration, tissue development and memory formation (15–17). A study indicated that phosphorylation levels of AKT and Stat3 are increased after humanin acts on the GP130/IL6ST receptor complex (11). Activated AKT and Stat3 participate in cytoprotective and neuroprotective mechanisms of humanin in multiple age-related diseases (11). Based on these data, it is reasonable to hypothesize that the PI3K/AKT pathway participates in the regulation of HNG on Jak2/Stat3 signaling. It was observed in the present study that Jak2 protein and Stat3 phosphorylation levels were decreased by the inhibition of either PI3K or AKT. The results indicated that the activation of Jak2/Stat3 signaling by HNG is related to the activation of the PI3K/AKT pathway.
In conclusion, to the best of our knowledge, the present study demonstrated for the first time that HNG has neuroprotective effects against OGD/R by reactivating Jak2/Stat3 signaling through the PI3K/AKT pathway. HNG activation of Jak2/Stat3 signaling inhibited oxidative stress and mitochondrial dysfunction-mediated apoptosis. The present study provided a theoretical basis that HNG may be a promising agent to antagonize stroke injury.
References
- 1.World Health Organization (WHO): The World Health report 2002 - Reducing Risks, corp-author. Promoting Healthy Life. WHO; Geneva: 2002. [Google Scholar]
- 2.Wu JY, Li M, Cao LJ, Sun ML, Chen D, Ren HG, Xia Q, Tao ZT, Qin ZH, Hu QS, Wang GH. Protease Omi cleaving Hax-1 protein contributes to OGD/R-induced mitochondrial damage in neuroblastoma N2a cells and cerebral injury in MCAO mice. Acta Pharmacol Sin. 2015;36:1043–1052. doi: 10.1038/aps.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Reeves B, Sparkenbaugh EM, Russell J, Soltys Z, Zhang H, Faber JE, Key NS, Kirchhofer D, Granger DN, et al. Protective and detrimental effects of neuroectodermal cell-derived tissue factor in mouse models of stroke. JCI Insight. 2016;1:e86663. doi: 10.1172/jci.insight.86663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang P, He Y, Li D, Han R, Liu G, Kong D, Hao J. Class I PI3K inhibitor ZSTK474 mediates a shift in microglial/macrophage phenotype and inhibits inflammatory response in mice with cerebral ischemia/reperfusion injury. J Neuroinflammation. 2016;13:192. doi: 10.1186/s12974-016-0660-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Zhu J, Liu Y, Chen X, Lei S, Li L, Jiang B, Tan L, Yu S, Zhao Y. Glycogen synthase kinase 3β influences injury following cerebral ischemia/reperfusion in rats. Int J Biol Sci. 2016;12:518–531. doi: 10.7150/ijbs.13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci USA. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY) 2016;8:796–809. doi: 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R, Hinton DR. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci. 2016;57:1238–1253. doi: 10.1167/iovs.15-17053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan L, Liu XJ, Han WN, Li QS, Wang ZJ, Wu MN, Yang W, Qi JS. [Gly14]-Humanin protects against amyloid β peptide-induced impairment of spatial learning and memory in rats. Neurosci Bull. 2016;32:374–382. doi: 10.1007/s12264-016-0041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charununtakorn ST, Apaijai N, Kerdphoo S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Humanin exerts cardioprotection against cardiac ischemia-reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc Ther. 2016;34:404–414. doi: 10.1111/1755-5922.12210. [DOI] [PubMed] [Google Scholar]
- 11.Kim SJ, Guerrero N, Wassef G, Xiao J, Mehta HH, Cohen P, Yen K. The mitochondrial-derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age-dependent signaling differences in the hippocampus. Oncotarget. 2016;7:46899–46912. doi: 10.18632/oncotarget.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Zhao W, Yang H, Zhang J, Ma J. S14G-humanin restored cellular homeostasis disturbed by amyloid-beta protein. Neural Regen Res. 2013;8:2573–2580. doi: 10.3969/j.issn.1673-5374.2013.27.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang T, Wang H. Identification of cross-contamination in SH-SY5Y cell line. Hum Cell. 2014;27:176–178. doi: 10.1007/s13577-014-0096-9. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y, Leng W, Zhang J. Protective effect of puerarin against oxidative stress injury of neural cells and related mechanisms. Med Sci Monit. 2016;22:1244–1249. doi: 10.12659/MSM.896058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y, Zhou J, Xu C, Lin H, Xiao J, Wang Z, Yang B. JAK/STAT and PI3K/AKT pathways form a mutual transactivation loop and afford resistance to oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem. 2008;21:305–314. doi: 10.1159/000129389. [DOI] [PubMed] [Google Scholar]
