

Abstract
A new class of hydrogels utilizing DNA (DNA quadruplex gel) has been constructed by directly and symmetrically coupling deoxynucleotide phosphoramidite monomers to the ends of polyethylene glycols (PEGs) in liquid phase, and using the resulting DNA‐PEG‐DNA triblock copolymers as macromonomers. Elongation of merely four deoxyguanosine residues on PEG, which produces typically ≈10 grams of desired DNA‐PEG conjugates in one synthesis, resulted in intelligent and biodegradable hydrogels utilizing DNA quadruplex formation, which are responsive to various input signals such as Na+, K+, and complementary DNA strand. Gelation of DNA quadruplex gels takes place within a few seconds upon the addition of a trigger, enabling free formation just like Ca+‐alginate hydrogels or possible application as an injectable polymer (IP) gel. The obtained hydrogels show good thermal stability and rheological properties, and even display self‐healing ability.
Keywords: DNA, gels, injectable polymers, polyethylene glycol, quadruplexes
Recent development of various hydrogels with unique properties is attracting scientists not only from material chemistry but also from broad research fields such as medical sciences to the field.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 DNA is one of popular materials used to prepare hydrogels, often with sophisticated functions, as a fruit of previous studies in DNA computing and structural DNA nanotechnology.12, 13 Most of them, however, are miniature material, since DNA used in such systems is prepared on solid‐phase DNA synthesizers, which only produces oligonucleotides in usually mg scales.14 For the applications of DNA hydrogels to practical purposes, it is desirable to overcome such scale barrier. Here, we present a versatile hydrogel system that consists of only two popular biocompatible materials, DNA and polyethylene glycol (PEG). DNA‐PEG‐DNA triblock conjugates, in which merely four or five consecutive nucleotides are symmetrically introduced, were prepared at least on a gram scale applying a solution‐phase large scale DNA synthesis, thus realizing intelligent and biodegradable hydrogels (DNA quadruplex gels). By utilizing efficient formation of G‐quadruplexes as the crosslinking points,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 DNA quadruplex gels shows sol‐to‐gel (or gel‐to‐sol) transition responsive to various input signals, such as Na+, K+, and a complementary DNA strand. DNA quadruplex gels can be instantly prepared and even show self‐healing properties.
Structures of typical DNA‐PEG‐DNA conjugates used in this study are presented in Figure 1. In L4.6k‐dG4, for example, DNA portions of d(GpGpGpGp) are attached to both of the ends of linear PEG4.6k. X10k‐dG4, on the other hand, bears four‐arm PEG10k in place of the linear PEG, and thus four dG4 portions at every end. To prepare such conjugates, we employed high‐efficiency liquid phase (HELP) synthesis of oligonucleotides developed by Bonora et al. with a few major modifications.26 Deoxyguanosine (dG) residues were directly and symmetrically coupled to the ends of either linear or branched PEG, instead of methoxy‐PEG used in the original HELP, which is used as a semi‐solution phase substrate soluble in a coupling reaction mixture and easily precipitable with a poor solvent for product recovery and washing (Figure 1 a and Scheme S1 in the Supporting Information). In addition, insertion of ester linkage between the PEG and DNA portions was omitted in the present modified HELP to obtain DNA‐PEG‐DNA conjugates as the final product. Other chemistry is almost the same as popular phosphoramidite DNA synthesis except that typically ≈10 g of protected conjugates with ≈70 % overall yield after four rounds of coupling can be obtained in one synthesis. As shown in the photograph in Figure 1 a, the final deprotection procedure using solid‐phase extraction and re‐precipitation gives the desired conjugates as macroscopic solid. Consecutive dG residues, just like those introduced in the conjugates, are known to form quite stable G‐quadruplex structures in the presence of Na+ or K+.27 The dG portion thus serves as four‐way crosslinking points to form a 3D network of PEG (Figure 1 b), resulting in efficient gelation of the conjugate solution upon Na+ or K+ addition. As shown in Figure 1 c and Movie S1, solutions of the conjugates turned into transparent hydrogels within a few seconds after the addition of 100 mm (final concentration) K+ or even Na+. Figure 1 d shows a phase diagram of the quadruplex gels with two conjugates, L4.6k‐dG4 or X10k‐dG4, in the presence of K+ or Na+. Quadruplex hydrogels prepared in the presence of K+ show overall a higher thermal stability compared to those with Na+, and for most of the conditions examined, the transition temperature T sol was sufficiently higher than 37 °C. The hydrogel formation was completely reversible across T sol so that repetitive phase transition between sol and gel states can be achieved by switching the temperature between 25 °C and 90 °C (Figure S5). Apparent critical gelation concentrations (CGCs), similarly to T sol, were roughly independent on the structure of the PEG portion and were 3 wt % for K+ systems and 4 wt % for those of Na+. Rheological studies of the quadruplex gels revealed that all of the present quadruplex gels are stiffer than most of the conventional DNA hydrogels with storage moduli (G′) around 1–10 kPa, and the X10k‐dG4/K+ gel does not turn into sol even at ≈90 °C (Figure 1 e). The ease of preparing highly concentrated macromonomer solution such as 5 wt % or even up to 30 wt % is one of the best advantage of the present system utilizing HELP. Conjugates with four‐arm PEG (X series), which quantitatively bear an additional chemical four‐way crosslinking point in the PEG portion, showed nearly a twice as high G′ compared to those with linear PEG (L series). Considering that G′ is theoretically proportional to the crosslinking density,28 the contribution of G‐quadruplexes, that is, physical four‐way crosslinking points, to the polymer 3D network in the quadruplex gels is almost comparable to the branching point in the X series. Conjugates bearing dG3 portions showed lower thermal stabilities compared to those bearing dG4 portions (Table S2, Supporting Information), probably representing the low stability of the corresponding G‐quadruplexes.
Figure 1.
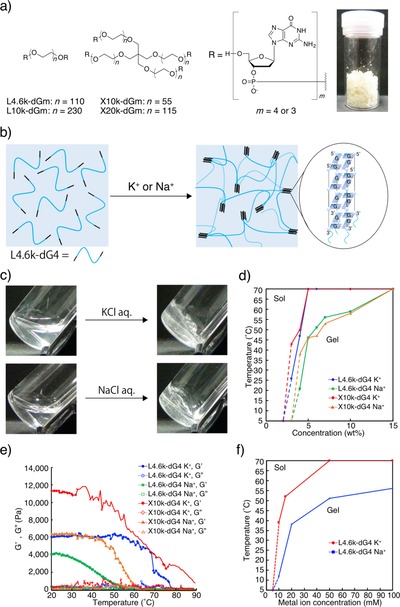
Metal‐ion‐responsive hydrogels utilizing G‐quadruplex formation between PEG–DNA conjugates. (a) Structure of PEG–DNA conjugates used in this study. Photograph: purified L4.6k‐dG4 (1 g) prepared by modified HELP. (b) Schematic representation of the system. Wavy blue lines represent PEG segments. Upon the addition of K+ or Na+ to a conjugate solution, DNA segments immediately form G‐quadruplexes to crosslink PEG segments into a 3D network. (c) Photographs of 10 wt % DNA‐PEG‐DNA conjugate in 0.2 m Tris/HCl before (left) and after (right) the addition of metal ion solutions (100 mm K+ or 100 mm Na+, final concentrations). (d) Temperature‐dependent phase diagram as a function of conjugate concentrations (0–15 wt %). Open symbols represent that no gelation was observed even at 5 °C. Apparent CGCs were 3 wt % for K+, and 4 wt % for Na+. (e) Temperature‐dependent rheological properties of 10 wt % DNA quadruplex gels. (f) Dependence of T sol of 7 wt % (≈10 mm) L4.6k‐dG4 gel on metal ion concentration.
The dependence of metal ion concentration over T sol was examined both for K+ and Na+ with 7 wt % (≈10 mm) L4.6k‐dG4 solution. Since K+ binds to the inter‐plane cavity between the two G‐tetrads whereas relatively smaller Na+ binds to the intra‐plane cavity, equivalent concentrations of each ion to G‐quadruplexes (5 mm) are 15 and 20 mm, respectively. Figure 1 f shows that CGCs for K+ and Na+ are both within a range between 5 mm and 10 mm. At the equivalent, T sol for L4.6k‐dG4/K+ is 51 °C and that for L4.6k‐dG4/Na+ is 38 °C. The T sol almost saturates around 50 mm both for K+ and Na+. These values also support nearly quantitative G‐quadruplex contribution in gelation at room temperature in the presence of 100 mm ions. Measurements of circular dichroism (CD) of diluted conjugate solutions revealed that DNA portions in all of the present conjugates exist as parallel quadruplexes in the presence of metal ions (Figure S6). Intensities of the positive cotton effect at 260 nm almost correlate with the thermal stability of the hydrogels. Similarly, melting temperatures (T m) determined by θ at 260 nm were comparable with corresponding T sol (Table S4).
