

Abstract
Objective
Patients with myotonia congenita have muscle hyperexcitability due to loss‐of‐function mutations in the ClC‐1 chloride channel in skeletal muscle, which causes involuntary firing of muscle action potentials (myotonia), producing muscle stiffness. The excitatory events that trigger myotonic action potentials in the absence of stabilizing ClC‐1 current are not fully understood. Our goal was to identify currents that trigger spontaneous firing of muscle in the setting of reduced ClC‐1 current.
Methods
In vitro intracellular current clamp and voltage clamp recordings were performed in muscle from a mouse model of myotonia congenita.
Results
Intracellular recordings revealed a slow afterdepolarization (AfD) that triggers myotonic action potentials. The AfD is well explained by a tetrodotoxin‐sensitive and voltage‐dependent Na+ persistent inward current (NaPIC). Notably, this NaPIC undergoes slow inactivation over seconds, suggesting this may contribute to the end of myotonic runs. Highlighting the significance of this mechanism, we found that ranolazine and elevated serum divalent cations eliminate myotonia by inhibiting AfD and NaPIC.
Interpretation
This work significantly changes our understanding of the mechanisms triggering myotonia. Our work suggests that the current focus of treating myotonia, blocking the transient Na+ current underlying action potentials, is an inefficient approach. We show that inhibiting NaPIC is paralleled by elimination of myotonia. We suggest the ideal myotonia therapy would selectively block NaPIC and spare the transient Na+ current. Ann Neurol 2017;82:385–395
Myotonia congenita is a member of a group of inherited skeletal muscle diseases known as the nondystrophic myotonias1, 2, 3 and results from a loss‐of‐function in the muscle chloride channel (ClC‐1).4, 5, 6 The debilitating slowed muscle relaxation experienced by patients with myotonia congenita is caused by involuntary firing of action potentials (APs; myotonia). Although it is well established that a decrease in ClC‐1 current causes muscle hyperexcitability, the excitatory events that trigger myotonic APs are not fully understood. Because of this, the mainstay of therapy remains avoiding activities that trigger myotonia,1 which can include running, walking, or other daily activities. Alternatively, drugs that block Na+ channels, such as mexiletine, are used to reduce excitability.1, 2, 3 However, many patients suffer side effects or incomplete symptom resolution. Thus, there is a need to advance our understanding of the currents that trigger myotonia if we are to develop improved therapies.
The currently accepted explanation for the generation of myotonia is that K+ exits the fiber during voluntary APs and accumulates in the transverse tubules (t‐tubules). T‐tubules are narrow invaginations of the surface membrane that are needed to conduct APs into the middle of muscle fibers.7 The resulting K+ buildup depolarizes the resting membrane potential8, 9, 10, 11 and is thought to open voltage‐gated Na+ channels that trigger the involuntary APs causing myotonia.12, 13 Normally, ClC‐1‐mediated chloride currents, which account for 70 to 80% of resting muscle membrane conductance, offset the depolarizing influence of K+ accumulation and prevent myotonia.6, 8, 14, 15
One shortcoming of only considering t‐tubular K+ buildup is that this model does not clearly explain the cessation of myotonia after a time course of seconds. Notably, we are not the first to question K+ buildup as the sole explanation for muscle hyperexcitability in myotonia congenita. In one of Dr Adrian's seminal papers on myotonia, he noted that “in the absence of a surface chloride conductance, tubular potassium accumulation could certainly contribute to the instability of the membrane; but it is clear that potassium accumulation is not the only reason for the instability of myotonic muscle fibres.”9 However, in the 40 years since this statement was published, no additional contributor to muscle hyperexcitability in myotonia congenita has been identified.
In neurons, repetitive firing is maintained by persistent inward currents (PICs), which are activated by long depolarizations and mediated by Na+ (Na+ persistent inward current [NaPIC]) or Ca2+.16, 17, 18, 19 A NaPIC has also been described in wild‐type (WT) skeletal muscle.20 However, since its discovery in 1989, little attention has been paid to the muscle NaPIC, and its function in WT muscle remains unknown. We hypothesized that this NaPIC, present in normal skeletal muscle, plays a central role in triggering pathologic repetitive firing during myotonia. In this report, we show that the activation and inhibition of skeletal muscle NaPIC can explain both the onset and termination of myotonic APs. Moreover, we demonstrate that the inhibition of NaPIC is paralleled by elimination of myotonic APs. Our findings lead to a new understanding of muscle hyperexcitability in myotonia congenita, potentially explain the time course for cessation of myotonic firing, and reveal a novel target for more effective therapies.
Materials and Methods
Mice
All animal procedures were performed in accordance with the policies of the Animal Care and Use Committee of Wright State University. Mice were bred from a colony of ClCn1adr‐mto2J mice and determined to be myotonic or unaffected by phenotype as previously described.21 Unaffected littermates were used as controls (WT). In both this and our previous study,21 male and female mice were found to have a similar severity of myotonia. Thus, both male and female mice were used from 2 months to 6 months of age. As mice with myotonia have difficulty climbing to reach food, symptomatic mice were supplied with moistened chow paste (Irradiated Rodent Diet, 2918; Harlan Teklad, Madison, WI) on the floor of the cage.
Electrophysiology
Current Clamp
Prior to removal of muscle for recording, mice were killed by CO2 inhalation followed by cervical dislocation. Both extensor digitorum longus (EDL) muscles were dissected out tendon‐to‐tendon. Experiments were done on the first muscle while the second muscle was maintained in oxygenated solution. Records from the second muscle began within 3 hours of dissection. Muscles were maintained and recorded at 21 to 23°C. Electrical properties of the muscle have been shown to be stable for 6 hours in vitro.21 The recording chamber was continually perfused with Ringer solution containing (in mM): 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4, 5.5 glucose, and maintained at pH 7.3–7.4 while aerated with 95% O2 and 5% CO2. The modified Ringer solution for high‐divalent cation concentration experiments contained (in mM): 118 NaCl, 3.5 KCl, 5 CaCl2, 2 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4, 5.5 glucose, and maintained at pH 7.3–7.4 while aerated with 95% O2 and 5% CO2.