- 16.Zhang LD, Chen L, Zhang M, Qi HJ, Chen L, Chen HF, Zhong MK, Shi XJ, Li QY. Downregulation of ERRα inhibits angiogenesis in human umbilical vein endothelial cells through regulating VEGF production and PI3K/Akt/STAT3 signaling pathway. Eur J Pharmacol. 2015;769:167–176. doi: 10.1016/j.ejphar.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, Xue J, Yang Y, Zhu H, Chen F, Wang J, Lou G, Liu Y, Shi Y, Yu Y, et al. Isoliquiritigenin inhibits interferon-γ-inducible genes expression in hepatocytes through down-regulating activation of JAK1/STAT1, IRF3/MyD88, ERK/MAPK, JNK/MAPK and PI3K/Akt signaling pathways. Cell Physiol Biochem. 2015;37:501–514. doi: 10.1159/000430372. [DOI] [PubMed] [Google Scholar]
- 18.Shi Z, Wu D, Yao JP, Yao X, Huang Z, Li P, Wan JB, He C, Su H. Protection against oxygen-glucose deprivation/reperfusion injury in cortical neurons by combining omega-3 polyunsaturated acid with lyciumbarbarum polysaccharide. Nutrients. 2016;8 doi: 10.3390/nu8010041. pii: E41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benchoua A, Guégan C, Couriaud C, Hosseini H, Sampaïo N, Morin D, Onténiente B. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001;21:7127–7134. doi: 10.1523/JNEUROSCI.21-18-07127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrer I. Apoptosis: Future targets for neuroprotective strategies. Cerebrovasc Dis. 2006;21(Suppl 2):S9–S20. doi: 10.1159/000091699. [DOI] [PubMed] [Google Scholar]
- 21.Muzumdar RH, Huffman DM, Calvert JW, Jha S, Weinberg Y, Cui L, Nemkal A, Atzmon G, Klein L, Gundewar S, et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler Thromb Vasc Biol. 2010;30:1940–1948. doi: 10.1161/ATVBAHA.110.205997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bachar AR, Scheffer L, Schroeder AS, Nakamura HK, Cobb LJ, Oh YK, Lerman LO, Pagano RE, Cohen P, Lerman A. Humanin is expressed in human vascular walls and has a cytoprotective effect against oxidized LDL-induced oxidative stress. Cardiovasc Res. 2010;88:360–366. doi: 10.1093/cvr/cvq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colón E, Strand ML, Carlsson-Skwirut C, Wahlgren A, Svechnikov KV, Cohen P, Söder O. Anti-apoptotic factor humanin is expressed in the testis and prevents cell-death in leydig cells during the first wave of spermatogenesis. J Cell Physiol. 2006;208:373–385. doi: 10.1002/jcp.20672. [DOI] [PubMed] [Google Scholar]
- 24.Takeshita Y, Hashimoto Y, Nawa M, Uchino H, Matsuoka M. SH3-binding protein 5 mediates the neuroprotective effect of the secreted bioactive peptide humanin by inhibiting c-Jun NH2-terminal kinase. J Biol Chem. 2013;288:24691–24704. doi: 10.1074/jbc.M113.469692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silachev DN, Plotnikov EY, Zorova LD, Pevzner IB, Sumbatyan NV, Korshunova GA, Gulyaev MV, Pirogov YA, Skulachev VP, Zorov DB. Neuroprotective effects of mitochondria-targeted plastoquinone and thymoquinone in a rat model of brain ischemia/reperfusion injury. Molecules. 2015;20:14487–14503. doi: 10.3390/molecules200814487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Wu J, Duan X, Tian X, Shen H, Sun Q, Chen G. NADPH oxidase: A potential target for treatment of stroke. Oxid Med Cell Longev. 2016;2016:5026984. doi: 10.1155/2016/5026984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolff V, Schlagowski AI, Rouyer O, Charles AL, Singh F, Auger C, Schini-Kerth V, Marescaux C, Raul JS, Zoll J, Geny B. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. Biomed Res Int. 2015;2015:323706. doi: 10.1155/2015/323706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith JK, Patil CN, Patlolla S, Gunter BW, Booz GW, Duhé RJ. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic Biol Med. 2012;52:1101–1110. doi: 10.1016/j.freeradbiomed.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazière C, Conte MA, Mazière JC. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic Biol Med. 2001;31:1334–1340. doi: 10.1016/S0891-5849(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 31.Kaur N, Lu B, Monroe RK, Ward SM, Halvorsen SW. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. J Neurochem. 2005;92:1521–1530. doi: 10.1111/j.1471-4159.2004.02990.x. [DOI] [PubMed] [Google Scholar]
- 32.Monroe RK, Halvorsen SW. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med. 2006;41:493–502. doi: 10.1016/j.freeradbiomed.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 33.Cai W, Yang X, Han S, Guo H, Zheng Z, Wang H, Guan H, Jia Y, Gao J, Yang T, et al. Notch1 pathway protects against burn-induced myocardial injury by repressing reactive oxygen species production through JAK2/STAT3 signaling. Oxid Med Cell Longev. 2016;2016:5638943. doi: 10.1155/2016/5638943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ten VS, Starkov A. Hypoxic-ischemic injury in the developing brain: The role of reactive oxygen species originating in mitochondria. Neurol Res Int. 2012;2012:542976. doi: 10.1155/2012/542976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumfu S, Charununtakorn ST, Jaiwongkam T, Chattipakorn N, Chattipakorn SC. Humanin prevents brain mitochondrial dysfunction in a cardiac ischaemia-reperfusion injury model. Exp Physiol. 2016;101:697–707. doi: 10.1113/EP085749. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y, Ruan Y, Xu A. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci Rep. 2016;6:26859. doi: 10.1038/srep26859. [DOI] [PMC free article] [PubMed] [Google Scholar]