The requirement of a reasonably low concentration of metal ions suggests that the gelation of present DNA quadruplex gels can be triggered by various body‐related fluids and not only by phosphate‐buffered saline (PBS). In fact, addition of one third the volume of either commercially available 1× PBS(−), fetal bovine serum, artificial tear, saliva, sweat, or even E‐MEM cell culture media to 10 wt % L4.6k‐dG4 solution all resulted in instant formation of DNA quadruplex gels, which are stable even above 37 °C (Figure S7). The present gels thus hold potential to be used for various healthcare purposes.
Prompt gelation of the present conjugates enabled easy preparation of hydrogel beads mimicking the process for calcium alginate gel beads, which involves dripping of an alginate solution to a Ca2+ solution. As expected, DNA quadruplex gel beads or even a gel string could be straightforwardly prepared by dripping or injecting a 10 wt % L4.6k‐dG4 solution to a 1 m K+ solution (Figure 2 a and Movie S2). No conjugate solution came out from the center when a gel bead was cut into half with a razor immediately after dripping, showing that the gelation takes place at once in whole drop. Since the conjugate solution injected to PBS also immediately turns into a gel (Movie S3), the present DNA quadruplex gel can be used as an injectable polymer (IP) gel, which is recently considered as one of the best candidates for drug/cell carrier.29, 30
Figure 2.

(a) Preparation of DNA quadruplex gel beads and string (see Supporting Movie S2). A conjugate solution (10 wt %) containing RB (23 μm) was added to 1 m K+ solution dropwise to form gel beads. Hydrogel string was also formed by injection of the conjugate solution. (b) Enzymatic digestion assay using hydrogel beads. Fluorescent DNA quadruplex gel beads were prepared with 10 μl conjugate solution, and immersed in PBS(−) with or without PDE II. After 3 days at 37 °C, only the hydrogel bead in the presence of the enzyme completely disappeared and the fluorescent polystyrene beads were released into the buffer.
The properties of the present DNA quadruplex gels were examined using 10 wt % L4.6k‐dG4/Na+ gel beads (10 μl) containing fluorescent polystyrene beads (d=0.5 μm) prepared as described above using 10× PBS(−). A gel bead subjected in a solution containing phosphodiesterase (PDE) II completely disappeared within 3 days at 37 °C, whereas a bead in a solution without the enzyme was almost intact both visually and quantitatively estimated by polystyrene‐beads leakage (Figure 2 b and Figure S6a). Addition of DNase I also resulted in the dissolution of bead (Figure S7), although PDE I was not able to digest the quadruplex gel probably because of the difference in their reaction mechanism. The digestion of the DNA quadruplex gel is unusually slow, presumably as a result of the exclusion volume effect from PEG portions and the structural rigidity of the G‐quadruplexes themselves, however, the present DNA quadruplex gels are nevertheless biodegradable.
According to the observation of fluorescent leakage from three L4.6k‐dG4/K+ gel beads containing d=0.5 μm, 0.1 μm, and 50 nm polystyrene beads (Figure S8 b–d), the mesh size of the polymer network is estimated to be somewhat larger than 100 nm, since significant leakage was observed for 50 nm polystyrene beads after 3 days while the leakage of 0.1 μm bead was only marginal. The largest 0.5 μm beads completely stayed in the L4.6k‐dG4/K+ gel during this period.