To prevent muscle contraction and allow for stable electrical recordings, muscles were loaded with 50μM N‐benzyl‐p‐toluenesulfonamide (BTS; Tokyo Chemical Industry, Tokyo, Japan) for 45 minutes prior to recording. BTS was dissolved in dimethylsulfoxide (DMSO) and added to the perfusate. Maximal DMSO concentration was kept under 0.15%, which has been found not to affect resting membrane properties of skeletal muscle.22
Staining of muscle with 10μM 4‐(4‐diethylaminostyrl)‐N‐methylpyridinium iodide (Molecular Probes, Eugene, OR) was performed as previously described.21 Muscle fibers were impaled with 2 sharp microelectrodes filled with 3M KCl solution containing 1mM sulforhodamine to visualize the electrodes with epifluorescence. Electrode resistance was 15 to 25MΩ. Capacitance compensation was optimized prior to recording. Fibers with resting potentials more depolarized than −74mV were excluded from analysis. AP voltage threshold was defined as the voltage at which the dV/dt was 10mV/ms.
Voltage Clamp
Flexor digitorum brevis (FDB) and interosseous muscle fibers were used, as they allow for better space clamp and voltage control. Isolation of fibers and recordings were performed as previously described.23, 24 Briefly, muscles were surgically removed, pinned to Sylgard‐bottomed Petri dishes, and enzymatically dissociated at 37°C under mild agitation for ∼1 hour using 1,000U/ml of collagenase type IV (Worthington Biochemical, Lakewood, NJ). Collagenase was dissolved in the extracellular solution used for recordings (below). Mechanical dissociation was completed using mild trituration in buffer with no collagenase. The fibers were allowed to recover at 21 to 23°C for 1 hour before being used for electrical measurements.
Fibers were visualized using an IX71 (Olympus, Tokyo, Japan) microscope equipped with ×10 and ×20 UPlanFLN objectives, and images were acquired with a CCD camera (ST‐7XMEI‐C1, Santa Barbara Instruments, Santa Barbara, CA). Electrical properties were measured under standard current and voltage clamp conditions at 21 to 23°C, using 2 borosilicate intracellular microelectrodes with a 1.5mm outside diameter and 0.86mm inside diameter (Sutter Instruments, Novato, CA), an Axoclamp 900A amplifier, a Digidata 1550 digitizer, and pCLAMP 10 data acquisition and analysis software (Molecular Devices, Sunnyvale, CA). Reference electrodes were placed in separate cups containing 3M KCl and connected to the extracellular solution via agar bridges. The voltage‐sensing electrode was connected with an Axoclamp HSx1 headstage. The current‐passing electrode was connected with an Axoclamp HSx10 headstage that was modified to have a 2MΩ output resistor (HSx5). Both the current‐passing and voltage‐sensing electrodes were filled with the same solutions (below) and had resistances of ∼15MΩ. After impalement, 20 minutes of hyperpolarizing current injection was allowed for equilibration of the electrode solution with the sarcoplasm before data acquisition. Data were acquired at 20kHz. Current and voltage records were low‐pass filtered with the internal Axoclamp 900A filters at 1kHz. The voltage clamp command signal was low‐pass filtered with an external Warner LFP‐8 at 1kHz.
Normal K+ Solutions
Internal solution (in mM) was as follows: 75 aspartate, 30 ethyleneglycoltetraacetic acid (EGTA), 15 Ca(OH)2, 5 MgCl2, 5 adenosine triphosphate (ATP) di‐Na, 5 phosphocreatine di‐Na, 5 glutathione, 20 4‐morpholinepropane sulfonic acid (MOPS), and pH 7.2 with KOH.
Extracellular solution (in mM) was as follows: 144 NaCl, 4 KCl, 1.2 CaCl2, 0.6 MgCl2, 5 glucose, 1 NaH2PO4, 10 MOPS, 0.02 nifedipine + 0.01 ouabain, and pH 7.4 with NaOH.
K+‐Free Solutions
Internal solution (in mM) was as follows: 75 aspartate, 30 EGTA, 15 Ca(OH)2, 5 MgCl2, 5 ATP di‐Na, 5 phosphocreatine di‐Na, 5 glutathione, 20 MOPS, and pH 7.2 with CsOH.
Extracellular solution (in mM) was as follows: 144 NaCl, 4 CsCl, 1.2 CaCl2, 0.6 MgCl2, 5 glucose, 1 NaH2PO4, 10 MOPS, 0.05 BaCl2, 0.02 nifedipine + 0.01 ouabain + 0.1 3,4‐diaminopyridine (3,4‐DAP), and pH 7.4 with NaOH.
K+‐Free Solutions with High Extracellular Divalent Cations
Internal solution (in mM) was as follows: 75 aspartate, 30 EGTA, 15 Ca(OH)2, 5 MgCl2, 5 ATP di‐Na, 5 phosphocreatine di‐Na, 5 glutathione, 20 MOPS, and pH 7.2 with CsOH.
Extracellular solution (in mM) was as follows: 144 NaCl, 4 CsCl, 5 CaCl2, 2 MgCl2, 5 glucose, 1 NaH2PO4, 10 MOPS, 0.05 BaCl2, 0.02 nifedipine + 0.01 ouabain + 0.1 3,4‐DAP, and pH 7.4 with NaOH.
Drugs
Drugs (in mM) were as follows: 0.4 9‐anthracenecarboxylic acid (Tocris Bioscience, Bristol, UK), 0.02 nifedipine (Sigma‐Aldrich, St Louis, MO), 0.001 tetrodotoxin (TTX; Alomone Labs, Jerusalem, Israel), 0.1 3,4‐DAP (Sigma‐Aldrich), 0.01 ouabain (Sigma‐Aldrich), 0.05 ranolazine (Sigma‐Aldrich).
Statistical Analyses
Differences between two datasets were analyzed using Student (unpaired) 2‐tailed t test, assuming unequal variance (Sigmaplot 13.0). For comparisons within the same fiber or myotonic runs, paired 2‐tailed t tests were used. All data are presented as mean ± standard error of the mean. p < 0.05 was considered to be significant. Sample sizes were chosen based on the laboratory's previous experiences in the calculation of experimental variability. The numbers of animals and fibers used are described in the corresponding figure legends and text. No blinding was used for the animal groups, as ClCn1adr‐mto2J mice are easily identifiable from their unaffected littermates phenotypically. This was not a significant issue for analysis of voltage clamp and current clamp data, as analysis is largely automated and thus not susceptible to subjective bias.
Study Approval
All animal procedures were performed in accordance with the policies of the Animal Care and Use Committee of Wright State University and the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals.
Experiments were approved by Wright State University's institutional animal care and use committee (#1082).
Results
Myotonic Firing
We used the Clcn1 adr‐mto2J (ClC) mouse model of myotonia congenita, in which affected mice are homozygous for a null mutation in the Clcn1 gene. As such, the ClC mice have a loss‐of‐function in the skeletal muscle chloride channel, ClC‐1, which is the main feature of all mutations that cause myotonia congenita. The ClC mice most closely recapitulate the recessive form of myotonia congenita (Becker disease). They have muscle stiffness due to myotonia and exhibit the warmup phenomenon, in which muscle performance improves with exercise.21, 25 Intracellular recordings of myotonic APs were performed under current clamp conditions in the EDL muscle, a standard muscle used for this procedure that can be removed with little perturbation and thus closely approximates in vivo conditions. Voluntary APs were triggered with 200‐millisecond step current pulses that depolarized the membrane potential by 15 to 25mV (Fig 1). Following the voluntary APs, we recorded trains of involuntary (myotonic) APs that were quite variable in length, lasting from 0.1 to 100 seconds. During the myotonic APs, the average maximum repolarization (Max Repol) was 16.2 ± 1.3mV (n = 51 fibers, 12 mice) more depolarized than the original resting potential and did not return to resting level until hundreds of milliseconds or >1 second after the myotonic APs (see Fig 1). This steady depolarization is consistent with K+ buildup in the t‐tubules.
Figure 1.
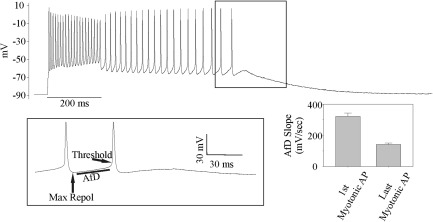
Myotonic firing in muscle from ClC mice due to both a steady depolarization and an afterdepolarization (AfD). Shown is the response of a ClC muscle fiber to a 200‐millisecond injection of stimulating current. The fiber fires multiple action potentials (APs) for the duration of the current injection, a normal behavior; it then also continues to fire APs after the cessation of current injection (myotonia). Contributors to myotonic firing of APs include a steady depolarization and an AfD occurring between APs. The AfD occurs over 20 to 50 milliseconds and brings the fiber to threshold. The inset shows the final 2 APs of the run of myotonia on an expanded timescale. Max Repol = the maximal repolarization following each AP; Threshold = AP threshold (dV/dt = 10mV/ms). An AfD is present following the last myotonic AP, but is too small to bring the fiber to threshold. Following the final AfD, there is a resolution of the steady depolarization as the fiber gradually returns to its resting membrane potential. Shown on the lower right is the mean value of the first and last AfD slopes (n = 51 fibers, 12 mice; p < 0.01, paired t test).
The average AP threshold, defined as the voltage when the AP rate of rise (dV/dt) was 10mV/ms, was 13.3 ± 0.5mV more depolarized than the Max Repol. After reaching the Max Repol between APs, the membrane potential would gradually depolarize over 20 to 50 milliseconds until the AP threshold was reached. We termed this slow depolarization the afterdepolarization (AfD). No explanation for the AfD has yet been proposed. During myotonia, the slope of AfD gradually decreased during the run of myotonia from 320.7 ± 20.9mV/s at the first myotonic AP to 141.9 ± 8.9mV/s by the last myotonic AP (see Fig 1, n = 51 fibers, 12 mice, p < 0.01, paired t test). Our current clamp data strongly suggest that the AfD triggers myotonic APs and that the decreasing slope of the AfD is responsible for the termination of myotonia. To determine which ionic current underlies the AfD, we applied slow depolarization under well‐controlled voltage clamp conditions.
Characteristics of NaPIC in Muscle from ClC Mice
In motor neurons, repetitive firing is triggered by PICs, which are carried by both Na+ and Ca+ channels.16, 17, 18, 19 A NaPIC has been described previously in skeletal muscle20; this NaPIC lacked fast inactivation and activated between −80 and −70mV, such that it would convert a steady depolarization from K+ buildup into myotonia. In motor neurons, PICs are commonly measured by applying slow ramp depolarizations to inhibit channels with fast inactivation. To determine whether PICs could contribute to the myotonic AfD, we applied slow ramp depolarization to dissociated FDB fibers. Because of their small size (400–500μm length × ∼40μm diameter), we were able to control whole FDB fibers under voltage clamp. We have previously used this technique to record larger and faster chloride and potassium currents.23, 24 During ramp depolarizations at a rate of 10mV/s in a normal K+ solution with 20μM nifedipine to block Cav1.1 channels (Fig 2A), we recorded an inward deflection in current consistent with the activation of a PIC in ClC FDB fibers (see Fig 2B, C). This current activated at −73.7 ± 1.1mV and was maximal at −34.7 ± 2.1mV (n = 3 mice, 21 fibers).