One major advantage of employing DNA as a component of a hydrogel is that various achievements in the fields of DNA computing and structural DNA nanotechnology are applicable to construct intelligent materials. In this study, we employed toe‐hold mediated strand exchange,31 one of the most popular method in these fields, to implement an oligonucleotide‐sensing capability to the DNA quadruplex gel (Figure 3). As shown in Figure 3 a, X20k‐dG3 with extra 5 nucleotides (X20k‐dG3toe) was synthesized for this experiment. This extra 5 nucleotides remain single‐stranded even after gel formation, and serve as a “toe‐hold” where a complementary strand first partially hybridizes as shown in Figure 3 b. The complementary strand then peels off the dG3 portion from the G‐quadruplex by forming a complete duplex. The stability of the G‐triplex structure, an intermediate of G‐quadruplex formation and dissociation, in the presence of K+ or Na+ is far lower than that of G‐quadruplexes.32 Accordingly, removal of one strand from a G‐quadruplex is expected to promote gel‐to‐sol phase transition. The X20k‐dG3toe/K+ bead immersed in K+ solution containing a matched complementary strand indeed disappeared within 12 hours at room temperature. When merely a one‐base mismatch was introduced to the input strand, on the other hand, the quadruplex gel bead still remained under the same conditions, although non‐negligible swelling of the bead and fluorescent leakage was observed. Since the DNA portion in the complexes can be freely elongated in the synthesis, further intelligent systems may be feasible by employing DNA aptamers or more sophisticated DNA logic gates developed to date.33, 34
Figure 3.
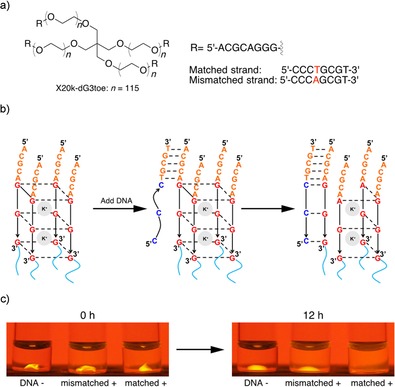
Sequence‐selective gel‐to‐sol transition utilizing toehold‐mediated strand displacement. (a) Structures of X20k‐dG3toe and the DNA strands used in this study. (b) Schematic illustration of the process. The complementary DNA strand added to the solution first hybridizes to the extended 5‐mer DNA portion (toehold) and peels off the G tract from the quadruplex. (c) Photographs of hydrogel beads before (left) and after (right) addition of 8‐mer DNA strands to the buffer solution.
Another feature of the present DNA quadruplex gels is their self‐healing property (Figure 4).35 As the formation of G‐quadruplexes is a reversible process, gradual exchange of the DNA portions in the crosslinking points as well as the diffusion of the conjugates realizes recovery of the defective 3D network. To demonstrate this, two cylindrical L4.6k‐dG4/Na+ gels, one colored in blue with bromophenol blue (BPB) and another in red with rose bengal (RB) were prepared, and cut into halves (Figure 4 a, photograph in the left). A pair of semicircles of different colors were pushed into each other and placed in a mold. After 1 day at room temperature, the two pieces fused into one cylindrical piece without any obvious defect (center). Two days later, the piece became almost uniform with a purple color as a mixture of the two dyes (right), showing that there is no boundary between the semicircles that prevents free diffusion of the dyes.
Figure 4.

Self‐healing properties of DNA quadruplex gel. (a) Cylindrical L4.6k‐dG4/Na+ gels (d=8 mm, h=3 mm) colored in blue with BPB and red with RB formed with PBS were cut into halves (left) and put into a template. After 1 day at room temperature, the two pieces fused into one cylindrical piece without any obvious defect (center). Two days later, the piece became almost uniform with a purple color as a mixture of two dyes (right). (b) Hydrogel strata made by gelating multiple quadruplex gels layer by layer (see Supporting Movie S3). On a red cylindrical/L4.6k‐dG4/Na+ gel, L4.6k‐dG4 solution without dye was added followed by PBS(−) to form the second transparent layer. After the third blue layer was formed, the resulting cylinder was sliced into thin pieces (left). After two days at room temperature, the slice became uniformly purple because of free diffusion of the dyes (right).
The recovery between freshly cut sections takes days and is rather slow, probably because of slow diffusion of the conjugates and/or the exclusion volume effect by the PEG portion.36 For immediate re‐adhesion, in situ gelation between the sections is effective as a filling. The L4.6k‐dG4/Na+ gel strata with tricolor layers presented in Figure 4 b were prepared by forming cylindrical gels in a mold layer by layer (Supporting Movie S4). The resulting gel cylinder was then sliced into thin pieces with a razor. Even if the whole process only took several minutes, the three layers responded to physical stress completely as unity. After two days at room temperature, the slice became uniformly purple again because of free diffusion of the dyes across the former interfaces. In combination with the above‐mentioned toehold method, reversible adhesion–separation between two gel pieces may be also feasible in the future.