Figure 2.
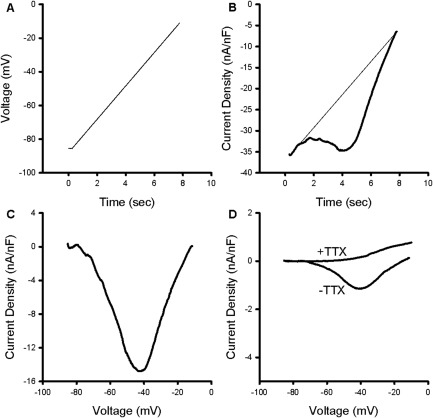
Characterization of a tetrodotoxin (TTX)‐sensitive persistent inward current in ClC muscle. (A) The voltage protocol used to identify persistent inward currents (PICs). From a holding potential of −85mV, fibers were depolarized to −10mV at a rate of 10mV/s. (B) The current trace generated by the ramp depolarization in normal K+ solution with 20μM nifedipine. A fit line (thin) is drawn using the first 0.5 seconds of the raw trace, representing the leak current. Deviations from the leak current/fit line in the negative direction are consistent with activation of a PIC. (C) Shown is a trace generated by subtracting the leak trace shown in B and plotting against voltage. (D) Plot of leak‐subtracted PIC in K+‐free solutions (−TTX). The PIC was blocked by the addition of 1μM TTX to the external solution (+TTX).
To determine whether this apparent PIC was due to a true inward current or a reduction in outward K+ leak current during ramp depolarization, we repeated recordings in K+‐free solutions that also contained 0.1mM 3,4‐DAP and 50μM Ba2+ in the external solution to block K+ channels. Under these conditions, the apparent PIC was reduced by 80% (see Fig 2D), suggesting that a reduction of outward K+ current is a major contributor to the apparent inward current. The PIC that remained in the K+‐free conditions activated at −71.4 ± 0.78mV, was maximal at −42.1 ± 2.0mV, and had a −1.4 ± 0.2nA/nF maximum current density. The current is likely mediated by Na+ channels, because it was eliminated by application of 1μM TTX (n = 4 mice, 23 fibers; see Fig 2D). Although the current is small, the depolarization caused by NaPIC would drive the closing of inwardly rectifying K+ (Kir) channels26, 27 in a small positive feedback cycle. Thus, NaPIC could be a key depolarizing trigger that drives myotonia.
We also performed voltage‐clamp ramp depolarizations on FDB fibers from unaffected WT siblings of ClC mice in K+‐free solutions. We used 400μM 9‐anthracenecarboxylic acid to block ClC‐1, an established method of pharmacologically inducing myotonia.28 In the WT FDB fibers, we recorded a NaPIC that activated at −73.3 ± 0.8mV, was maximal at −51.1 ± 1.8mV, and had a −0.9 ± 0.2nA/nF maximum current density.
As described above, the slope of AfD gradually declines during myotonia (see Fig 1). The slope of the AfD is determined by the net current resulting from subthreshold Na+, K+, and Cl− currents. These currents in turn depend on the number of channels open and the driving force for current flow through each ion channel. Thus, although a number of factors likely contribute to the AfD slope, our data suggest NaPIC is an important contributor. The previous study that identified NaPIC in muscle20 did not determine whether the current inactivated over seconds. To examine slow inactivation, we modified our ramp protocols to slowly return to the baseline holding potential before repeating the slow depolarization (Fig 3). The mean current density of NaPIC on the descending ramp was reduced by 86% in ClC FDB fibers (−1.4 ± 0.2nA/nF on ascending ramp vs −0.2 ± 0.1nA/nF on descending ramp, n = 20 fibers; 6 mice, p < 0.01, paired t test). This decrease in current density on the descending ramp is consistent with inactivation of NaPIC. On a subsequent ascending ramp, the mean NaPIC amplitude was −1.3 ± 0.2nA/nF, suggesting that the inactivation of NaPIC was relieved by repolarization. Thus, although NaPIC lacks fast inactivation, it appears to undergo a reversible slow inactivation that could contribute to the reduction in the slope of the AfD during runs of myotonia. These findings suggested that slow inactivation of NaPIC contributes to the termination of myotonic firing.
Figure 3.
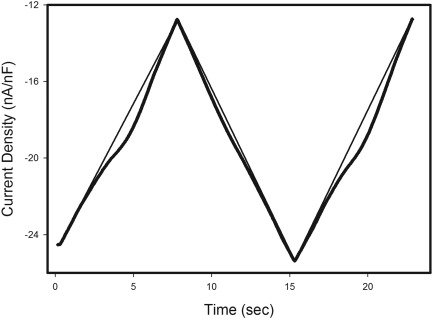
Current recorded during a voltage‐clamp ramp protocol with repeated slow depolarization. Current recorded during a voltage‐clamp ramp protocol in which the fiber was slowly depolarized from a holding potential of −85mV to −10mV, repolarized to −85mV, and again depolarized to −10mV is shown. The larger persistent inward current during the depolarizing ramps than the repolarizing ramp shows that the muscle Na+ PIC undergoes slow inactivation. The recording was performed in K+‐free solutions. The fit lines for linear leak currents are superimposed on all 3 ramps.
Reduction in the Slope of AfD Is Responsible for Termination of Myotonia
In a NaPIC model of myotonia, slow inactivation of NaPIC during a run of myotonia will cause a gradual decrease in the slope of the AfD. Furthermore, if inhibition of NaPIC underlies the termination of myotonia, the slope of the AfD should be at its minimum prior to termination of myotonia. This was the case. Before the first myotonic AP, the average AfD slope was 320.7 ± 20.9mV/s; after the last myotonic AP, the average AfD slope was 141.9 ± 8.9mV/s, its lowest value (51 of 51 fibers, 12 mice). Figure 4 shows a plot of the slope of the AfD during myotonia from a representative fiber. These data suggest that slow inactivation of NaPIC contributes to termination of myotonia.
Figure 4.
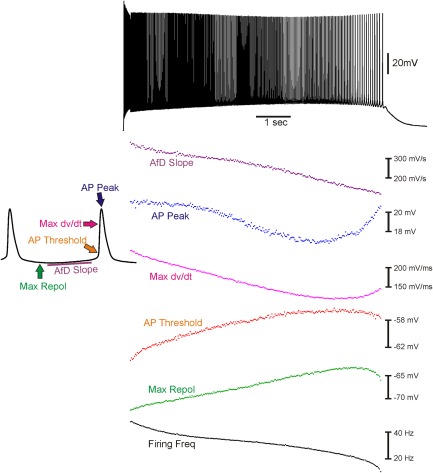
Changes in action potential (AP) parameters during a myotonic run. Shown at the top is a 10‐second run of myotonia triggered by a 200‐millisecond stimulation. On the lower left is a blowup of 2 APs from the middle of the myotonic run to demonstrate the AP parameters measured. On the lower right is a plot of the values of the various parameters, time‐matched to the run of myotonia shown above. Termination of myotonia correlates with a reduction in the slope of the afterdepolarization (AfD). Max dV/dt = the maximal rate of rise of the AP; Max Repol = the maximal repolarization reached between APs.
We also examined whether slow inactivation of the transient Na+ current contributed to resolution of myotonia, as has recently been proposed.21, 29 If the transient Na+ channel availability decreases, the AP peak and dV/dt will decrease, and the AP threshold will increase. By these measures, a decrease in transient Na+ channel availability occurred in only 13.7% of fibers prior to termination of myotonia, there was no change in 27.5% of fibers, and there was an increase in 58.8% of fibers (see Fig 4). The lack of correlation between transient Na channel availability and termination of myotonia argues strongly that inactivation of transient Na+ currents does not play a central role in termination of myotonia. Another possibility is the resolution of t‐tubular K+ buildup. However, arguing against this idea is the finding that the maximal repolarization following each myotonic AP did not correlate with termination of myotonia (see Fig 4). The only parameter that closely correlated with termination of myotonia was reduction in the slope of the AfD.
Treatments That Eliminate Myotonia Reduce NaPIC
The tight correlation between reduction in the slope of the AfD and termination of myotonia suggests that pharmacologically inhibiting NaPIC would prevent myotonia. We studied the mechanism of action of 2 different manipulations that eliminate myotonia (Fig 5A). The first was the drug ranolazine, which we previously demonstrated was effective in treating myotonia.21 With no exposure to ranolazine, myotonia was detected in 100% of ClC EDL fibers (n = 217 fibers, 28 mice); in the presence of 50μM ranolazine, 0% of ClC EDL fibers exhibited myotonia (n = 35 fibers, 4 mice). The second treatment we studied was elevated extracellular Ca2+ and Mg2+, which has been shown to prevent myotonia.13, 30 We recorded from ClC EDL muscle in the presence of physiological (1.5mM Ca2+ and 1.4mM Mg2+) and elevated (5mM Ca2+ and 2mM Mg2+) extracellular divalent cation concentrations. Myotonia was detected in only 8.33% of fibers in the presence of high divalent cations (4 of 48 fibers, 4 mice).
Figure 5.
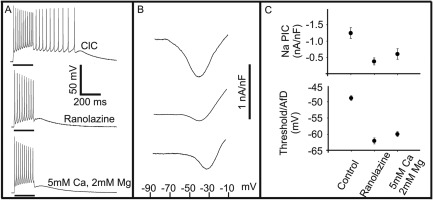
Elimination of myotonia and parallel reduction of Na+ persistent inward current (NaPIC) by ranolazine, and divalent cation‐treated ClC muscle. (A) In untreated ClC muscle (top), myotonia is triggered with a 200‐millisecond depolarizing current pulse, whereas 50μM ranolazine treatment (middle) and high extracellular divalent solutions (bottom) eliminate myotonia. (B) Representative NaPIC traces evoked by slow‐depolarizing voltage ramps (similar to those shown in Fig 2D) for untreated (top), ranolazine (middle), and elevated cation (bottom) groups. NaPIC current density is reduced by both ranolazine and elevated extracellular divalent concentration. (C) Top plot: ClC muscle (untreated) shows a maximal PIC current density of −1.25 ± 0.17nA/nF (n = 23 fibers, 4 mice) versus treatment with 50μM ranolazine (maximal PIC current density is reduced to −0.38 ± 0.11nA/nF; n = 20 fibers, 3 mice), and versus elevated cation solutions (maximal PIC current density is decreased to −0.61 ± 0.16nA/nF; n = 25 fibers, 3 mice). Lower plot: Mean threshold of the first AP of a myotonic run of APs in ClC muscle (control) compared to the peak afterdepolarization (AfD) following treatment with ranolazine and high divalent solution. Both treatments that eliminate myotonia cause the AfD to fail to reach the threshold at which myotonic firing is triggered in untreated, control muscle.
We next studied whether both treatments reduced NaPIC. Voltage clamp recordings in the presence of 50μM ranolazine revealed an 80% reduction in NaPIC current density (n =20 fibers, 3 mice; see Fig 5B, C). When NaPIC was recorded in the presence of elevated extracellular divalent cations, the current density was reduced by 55% (n = 25 fibers, 3 mice; see Fig 5B). Thus, both ranolazine and the elevation of extracellular divalent cations eliminate myotonia and reduce NaPIC.
If NaPIC plays a central role in triggering the AfD, elimination of myotonia by both treatments should be paralleled by reduction in the slope of the AfD. The slope of the AfD in the presence of 50μM ranolazine was 54.7 ± 8.6mV/s, a value much lower than the slope of the AfD in untreated muscle just prior to termination of myotonia (141.9 ± 8.9mV/s). Elevation of divalent cation reduced the slope of the AfD to 38.1 ± 5.3mV/s. These data are consistent with the possibility that a reduction of NaPIC causes a reduction in AfD and this is the mechanism underlying the efficacy of both treatments against myotonia.
That NaPIC and AfD are key determining factors in the onset and termination of myotonia is a dramatic shift from current views on myotonia. Hitherto, it was thought that treatments eliminate myotonia via effects on transient Na+ channels that lead to elevation of AP threshold.1, 31 Moreover, the previous model did not address termination of myotonia. To assess the previous model, we compared the AP threshold to the AfD in our ranolazine and high divalent records. If treatments eliminate myotonia by raising AP threshold and not altering AfD, the AfD following treatment should reach the pretreatment AP threshold, but not trigger myotonia. The AP threshold of the first myotonic AP measured in ClC muscle was −48.9 ± 0.8mV (n = 51 fibers, 12 mice; see Fig 5C). The peak of the AfD following a 200‐millisecond stimulation in the presence of 50μM ranolazine averaged −62.1 ± 0.9mV (n = 35 fibers, 4 mice), a value significantly more negative than the pretreatment AP threshold. In the presence of elevated extracellular divalent cation, the peak voltage reached by the AfD averaged −59.9 ± 0.8mV (n = 4 mice, 42 fibers). Thus, although both treatments elevate AP threshold, in neither case does this appear to be the mechanism by which they eliminate myotonia. In contrast, the onset, termination, and treatment of myotonia can be understood by considering AfD and NaPIC.
Discussion
A New Model of Myotonic Muscle Hyperexcitability
In both the autosomal dominant (Thomsen) and recessive (Becker) forms of myotonia congenita, loss of function mutations in the ClC‐1 gene (CLCN1) cause muscle hyperexcitability.1, 2, 3 However, it has remained unclear what excitatory events trigger myotonic APs in the absence of the stabilizing ClC‐1 current. Prior to this study, the only excitatory factor implicated in the generation of involuntary APs in myotonia congenita was a steady membrane depolarization from K+ buildup in t‐tubules8, 9, 10, 11 that activated the transient Na+ channels responsible for APs.12, 13 It was posited >4 decades ago that some additional factor was necessary to account for the spontaneous firing of APs during myotonia9; however, until now, no such factor had been identified. While closely examining APs in the Clcn1adr‐mto2J (ClC) mouse model of myotonia congenita, we observed 2 possible contributors to the depolarization that brings fibers to threshold during runs of myotonic APs: (1) a blunted repolarization between APs, presumably from K+ buildup; and (2) a slow depolarization after each myotonic AP, which we termed the AfD. We found that the onset of myotonia is accompanied by a steep AfD slope and the termination of myotonia follows a decrease in the AfD slope. Moreover, effective treatments reduced the AfD slope. No other AP characteristic predicted the presence or end of myotonia. We further posited that a current, unmasked in the absence of ClC‐1, underlays the AfD. A NaPIC previously identified in WT skeletal muscle20 exhibited characteristics that could generate the AfD. We found a NaPIC in muscle from ClC mice and WT siblings with properties similar to those initially described by Gage et al.20 Notably, the voltage‐dependent activation and very slow inactivation describe an excitatory current that would drive the closure of Kir channels that contributes to the AfD. Based on characteristics of the NaPIC, we present a modified model of the generation of myotonia in Figure 6.
Figure 6.
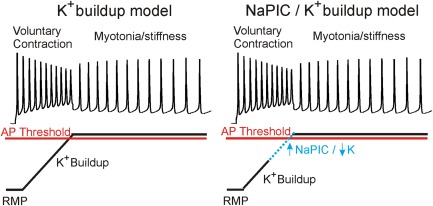
A new model of generation of myotonia. In the K+ buildup model on the left, myotonia is triggered solely by a steady depolarization caused by K+ buildup in t‐tubules, which is sufficient to depolarize the membrane to the action potential (AP) threshold and trigger myotonia. In the Na+ persistent inward current (NaPIC) and K+ buildup model on the right, K+ buildup is insufficient to trigger myotonia in isolation, but rather depolarizes the membrane sufficiently to activate NaPIC, which then contributes to the final depolarization that triggers myotonic APs. RMP = resting membrane potential.
In both models, the absence of a stabilizing chloride current through ClC‐1 unmasks the depolarizing events. This occurs because the equilibrium potential for chloride is at or near the resting membrane potential in skeletal muscle (−80 to −90mV).8, 14 In the K+ buildup model (see Fig 6, left panel) the only depolarizing event is K+ buildup in t‐tubules, which is sufficient to drive the membrane to the AP threshold. The role of K+ buildup in myotonia is well established from experiments showing that detubulation of muscle fibers eliminated myotonia.8, 9 Arguing against this model fully explaining myotonia, we show that the average maximum repolarization between myotonic APs, which is driven primarily by the K+ gradient, is 13mV more negative than the AP threshold. This strongly suggests that K+ buildup alone could not trigger myotonia.
In the NaPIC model presented here, t‐tubular K+ buildup provides the initial depolarizing event, which explains the steady depolarization during myotonia. The maximum repolarization between APs reflects the magnitude of this depolarizing effect. Because the membrane potential remains subthreshold without an additional depolarizing event, the membrane potential would return to resting values. However, K+ buildup is sufficient to activate NaPIC. In response to NaPIC, the membrane potential begins to depolarize past the maximum repolarization to generate the AfD. Although NaPIC is small, we propose that the resulting depolarization will begin to close Kir channels. The steep voltage‐dependence of Kir channels is such that they close at potentials slightly depolarized relative to the K+ equilibrium potential.26, 27
NaPIC in WT Muscle
Our demonstration that NaPIC is present in WT muscle leads to the idea that the excitatory currents triggering myotonia are simply unmasked by the absence of ClC‐1. Importantly, it also implies that NaPIC has a role in normal muscle function. One role of PICs in neurons is to convert steady depolarizations caused by the asynchronous firing of many weak synaptic inputs into repetitive spiking.16, 17, 18, 19 However, because skeletal muscle fibers receive only one synaptic input, there is no need for a PIC to convert multiple asynchronous synaptic firings into repetitive muscle APs. The normal function of NaPIC in muscle might be to improve the fidelity of neuromuscular transmission during periods of intense stimulation; in addition to enabling repetitive firing, PICs in neuronal dendrites serve as amplifiers for weak synaptic inputs.18, 32 During intense exertion, the neuromuscular junction is stimulated at rapid rates, resulting in depression of the endplate potential, such that synaptic transmission might fail.33 The NaPIC in muscle could thus amplify subthreshold endplate potentials during strenuous activity.
Molecular Identity of NaPIC
That NaPIC is sensitive to low doses of TTX in ClC and WT muscle suggests NaPIC is mediated by the normal muscle Na+ channel that drives the AP rising phase, namely Nav1.4.34 It is likely that NaPIC derives from a small subset of Nav1.4 channels that are in a different conformation. This understanding is based on recordings from frog skeletal muscle, as well as rat and cat neurons, showing that single Na+ channels shift modes between a normal, fast‐inactivating mode and a mode lacking fast inactivation.35, 36 For simplicity, we refer to the currents through the Nav1.4 channels that lack fast inactivation as NaPIC and the currents through the normal fast‐inactivating Nav1.4 channels as the transient Na+ currents. We hypothesize that some Na+ channel mutations that trigger myotonia do so by promoting the channel conformation that generates PIC. Mechanisms that have been identified in neurons as promoting NaPIC include cleavage by calpain and association of Na+ channels with calmodulin.37, 38
We should note that the NaPIC present in ClC muscle is distinct from the pathological persistent inward currents arising from mutations in Na+ or Ca+ channels that trigger periodic paralysis, sodium channel myotonia, or paramyotonia congenita.1, 2, 3 The NaPIC normally present is too small to cause depolarization sufficient to cause muscle inexcitability and periodic paralysis. Also, it inactivates over seconds and thus could not cause the sustained depolarization that underlies periodic paralysis. In addition, it is too small to trigger myotonia when Cl conductance is normal and thus only becomes pathologic when Cl conductance is low. However, when NaPIC is increased by mutations in the Nav1.4 sodium channel, it does become large enough to trigger myotonia despite normal muscle Cl conductance.1, 2, 3
Treating ClC‐1 Disorders by Targeting NaPIC
Our model of NaPIC in myotonia reveals an attractive target for developing novel therapies with minimized side effects. Previously, the only ion channel target available for treatment was the transient Na+ current responsible for the AP. Blocking this critical current poses serious difficulties, such as how to provide sufficient relief from involuntary APs while minimizing interference with voluntary APs, which can produce weakness and paralysis. Highlighting the importance of NaPIC to myotonia, we demonstrate that 2 different treatments that are effective against myotonia both reduce NaPIC. Our work suggests that reduction of NaPIC contributes to the efficacy of both treatments. Prior to our study, treatment with ranolazine and elevation of extracellular divalent cations were thought to eliminate myotonia by different mechanisms. As we had not yet identified NaPIC, we proposed that ranolazine was eliminating myotonia by enhancing slow inactivation of transient Na+ currents.21 Elevation of extracellular divalent cations was suggested to eliminate myotonia by a rightward shift in the voltage dependence of transient Na+ activation, such that the AP threshold shifted to more depolarized potentials.13, 30 In the current study, we found that the elimination of myotonia by both ranolazine and elevated divalent cations coincided with NaPIC inhibition and a parallel reduction in the slope of AfD such that the threshold for triggering myotonic APs was not reached. Thus, although both ranolazine and elevation of extracellular divalent cations may raise AP threshold through effects independent of NaPIC, this does not appear to be the mechanism underlying their efficacy against myotonia. These findings lead to the conclusion that targeting NaPIC may yield the most effective therapy for myotonia.
Author Contributions
All authors contributed to the conception and design of the study, drafting the text, and preparing the figures; A.A.H. performed the acquisition and analysis of data.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This work was supported by the NIH/NINDS (R01NS082354, M.M.R.), and Muscular Dystrophy Association (378033, M.M.R. A.A.V.).
Contributor Information
Andrew A. Voss, Email: andrew.voss@wright.edu.
Mark M. Rich, Email: mark.rich@wright.edu.
References
- 1. Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol 2015;5:761–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trivedi JR, Cannon SC, Griggs RC. Nondystrophic myotonia: challenges and future directions. Exp Neurol 2014;253:28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lehmann‐Horn F, Jurkat‐Rott K, Rudel R. Diagnostics and therapy of muscle channelopathies—Guidelines of the Ulm Muscle Centre. Acta Myol 2008;27:98–113. [PMC free article] [PubMed] [Google Scholar]
- 4. Lipicky RJ, Bryant SH, Salmon JH. Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. J Clin Invest 1971;50:2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992;257:797–800. [DOI] [PubMed] [Google Scholar]
- 6. Steinmeyer K, Klocke R, Ortland C, et al. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 1991;354:304–308. [DOI] [PubMed] [Google Scholar]
- 7. Peachey LD. Structure and function of the T‐system of vertebrate skeletal muscle In: Tower DB, ed. The nervous system. Vol 1 New York, NY: Raven Press, 1975:81–89. [Google Scholar]
- 8. Adrian RH, Bryant SH. On the repetitive discharge in myotonic muscle fibres. J Physiol 1974;240:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adrian RH, Marshall MW. Action potentials reconstructed in normal and myotonic muscle fibres. J Physiol 1976;258:125–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wallinga W, Meijer SL, Alberink MJ, et al. Modelling action potentials and membrane currents of mammalian skeletal muscle fibres in coherence with potassium concentration changes in the T‐tubular system. Eur Biophys J 1999;28:317–329. [DOI] [PubMed] [Google Scholar]
- 11. Fraser JA, Huang CL, Pedersen TH. Relationships between resting conductances, excitability, and t‐system ionic homeostasis in skeletal muscle. J Gen Physiol 2011;138:95–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burge JA, Hanna MG. Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr Neurol Neurosci Rep 2012;12:62–69. [DOI] [PubMed] [Google Scholar]
- 13. Skov M, Riisager A, Fraser JA, et al. Extracellular magnesium and calcium reduce myotonia in ClC‐1 inhibited rat muscle. Neuromuscul Disord 2013;23:489–502. [DOI] [PubMed] [Google Scholar]
- 14. Palade PT, Barchi RL. Characteristics of the chloride conductance in muscle fibers of the rat diaphragm. J Gen Physiol 1977;69:325–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 1991;354:301–304. [DOI] [PubMed] [Google Scholar]
- 16. Harvey PJ, Li Y, Li X, Bennett DJ. Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J Neurophysiol 2006;96:1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heckman CJ, Johnson M, Mottram C, Schuster J. Persistent inward currents in spinal motoneurons and their influence on human motoneuron firing patterns. Neuroscientist 2008;14:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heckman CJ, Enoka RM. Motor unit. Compr Physiol 2012;2:2629–2682. [DOI] [PubMed] [Google Scholar]
- 19. Powers RK, Nardelli P, Cope TC. Estimation of the contribution of intrinsic currents to motoneuron firing based on paired motoneuron discharge records in the decerebrate cat. J Neurophysiol 2008;100:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gage PW, Lamb GD, Wakefield BT. Transient and persistent sodium currents in normal and denervated mammalian skeletal muscle. J Physiol 1989;418:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Novak KR, Norman J, Mitchell JR, et al. Sodium channel slow inactivation as a therapeutic target for myotonia congenita. Ann Neurol 2015;77:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pedersen TH, de Paoli FV, Flatman JA, Nielsen OB. Regulation of ClC‐1 and KATP channels in action potential‐firing fast‐twitch muscle fibers. J Gen Physiol 2009;134:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waters CW, Varuzhanyan G, Talmadge RJ, Voss AA. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci U S A 2013;110:9160–9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miranda DR, Wong M, Romer SH, et al. Progressive Cl‐ channel defects reveal disrupted skeletal muscle maturation in R6/2 Huntington's mice. J Gen Physiol 2017;149:55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mehrke G, Brinkmeier H, Jockusch H. The myotonic mouse mutant ADR: electrophysiology of the muscle fiber. Muscle Nerve 1988;11:440–446. [DOI] [PubMed] [Google Scholar]
- 26. Standen NB, Stanfield PR. Rubidium block and rubidium permeability of the inward rectifier of frog skeletal muscle fibres. J Physiol 1980;304:415–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Struyk AF, Cannon SC. Paradoxical depolarization of BA2+‐treated muscle exposed to low extracellular K+: insights into resting potential abnormalities in hypokalemic paralysis. Muscle Nerve 2008;37:326–337. [DOI] [PubMed] [Google Scholar]
- 28. Palade PT, Barchi RL. On the inhibition of muscle membrane chloride conductance by aromatic carboxylic acids. J Gen Physiol 1977;69:879–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lossin C. Nav 1.4 slow‐inactivation: is it a player in the warm‐up phenomenon of myotonic disorders? Muscle Nerve 2013;47:483–487. [DOI] [PubMed] [Google Scholar]
- 30. Skov M, De Paoli FV, Lausten J, et al. Extracellular magnesium and calcium reduce myotonia in isolated ClC‐1 chloride channel‐inhibited human muscle. Muscle Nerve 2015;51:65–71. [DOI] [PubMed] [Google Scholar]
- 31. Desaphy JF, Carbonara R, Costanza T, Conte Camerino D. Preclinical evaluation of marketed sodium channel blockers in a rat model of myotonia discloses promising antimyotonic drugs. Exp Neurol 2014;255:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee RH, Heckman CJ. Adjustable amplification of synaptic input in the dendrites of spinal motoneurons in vivo. J Neurosci 2000;20:6734–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rich MM. The control of neuromuscular transmission in health and disease. Neuroscientist 2006;12:134–142. [DOI] [PubMed] [Google Scholar]
- 34. Yang JS, Sladky JT, Kallen RG, Barchi RL. TTX‐sensitive and TTX‐insensitive sodium channel mRNA transcripts are independently regulated in adult skeletal muscle after denervation. Neuron 1991;7:421–427. [DOI] [PubMed] [Google Scholar]
- 35. Patlak JB, Ortiz M. Two modes of gating during late Na+ channel currents in frog sartorius muscle. J Gen Physiol 1986;87:305–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. J Neurosci 1993;13:660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brocard C, Plantier V, Boulenguez P, et al. Cleavage of Na(+) channels by calpain increases persistent Na(+) current and promotes spasticity after spinal cord injury. Nat Med 2016;22:404–411. [DOI] [PubMed] [Google Scholar]
- 38. Yan H, Wang C, Marx SO, Pitt GS. Calmodulin limits pathogenic Na+ channel persistent current. J Gen Physiol 2017;149:277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]