Interestingly, addition of unmodified PEG4.6k to the solution of L4.6k‐dG4 significantly enhanced the stability of the resulting DNA quadruplex gel (Table S5). This result suggests that the PEG portions in the conjugates act not only as the matrix of the 3D polymer network but also as a molecular crowding agent, which is known to stabilize both G‐quadruplexes but rather destabilize B‐type DNA duplexes.37
In summary, we have successfully developed a new class of DNA hydrogels. The present system consists of only DNA and PEG portions, which are both biocompatible and popular biomaterials. Considering their numerous advantages including rapid gelation triggered by physiological Na+ solutions, the present DNA quadruplex gels should be useful, for example, in medicine‐ or biology‐related fields as a potential drug carrier or 3D cell culture scaffold.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
We thank T. Sakai for synthesizing DNA‐PEG complexes, and Prof. T. Kato for valuable comments. The 4‐arm PEGs used in this study were kindly provided by NOF Corporation. This work was supported by Precursory Research for Embryonic Science and Technology (PRESTO), “Molecular Technology and Creation of New Functions” from Japan Science and Technology Agency (JST) (JPMJPR12K4). Supports from Private University Research Branding Project (2016–2021), Grant‐in‐Aid for Scientific Research (A) (16H01854), (B) (24350088), Grant‐in‐Aid for Scientific Research on Innovative Areas “Molecular Robotics” (24104004) from the Ministry of Education, Science, Sports, Culture and Technology, Japan, and from Nihon L′Oréal are also acknowledged.
S. Tanaka, K. Wakabayashi, K. Fukushima, S. Yukami, R. Maezawa, Y. Takeda, K. Tatsumi, Y. Ohya, A. Kuzuya, Chem. Asian J. 2017, 12, 2388.
Contributor Information
Prof. Dr. Yuichi Ohya, Email: yohya@kansai-u.ac.jp.
Prof. Dr. Akinori Kuzuya, Email: kuzuya@kansai-u.ac.jp.
References
- 1. Ahmed E. M., J. Adv. Res. 2015, 6, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffman A. S., Adv. Drug Delivery Rev. 2012, 64, 18–23. [Google Scholar]
- 3. Xiong X., Wu C., Zhou C., Zhu G., Chen Z., Tan W., Macromol. Rapid Commun. 2013, 34, 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao Y., Jia H., Cao T., Liu D., Acc. Chem. Res. 2017, 50, 659–668. [DOI] [PubMed] [Google Scholar]
- 5. Kahn J. S., Hu Y., Willner I., Acc. Chem. Res. 2017, 50, 680–690. [DOI] [PubMed] [Google Scholar]
- 6. Um S. H., Lee J. B., Park N., Kwon S. Y., Umbach C. C., Luo D., Nat. Mater. 2006, 5, 797–801. [DOI] [PubMed] [Google Scholar]
- 7. Umeno D., Kano T., Maeda M., Anal. Chim. Acta 1998, 365, 101–108. [Google Scholar]
- 8. Liedl T., Dietz H., Yurke B., Simmel F., Small 2007, 3, 1688–1693. [DOI] [PubMed] [Google Scholar]
- 9. Qi H., Ghodousi M., Du Y., Grun C., Bae H., Yin P., Khademhosseini A., Nat. Commun. 2013, 4, 2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song J., Im K., Hwang S., Hur J., Nam J., Ahn G.-O., Hwang S., Kim S., Park N., Nanoscale 2015, 7, 9433–9437. [DOI] [PubMed] [Google Scholar]
- 11. Bomboi F., Romano F., Leo M., Fernandez-Castanon J., Cerbino R., Bellini T., Bordi F., Filetici P., Sciortino F., Nat. Commun. 2016, 7, 13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adleman L. M., Science 1994, 266, 1021–1024. [DOI] [PubMed] [Google Scholar]
- 13. Seeman N. C., J. Theor. Biol. 1982, 99, 237–247. [DOI] [PubMed] [Google Scholar]
- 14.E. Ellington, J. D. Pollard, Curr. Protoc. Mol. Biol 2001, https://doi.org/10.1002/0471142727.mb0211s42. [DOI] [PubMed]
- 15. Wei B., Cheng I., Luo K. Q., Mi Y., Angew. Chem. Int. Ed. 2008, 47, 331–333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 337–339. [Google Scholar]
- 16. Lu C.-H., Qi X.-J., Orbach R., Yang H.-H., Mironi-Harpaz I., Seliktar D., Willner I., Nano Lett. 2013, 13, 1298–1302. [DOI] [PubMed] [Google Scholar]
- 17. Kahn J. S., Trifonov A., Cecconello A., Guo W., Fan C., Willner I., Nano Lett. 2015, 15, 7773–7778. [DOI] [PubMed] [Google Scholar]
- 18. Lu C.-H., Guo W., Qi X.-J., Neubauer A., Paltiel Y., Willner I., Chem. Sci. 2015, 6, 6659–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu C.-H., Guo W., Hu Y., Qi X.-J., Willner I., J. Am. Chem. Soc. 2015, 137, 15723–15731. [DOI] [PubMed] [Google Scholar]
- 20. Shao Y., Li C., Zhou X., Chen P., Yang Z., Li Z., Liu D., Acta Chim. Sin. 2015, 73, 815–818. [Google Scholar]
- 21. Xiang B., He K., Zhu R., Liu Z., Zeng S., Huang Y., Nie Z., Yao S., ACS Appl. Mater. Interfaces 2016, 8, 22801–22807. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Z., Liu B., Liu J., Small 2016, 17, 1602730. [Google Scholar]
- 23. Zhao H., Jiang G., Weng J., Ma Q., Zhang H., Ito Y., Liu M., J. Mater. Chem. B 2016, 4, 4648–4651. [DOI] [PubMed] [Google Scholar]
- 24. Hasuike E., Akimoto A. M., Kuroda R., Matsukawa K., Hiruta Y., Kanazawa H., Yoshida R., Chem. Commun. 2017, 53, 3142–3144. [DOI] [PubMed] [Google Scholar]
- 25. Huang Y., Xu W., Liu G., Tian L., Chem. Commun. 2017, 53, 3038–3041. [DOI] [PubMed] [Google Scholar]
- 26. Bonora G. M., Zaramella S., Veronese F. M., Biol. Proced. Online 1998, 1, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burge S., Parkinson G. N., Hazel P., Todd A. K., Neidle S., Nucleic Acids Res. 2006, 34, 5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang H., Su W., Mather P. T., Bunning T. J., Polymer 1999, 40, 4593–4602. [Google Scholar]
- 29. Li Y., Rodrigues J., Tomás H., Chem. Soc. Rev. 2012, 41, 2193–2221. [DOI] [PubMed] [Google Scholar]
- 30. Nishikawa M., Ogawa K., Umeki Y., Mohri K., Kawasaki Y., Watanabe H., Takahashi N., Kusuki E., Takahashi R., Takahashi Y., Takakura Y., J. Controlled Release 2014, 180, 25–32. [DOI] [PubMed] [Google Scholar]
- 31. Yurke B., Turberfield A. J., Mills A. P., Simmel F. C., Neumann J. L., Nature 2000, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- 32. Jiang H.-X., Cui Y., Zhao T., Fu H.-W., Koirala D., Punnoose J. A., Kong D.-M., Mao H., Sci. Rep. 2015, 5, 9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellington A. D., Szostak J. W., Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 34. Qian L., Winfree E., Science 2011, 332, 1196–1201. [DOI] [PubMed] [Google Scholar]
- 35. Taylor D. L., in het Panhuis M., Adv. Mater. 2016, 28, 9060–9093. [DOI] [PubMed] [Google Scholar]
- 36. Li C., Chen P., Shao Y., Zhou X., Wu Y., Yang Z., Li Z., Weil T., Liu D., Small 2015, 11, 1138–1143. [DOI] [PubMed] [Google Scholar]
- 37. Nakano S., Miyoshi D., Sugimoto N., Chem. Rev. 2014, 114, 2733–2758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary