

Abstract
A mechanistic study of arylations of aliphatic alcohols and hydroxide with diaryliodonium salts, to give alkyl aryl ethers and diaryl ethers, has been performed using experimental techniques and DFT calculations. Aryne intermediates have been trapped, and additives to avoid by‐product formation originating from arynes have been found. An alcohol oxidation pathway was observed in parallel to arylation; this is suggested to proceed by an intramolecular mechanism. Product formation pathways via ligand coupling and arynes have been compared, and 4‐coordinated transition states were found to be favored in reactions with alcohols. Furthermore, a novel, direct nucleophilic substitution pathway has been identified in reactions with electron‐deficient diaryliodonium salts.
Keywords: aryl ethers, arynes, hypervalent iodine, mechanistic study, oxidation
Introduction
The interest in hypervalent iodine chemistry has dramatically increased over the past decade, and a plethora of novel transformations has appeared under both metal‐free and metal‐catalyzed conditions.1 Apart from being excellent oxidants, hypervalent iodine reagents can be used in carbon ligand transfer to a variety of nucleophiles. Several interesting mechanistic studies have recently been reported on reactions with iodine(III) reagents, giving new insights in some of the key aspects of these reactions.2 The knowledge gained from these experimental and theoretical studies will certainly be highly important for further developments in the field.
Diaryliodonium salts (diaryl‐λ3‐iodanes) are increasingly applied in organic synthesis as efficient electrophilic arylation reagents with a wide range of nucleophiles.3 Their straightforward synthesis using one‐pot reactions, combined with high bench‐stability and low toxicity, has made them desirable in a growing number of transformations. In metal‐free reactions with nucleophiles, the generally accepted mechanism for these reagents proceeds via a ligand exchange to T‐shaped Nu‐I intermediates that subsequently react via a ligand coupling mechanism to provide the arylated nucleophile and the corresponding iodoarene in a regiospecific fashion (Scheme 1 a).4
Scheme 1.
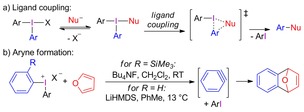
Mechanistic pathways in metal‐free arylations.
Aryl transfer via single electron transfer (SET) reactions is preferred under certain conditions, which has been investigated in detail by Kita and co‐workers.5 Less studied pathways include radical reactions and aryne formation, which have been suggested to explain by‐product formation in certain reactions with iodonium salts.6 While aryne intermediates have been applied in cycloadditions employing ortho‐silylated diaryliodonium salts or strongly basic conditions (Scheme 1 b), the mechanism for aryne formation from diaryliodonium salts remains unexplored.7
Our research group has gained considerable experience in metal‐free arylations with these reagents using O‐, N‐ and C‐centered nucleophiles.8 While most of these have been efficiently arylated by the ligand coupling pathway, several intriguing indications on alternative reaction pathways have been observed, for example, by formation of regioisomeric products and oxidized byproducts. Mechanistic insights to these fascinating observations would aid further developments with diaryliodonium salts and other iodine(III) reagents, and we thus initiated an investigation on competing pathways in O‐arylations using both experimental techniques and DFT calculations. Herein we report evidence for aryne intermediates as the origin of several products, as well as the use of amine additives to suppress their formation. A mechanism has been outlined to explain the unexpected oxidation of alcohols to carbonyl compounds with diaryliodonium salts, which are generally not used as oxidants. Furthermore, a novel, direct nucleophilic substitution pathway has been identified for reactions with electron‐deficient iodonium salts.
The mechanistic investigations were focused on O‐arylations of hydroxide and aliphatic alcohols,9 as the synthesis of aryl ethers is an important quest due to their abundance in natural products and pharmaceutically active compounds.10 Already sixty years ago, diaryliodonium salts were used to arylate alkoxides in moderate yields and with poor scope,11 and the reactions were later observed to suffer from severe by‐product formation.6a We recently developed an efficient and general synthesis of diaryl ethers by arylation of phenols (Scheme 2 a).9d, 9e Double arylation of sodium hydroxide with strongly electron‐withdrawing salt 1 a yielded diaryl ether 2 a (Scheme 2 b),9d and mild conditions were reported for the arylation of aliphatic alcohols 3 to aryl ethers 4 (Scheme 2 c).9c, 9d Contrary to the phenol arylation, the reactions with aliphatic alcohols proved sensitive to steric hindrance, and were most efficient for primary alcohols and electron‐deficient aryl groups. Unexpected oxidation of the alcohol substrate to the corresponding aldehyde/ketone or carboxylic acid was observed (Scheme 2 d), as well as formation of regioisomeric product mixtures in arylations with electron‐donating iodonium salts.
Scheme 2.
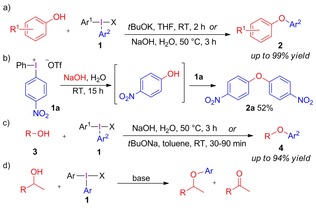
Synthesis of aryl ethers and oxidation.
Results and Discussion
Arylation of hydroxide
Based on the conditions in Scheme 2 b, a small screening with other diaryliodonium salts 1 was performed. The reactions generally required elevated temperature to proceed in water, whereas room temperature proved sufficient for reactions in dichloromethane.12 Surprisingly, a mixture of regioisomeric diaryl ethers 2, as well as iodo‐substituted by‐products 5 was obtained with salts 1 lacking strong electron‐withdrawing group (EWG) substituents in both solvents, as exemplified with di(4‐tolyl)iodonium triflate (1 b) in Scheme 3 a. As the ligand coupling mechanism is regiospecific, the formation of regioisomeric products 2 b–d indicated that another mechanism was operating.
Scheme 3.
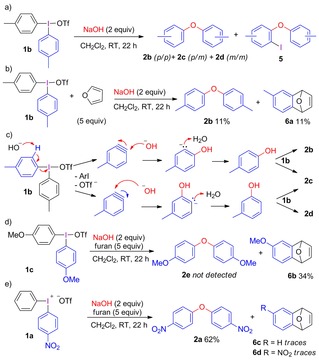
Aryne trapping and mechanistic proposal.
Aryne intermediates are known to react with nucleophiles with poor regioselectivity in the absence of strong directing groups,13 and aryne formation was hence hypothesized to explain the observed regioisomeric mixture of 2. This was supported by a trapping experiment with excess furan, giving Diels–Alder adduct 6 a together with diaryl ether 2 b as a single regioisomer (Scheme 3 b). Furthermore, the ortho‐iodo products 5 were no longer formed. Scheme 3 c depicts a plausible aryne mechanism leading to diaryl ethers 2 b–d, where deprotonation at the ortho‐position with elimination of the iodoarene gives the aryne. Unselective reaction with hydroxide would yield two regioisomeric anionic intermediates that are quickly protonated to the phenols. Subsequent arylation of the phenol9e, 14 by ligand coupling (cf. Scheme 1 a) would furnish diaryl ethers 2 b and 2 c. The formation of 2 d could originate from the attack of a phenoxide on the aryne, which could also lead to 2 c. The clean formation of 2 b in the presence of furan can be rationalized by efficient trapping of the arynes, shutting down the aryne pathway to aryl ether products, leaving ligand coupling as the only pathway leading to diaryl ether.
The substituents on the diaryliodonium salt 1 proved to strongly influence the outcome in reactions with hydroxide. With strongly electron‐donating methoxy substituents on the iodonium salt (1 c), only the aryne pathway operated and Diels–Alder adduct 6 b was the sole product (Scheme 3 d). On the contrary, electron‐deficient nitro salt 1 a gave diaryl ether 2 a as the main product with only traces of 6 c,d (Scheme 3 e).
The formation of by‐products 5, with iodine incorporated in the ortho‐position, was intriguing. While ortho‐iodinated diaryl ethers recently have been obtained from phenols with other iodine(III) reagents,15 their synthesis from diaryliodonium salts has not been reported. To simplify the analysis, diphenyliodonium triflate (1 d) was employed in the investigations into the formation of this product. Treatment of sodium hydroxide with salt 1 d resulted in formation of an inseparable mixture of diaryl ethers 2 f and 5 a (Table 1, entry 1). The product ratio was increased, at the cost of sharply reduced yield of 2 f, when only 1 equivalent NaOH was used (entry 2) The addition of furan as aryne trap delivered ether 2 f in diminished yield, together with the cycloaddition product 6 c (entry 3). By‐product 5 a was only obtained in trace amount, indicating that 5 a forms via an aryne pathway, whereas 2 f forms both via ligand coupling and via arynes.
Table 1.
Trapping of benzyne.
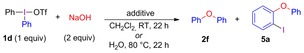
| |||||
|---|---|---|---|---|---|
| Entry | Conditions | Additive (equiv) | 2 f [%][a] | 5 a [%][a] | Other products[a] |
| 1 | CH2Cl2, RT | – | 40 | 25 | – |
| 2[b,c] | CH2Cl2, RT | – | 13 | 3 | – |
| 3 | CH2Cl2, RT | furan (5) | 23 | 2 |
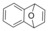
|
| 6 c 20 % | |||||
| 4 | CH2Cl2, RT | piperidine (0.5) | 27 | 0 |
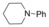
|
| 7 a 26 % | |||||
| 5 | H2O 80 °C | – | 56 | 3 | – |
| 6[c] | H2O 80 °C | piperidine (1.2) | 17 | 0 | 7 a 4 % |
[a] Isolated yields. [b] 1 equiv NaOH. [c] 1H NMR yields with 1,3,5‐trimethoxybenzene (TMB) as internal standard.
Other aryne scavengers were also considered, as furan did not completely suppress the formation of 5 a. Amines were deemed suitable as they readily react with arynes,16 but are difficult to arylate with diaryliodonium salts under metal‐free conditions.17 Piperidine was thus added in sub‐stoichiometric amounts, which indeed inhibited the formation of 5 a (entry 4). Likewise, addition of piperidine to the reaction of tolyl salt 1 b with hydroxide resulted in facile isolation of diaryl ether 2 b without concomitant formation of 2 c, 2 d and 5.12 In both cases, the formed N‐arylated trap 7 was easily separated from 2, making this additive useful in avoiding by‐products resulting from arynes at the expense of lower isolated yield of 2.
The product distribution was also investigated in other solvents. Reactions in water required elevated temperature to proceed, but resulted in increased yield of 2 f (entry 5). Interestingly, by‐product 5 a was only formed in trace amounts under these conditions. The addition of piperidine considerably suppressed formation of 2 f (entry 6).
When salt 1 d was treated with NaOD in deuterated water, diphenyl ether 2 f‐D was isolated as the only deuterated diaryl ether according to NMR and GC‐MS (Scheme 4).12 Deuterium‐free product 2 f was not detected, and by‐product 5 a was formed in 4 %, corresponding well to the reaction using NaOH (Table 1, entry 5). The labeling outcome is consistent with one of the two arylations taking place mainly via the aryne pathway in water.12
Scheme 4.
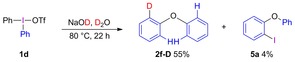
Deuterium labeling experiment.
Previous arylations of phenols in water did not suffer from by‐product formation (cf Scheme 2 a),9d indicating that the arylation of hydroxide proceeds via arynes, followed by regiospecific phenoxide arylation by ligand coupling. Phenolic products were not detected in either solvent, supporting the hypothesis that arylation of phenol intermediates is fast compared to arylation of hydroxide.
The competition between ligand coupling and aryne formation in arylations of hydroxide with diaryliodonium salts 1 a, 1 c, and 1 d was studied by DFT calculations using two different functionals (B3LYP‐D3, M06‐2X) commonly used for hypervalent iodine reactions.2a, 8b, 15c, 18 We started to investigate the reaction between 1 d and hydroxide in CH2Cl2 (Figure 1).19 Ligand exchange in iodine(III) compounds is considered to be facile,2h, 4e, 20 and formation of intermediate 1 d‐OH was followed by a large decrease in energy (−69 kJ mol−1), as expected by the different pK a of TfOH and H2O. Reactions via 4‐coordinated complexes have rarely been investigated in iodine(III) reactions,2b, 2e, 4e, 21 but were deemed interesting due to the stoichiometry of this transformation. Indeed, addition of a second hydroxide led to the more stable, 4‐coordinated iodonium complex 1 d‐(OH)2 (−73 kJ mol−1), whereas the mixed 4‐coordinated complex 1 d‐(OH)OTf was higher in energy.22
Figure 1.
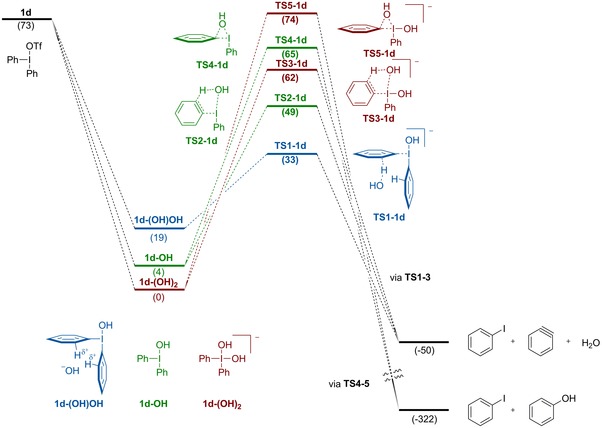
Free energy surface for the reaction between 1 d and hydroxide in CH2Cl2 using B3LYP‐D3. Dissociated OH and OTf are omitted for clarity. Energies are given in kJ mol−1.
Also complex 1 d‐(OH)OH, with a hydroxide coordinated to the ortho‐protons of 1 d‐OH, is higher in energy. Due to the fast equilibrium between these three intermediates, the transition state energies are calculated with respect to 1 d‐(OH)2 according to the Curtin–Hammett principle.23 The possible transitions states for deprotonation and elimination to the aryne are lower in energy than the ligand coupling transition states. External deprotonation (TS1‐1 d, +33 kJ mol−1) is favored over internal 3‐coordinated (TS2‐1 d), 4‐coordinated deprotonation (TS3‐1 d) or direct deprotonation of 1 d, as depicted in Scheme 3 b. The 3‐coordinated ligand coupling (TS4‐1 d) and the 4‐coordinated ligand coupling (TS5‐1 d) are significantly higher in energy. While the experimental results also show a preference for aryne formation, the calculated energy differences between the aryne formation and the ligand coupling are higher than expected with this diaryliodonium salt.
The results with methoxy‐substituted salt 1 c are similar to those with 1 d, with three intermediates in fast equilibrium, although the 3‐coordinated 1 c‐OH is slightly more stable than 1 c‐(OH)2 (Figure 2 a). The transition states energies for the aryne pathway are lowest in energy, and the external deprotonation via TS1‐1 c (+36 kJ mol−1) is again slightly favored over the internal deprotonation via TS2‐1 c (+46 kJ mol−1). The 4‐coordinated deprotonation (TS3‐1 c), the 3‐coordinated ligand coupling (TS4‐1 c) and the 4‐coordinated ligand coupling (TS5‐1 c), are all much higher in energy, in agreement with the experimental results where coupling product 2 e was not detected (Scheme 3 d).
Figure 2.
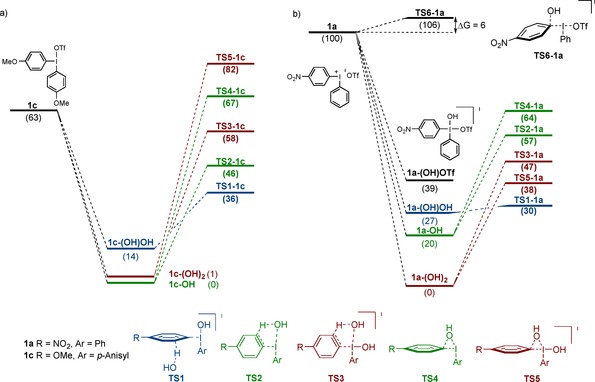
Free energy surface for the reaction between 1 a or 1 c and hydroxide in CH2Cl2. OH and OTf are omitted for clarity. Energies are given in kJ mol−1.
The reaction of nitro salt 1 a with hydroxide shows a rather different energy profile, with a clear preference for the 4‐coordinated intermediate 1 a‐(OH)2 over the 3‐coordinated 1 a‐OH (Figure 2 b). Surprisingly, the obtained transition state energies did not match the experimental results, where formation of product 2 a is strongly favored (Scheme 3 e). Instead, the external deprotonation via TS1‐1 a (+30 kJ mol−1) is slightly favored over the 4‐coordinated ligand coupling (TS5‐1 a, +38 kJ mol−1). While the barriers between the pathways are more similar than for 1 c and 1 d, aryne formation is still favored according to these calculations.
This unexpected finding inspired us to search for alternative mechanisms with salt 1 a, and a direct C‐attack of the hydroxide on the iodonium salt 1 a, without prior coordination to the iodine, indeed proved to have a very low barrier. TS6‐1 a (r O−I=3.2 Å) is only 6 kJ mol−1 higher in energy than 1 a, which explains the product distribution seen in Scheme 3 e. This is not a normal nucleophilic aromatic substitution (SNAr), as no Meisenheimer complex could be identified. Instead, TS6‐1 a directly leads to the phenol in a concerted nucleophilic aromatic substitution (CSNAr),24 similar to previous reports for vinyliodonium salts and ethynylbenziodoxolones.2c, 25
To fully compare this pathway to the alternative paths in Figure 2, we would need to calculate the barrier to attack of hydroxide directly on iodine, leading to 1 a‐(OH)OTf. We have been unable to locate this transition state; the energy change on approach is monotonous, indicating that this is a diffusion‐controlled reaction. Harvey and co‐workers have estimated that the free energy barrier corresponding to diffusion control is ca. 20 kJ mol−1.26 Since this is higher than the free energy barrier calculated for TS6‐1 a, we postulate that both reactions are under diffusion control, with the reaction preference determined by a branching point that is not connected to the actual free energy transition state. Such situations can be addressed, for example by dynamic simulations with randomized approach vectors,27 but such calculations are currently beyond the scope of our computational resources. We are satisfied that our calculations have revealed that both reaction paths are plausible, and the experiments clearly show that the preference is for attack on the ipso carbon. All attempts to find this type of TS for salt 1 c and 1 d were unsuccessful as they both showed preference for coordination to the iodine.12
Based on the experimental and theoretical results, we propose that the formation of diaryl ethers 2 from hydroxide takes place via three competing mechanisms. Ethers 2 are partly formed via the traditional ligand coupling mechanism that is depicted in Scheme 5 a, with ligand exchange to 1 d‐OH followed by regiospecific ligand coupling (LC). The intermediate phenol (A) quickly undergoes another arylation by LC. Diaryliodonium salts lacking strong EWG substituents partly react via the aryne mechanism shown in Scheme 5 b, and this pathway dominates with electron‐donating salts such as 1 c. In this pathway, a hydroxide deprotonates intermediate 1 d‐OH, (rather than salt 1 d directly, cf. Scheme 3 c), with concomitant elimination of iodobenzene to give benzyne B, which is attacked by a hydroxide to form anionic intermediate C.
Scheme 5.
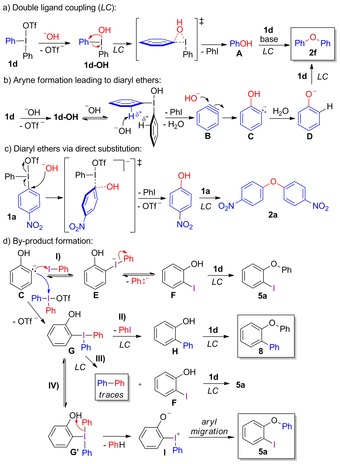
Proposed mechanisms for reactions with hydroxide.
A solvent‐mediated proton shift delivers phenoxide (D) followed by facile O‐arylation via LC to yield diaryl ether 2 f. Contrary to the ligand coupling, the intermediate and TS for the aryne pathway involves two hydroxides, which is in line with the observed product ratio variation with the stoichiometry of the reaction (cf. Table 1, entries 1–2). The competition between these pathways is solvent dependent, and reactions in CH2Cl2 proceed via both mechanisms whereas product formation in water takes place mainly via arynes.28 Reactions with strongly electron‐deficient salts result in regiospecific product formation, which could either proceed via ligand coupling, or via a low energy, direct substitution mechanism. This pathway was only found for nitro salt 1 a (Scheme 5 c), explaining the facile reactions with this salt.
The observed diaryl ether by‐products could be formed via several aryne pathways from intermediate C, as illustrated in Scheme 5 d. Aryne‐derived intermediates similar to C have been reported to attack the iodine of iodobenzene.29 Should C react with PhI, which is produced in the aryne formation step, the hypervalent iodine (ate) complex E could fragment to iodophenol F and a high‐energy phenyl carbanion. F could then be arylated by LC to give 5 a. However, the addition of another iodoarene did not alter the ratio between 2 f and 5 a,12 and as a high energy intermediate would be formed, this pathway seems unlikely.
Alternatively, C could attack the much better electrophile 1 d to give the T‐shaped triaryl intermediate G.30 Ligand coupling from G would deliver phenol H and iodobenzene, as depicted in pathway II.
Subsequent arylation of H by LC would yield ether 8, which has indeed been detected in minor amounts by GC‐MS. The other possible ligand coupling from triaryl intermediate G would give iodophenol F and biphenyl (pathway III). Subsequent arylation of F with 1 d by a ligand coupling would give 5 a. As only a trace amount of biphenyl has been detected, this cannot be the major pathway to 5 a.
To account for the observed formation of iodo ethers 5, we instead propose the novel mechanism depicted in pathway IV. Triaryl intermediate G is in fast equilibrium with the other T‐shaped intermediate G′, having the phenolic moiety in the equatorial position. In this conformation, a facile collapse of G′ into benzene and the zwitterionic intermediate I is plausible. Such iodonium phenolates are well known to undergo intramolecular rearrangement leading to ortho‐iodo substituted diaryl ethers like 5 a under mild conditions.31 The suppressed formation of 5 a in water compared to CH2Cl2 (Table 1, entries 1, 5) is explained by the facile proton shift from C to D in aqueous media, leading to product 2 f rather than 5 a.32
Arylation of aliphatic alcohols
The competition between ligand coupling and aryne formation was also found when primary aliphatic alcohols were arylated with electron‐donating diaryliodonium salts. This is exemplified by the arylation of 1‐pentanol (3 a) with p‐tolyl salt 1 b under our reported conditions,9c which resulted in a regioisomeric mixture of ethers 4 a–b (Table 2, entry 1). Contrary to reactions with hydroxide, only traces of iodo‐substituted by‐products were formed. The product ratio between 4 a and 4 b indicates that ligand coupling and aryne intermediates yield ethers in a 60:40 ratio. Diels–Alder adduct 6 a was indeed formed upon addition of furan to the reaction mixture, supporting the presence of arynes (entry 2).12 As by‐product 4 b could still be detected, the reaction was examined with amine additives to suppress the by‐product formation.
Table 2.
Arylation of 1‐pentanol.
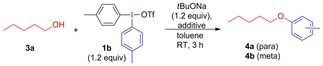
| ||||
|---|---|---|---|---|
| Entry | Additive (equiv) | 4 a+4 b [%][a] | Ratio 4 a:4 b | Other products[a] |
| 1 | – | 51 | 80:20 | – |
| 2[b] | furan (5) | 36 | nd | 6 a 9 % |
| 3 | piperidine (0.5) | 27 | 100:0 |
|
| 7 b (m)+7 c (p) 31 % | ||||
| 4[c] | piperidine (1.2) | 0 | – | – |
| 5 | NaHMDS (1.2) | 52[b] | 80:20 | – |
[a] Isolated yields. [b] 1H NMR yield with TMB as internal standard. [c] No tBuONa was added. nd=not determined due to overlapping peaks, mainly 4 a.
The use of piperidine changed the reaction outcome, and ether 4 a was isolated as the only regioisomer (entry 3). Other amines were also screened, but piperidine proved best.12 When piperidine was employed as both base and aryne trap, no ether was formed (entry 4).33 The use of the stronger base NaHMDS, which gives an amine after the initial deprotonation and might act as an internal trap, unfortunately delivered a regioisomeric mixture of the product (entry 5).
Similar types of by‐products, derived from aryne intermediates, were detected in arylations of both secondary and tertiary alcohols with electron‐donating diaryliodonium salts. Furthermore, primary and secondary alcohols suffered from partial oxidation of the alcohol to the corresponding aldehyde/ketone or carboxylic acid.9d Diaryliodonium salts are generally poor oxidants, in contrast to iodine(III) reagents with two heteroaromatic ligands,1b and McEwen and co‐workers suggested a radical pathway to explain the observed oxidation products in this type of transformation.6a
We envisioned that the oxidation could either proceed via radicals, arynes or the T‐shaped intermediate, which is formed prior to the ligand coupling. The oxidation was especially prominent with benzylic and allylic alcohols,9c, 9d and the reaction was hence investigated using secondary benzylic alcohol 3 b (Table 3). Ketone 9 was indeed formed as major product in the attempts to arylate 3 b with salt 1 d, delivering ether 4 c in poor yield under the reaction conditions optimized for primary9c and tertiary alcohols9a (entries 1 and 3). Only traces of an ortho‐iodo by‐product was identified, and the Diels–Alder adduct 6 d could barely be detected (<5 %) upon addition of furan.12 Addition of piperidine to the reaction only slightly changed the reaction outcome, (entries 2 and 4), indicating that arynes are not the main pathway in the oxidation mechanism.
Table 3.
Phenylation and oxidation of alcohol 3 b.[a]
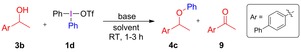
| |||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Additive (equiv) | 4 c [%][b] | 9 [%][b] |
| 1 | tBuONa | toluene | – | 21 | 60 |
| 2 | tBuONa | toluene | piperidine (1.2) | 14 | 54 |
| 3 | NaHMDS | pentane | – | 9 | 35 |
| 4 | NaHMDS | pentane | piperidine (1.2) | 5 | 30 |
| 5[c] | NaH | TBME | – | 30 | 53 |
| 6[c,d] | NaH | TBME | – | 11 | 65 |
| 7 | tBuONa | THF | – | 10 | 31 |
| 8 | tBuONa | THF | DPE (1.2) | 21 | 39 |
| 9 | tBuONa | THF | TEMPO (1.2) | 24 | 43 |
[a] Reaction conditions: see the Supporting Information. [b] 1H NMR yields using TMB as internal standard. [c] 3 b in excess, 50 °C for 1 h, isolated yields. [d] Ph(Mes)IBr was used instead of 1 d, chemoselectivity Ph:Mes 2:1.
In Stuart's recent methodology to arylate primary and secondary aliphatic alcohols, benzyl alcohol was arylated using aryl(mesityl)iodonium salts with only trace formation of benzaldehyde.9b However, submission of 3 b to these conditions, with either salt 1 d or unsymmetric Ph(Mes)IBr, delivered the oxidized product 9 as a major product and ether 4 c as a minor product (entries 5–6). Furthermore, the arylation chemoselectivity was only 2:1 in favor of phenyl over mesityl, illustrating the need for electron‐deficient aryl groups when using the mesityl as dummy group.
The effect of radical traps was examined in THF, as traps in toluene had no effect.9c, 12 While the yield of ether 4 c increased in the presence of 1,1‐diphenylethylene (DPE) or TEMPO (entries 7–9), the oxidation product 9 formed in similar amounts. This indicates that the oxidation does not take place via radicals, while radical pathways leading to other by‐products could play a role in THF.
Reactions between alcohol 3 b and ortho‐blocked dimesityl salt 1 e resulted in considerably higher arylation yields than with salts having ortho‐protons (Scheme 6 a). This could be rationalized by facilitated ligand coupling due to the ortho‐effect and aryne formation being impossible with 1 e.9a Importantly, the oxidation to ketone 9 still competed with the ether formation, and the combined yield of ether 4 d and 9 corresponds well to reactions with salt 1 d.
Scheme 6.
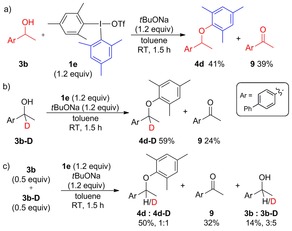
Arylations with ortho‐blocked salt 1 e.
The deuterated substrate 3 b‐D was arylated to ether 4 d‐D in higher yield, with less of ketone 9 compared to reactions with 3 b (Scheme 6 b). Deuterated mesitylene was detected by GC‐MS. A reaction containing both alcohols 3 b and 3 b‐D gave a good average yield of ethers 4 d and 4 d‐D in a 1:1 ratio, whereas the unreacted starting materials contained more of 3 b‐D (Scheme 6 c). The yield of 9 was affected in a similar fashion.
Having ruled out both arynes and radicals as oxidation promoters, we envisioned two possible mechanisms for the oxidation (Scheme 7). Both pathways go via the T‐shaped intermediate J, which forms from the deprotonated alcohol and salt 1 d. The benzylic proton could either be transferred to the phenyl ring in an intramolecular, concerted fashion to release 9, iodobenzene and benzene (Scheme 7 a), similar to mechanisms involving iodine(V) reagents.34
Scheme 7.
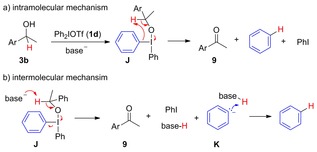
Plausible oxidation mechanisms.
Alternatively, intermediate J could be deprotonated by an external base, releasing 9, iodobenzene and benzene carbanion K, which is quickly protonated to benzene (Scheme 7 b). Assuming that both coupling and oxidation occurs from common intermediate J, the results in Scheme 6 clearly show that the proton transfer is subject to a primary kinetic isotope effect, which also implies that the deprotonation is rate limiting for the oxidation. As no increase in deuterium content of 4 d was observed, the intermediate must be in rapid equilibrium with free alcohol/alkoxide.
To distinguish between these mechanisms, excess base was added to the reaction of alcohol 3 b with mesityl salt 1 e. This neither affected the yield nor the ratio of 4 d and 9. The amount of oxidized product 9 also remained the same upon dilution of the reaction. The reaction mixtures were heterogeneous in toluene, but comparable ratios were obtained in homogenous solution (toluene/DMF).12 This indicates that the two processes are of the same order. As the ligand coupling mechanism is considered to be a concerted process (see Scheme 1 a), the oxidation should proceed via the intramolecular pathway shown in Scheme 7 a.
Further investigations were conducted using DFT. The arylation of 1‐phenylethoxide with mesityl salt 1 e was investigated, to avoid alternative pathways via arynes. The 4‐coordinated intermediate 1 e‐(OR)2 was found to be considerably lower in energy than the 3‐coordinated 1 e‐OR (Figure 3 a). Likewise, the TS corresponding to the 4‐coordinated coupling (TS7‐1 e) is strongly favored over any other TS corresponding to coupling or oxidation (TS8‐1 e to TS11‐1 e). The internal oxidation (TS9‐1 e) is much more favorable compared to the external pathway (TS11‐1 e). The same trends were observed using both the B3LYP‐D3 and the M06‐2X functional.12
Figure 3.
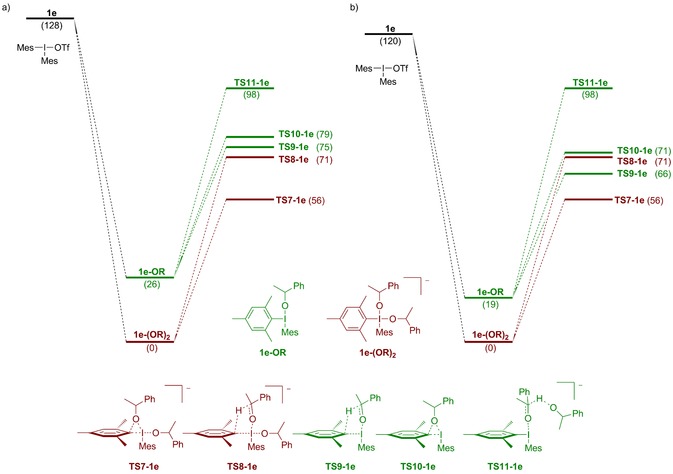
(a) Free energy surface for the reaction between 1 e and 1‐phenylethoxide in toluene using the B3LYP‐D3 functional (b) Free energy surface with an applied standard state correction. Free alkoxides and triflates are omitted for clarity. Energies are given in kJ mol−1.
Considering that the experimental conditions (0.1 m) diverge from the standard state conditions (1 m), this accounts for an energy penalty of around 7.9 kJ mol−1 for the reactions that are bimolecular with respect to the alkoxide.23, 35 Taking this into account, the energy for the 4‐coordinated TSs and intermediates increase and hence the gap between ligand coupling and oxidation (TS7‐1 e vs. TS9‐1 e) decreases to 10 kJ mol−1 using B3LYP‐D3 (Figure 3 b). This difference indicates a selectivity of >10:1 in favor of coupling, which is not experimentally observed.
The deviations between the experimental results (Scheme 6) and the DFT investigations could be caused by the chosen DFT method being insufficiently accurate in the current context. Discrepancies between DFT and experimental results are not uncommon. In particular, continuum solvation models can be unreliable for anions.36 In the current case, the relative energies of small anions (like hydroxide) and distributed anions (like square planar iodine(III) complexes) would be expected to be unreliable. Calculations with explicit water molecules around the nucleophile could provide more reliable energies,37 but were beyond our computational capacity. Within a set of similar compounds, errors will cancel to a large extent, but between sets, we have relied on experimental results to judge which of several manifolds that best describe the studied transformations.
Based on the combination of experimental and theoretical results, we believe that 4‐coordinated intermediates and transition states are favored in reactions with alcohols and diaryliodonium salts to give aryl ethers (Scheme 8). The ligand coupling pathway proceeds regiospecifically via intermediate 1 d‐(OR)2 (Scheme 8 a). Arynes were formed in reactions with diaryliodonium salts lacking EWG substituents. This pathway also yields aryl ethers, albeit with poor regioselectivity when substituted salts are employed. In another competing reaction pathway, the alcohol is oxidized to the corresponding ketone or aldehyde via an intramolecular mechanism (Scheme 8 b). The computational barriers for the 4‐ and 3‐coordinated TSs are similar. Still, we suggest that the oxidation mainly proceeds via the 4‐coordinated TS, since the experimental results show that oxidation and ligand coupling are of the same order. This side reaction is most prominent with allylic and benzylic alcohols.
Scheme 8.
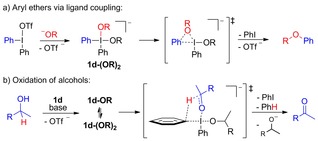
Mechanistic summary with alcohols.
Conclusion
A mechanistic investigation of O‐arylations of hydroxide and aliphatic alcohols with diaryliodonium salts under metal‐free conditions has been performed to understand the pathways that compete with ligand coupling. The study involved both experimental techniques and DFT calculations to understand the different reaction outcomes depending on the electronic properties of the diaryliodonium salt. Trapping experiments with furan were consistent with the formation of aryne intermediates under mild conditions. Piperidine proved to be a more efficient trap to avoid by‐products, which demonstrates an important proof of principle. While the aryne formation presently cannot be avoided in arylation of hydroxide and aliphatic alcohols with electron‐donating salts, the use of piperidine enables the synthesis and easy isolation of aryl ethers as single regioisomers in moderate isolated yields.
The oxidation of alcohols by diaryliodonium salts was found to proceed via a novel cyclic transition state, ruling out the involvement of arynes and radicals. Small differences in substrate and reagent structures were found to have large impacts on the reaction outcome with competing pathways via arynes, ligand coupling and oxidation. Furthermore, a direct substitution mechanism was found for arylations with nitrophenyl salt 1 a, explaining the facile synthesis of aryl ethers with this salt. We believe that the mechanistic insights presented herein, as well as the piperidine trap technique, can be utilized to develop novel reactions with iodine(III) reagents and to overcome the present limitations with these reagents.
Experimental Section
Arylation of hydroxide
Sodium hydroxide (1.6 mmol, 2 equiv) was added to a 10–20 mL or 2–5 mL microwave vial and CH2Cl2 or deionized H2O (3 mL) was added. The indicated additive was added followed by immediate addition of diaryliodonium salt 1 (0.8 mmol, 1 equiv). The reagents were rinsed down from the walls of the vial with solvent (1 mL). The vial was capped and stirred at RT for 22 h, or stirred in a pre‐heated oil bath at 80 °C for 22 h. The reaction mixture was quenched with sat. NH4Cl and then extracted with CH2Cl2 (×3). The combined organic phases were dried over MgSO4, filtrated and the solvent removed in vacuo. The crude mixture was then submitted to column chromatography.
Arylation of alcohols
A dry 10 mL Schlenk tube was evacuated and backfilled with argon three times. tBuONa (58 mg, 0.6 mmol, 1.2 equiv) was added, followed by anhydrous toluene (1.5 mL). The mixture was cooled to 0 °C and 1‐pentanol (54 μL, 0.5 mmol, 1 equiv) was added and rinsed down using toluene (0.5 mL). After stirring at RT for 15 min the mixture was cooled to 0 °C and additive (0.3–5.0 equiv) was added followed by salt 1 (0.6 mmol, 1.2 equiv). After rinsing down using toluene (0.5 mL) the mixture was left to stir at RT for 3 hours. The reaction was quenched using sat. NH4Cl, extracted with CH2Cl2 (3×10 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude was then submitted to flash column chromatography to obtain the product.
Computational methods
Geometry optimizations and energy calculations were carried out with the Becke Three‐Parameter Lee–Yang–Parr functional38 with the dispersion correction by Grimme39 (B3LYP‐D3) or the Minnesota functional, M06‐2X,12, 40 as implemented by Gaussian09. The SDD basis set with an applied effective core potential (MWB46) was used for iodine,41 Pople's triple‐zeta basis set with added polarization and diffuse functions (6–311+G(d,p))42 were used for N, O, F, Cl and Na atoms while the triple‐zeta basis set with added polarization (6–311G(d,p))42b, 42c were used for the H, C and S atoms. Several iodine(III) reactions have recently been studied by DFT using similar setups.2a, 8b, 15c, 18 The systems were studied in both CH2Cl2 and H2O using the polarizable continuum model (PCM, Surface=SES, Radii=UFF),43 and the results in H2O are given in the Supporting Information. Structures were optimized with the applied solvation model and true transition states were verified via frequency analysis and the presence of one, and only one, imaginary frequency.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Swedish Research Council (621‐2011‐3608 and 2015‐04404) is kindly acknowledged for financial support. This research was conducted using the resources of High Performance Computing Center North (HPC2N, SNIC 2016/3‐66). Dr. R. Ghosh is gratefully acknowledged for initial results.
E. Stridfeldt, E. Lindstedt, M. Reitti, J. Blid, P.-O. Norrby, B. Olofsson, Chem. Eur. J. 2017, 23, 13249.
Contributor Information
Dr. Elin Stridfeldt, http://www.organ.su.se/bo/
Prof. Berit Olofsson, Email: berit.olofsson@su.se.
References
- 1.
- 1a. Wirth T., in Top. Curr. Chem. Vol. 373, Springer International Publishing, Cham, 2016; [Google Scholar]
- 1b. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328–3435; [DOI] [PubMed] [Google Scholar]
- 1c. Zhdankin V. V., Hypervalent Iodine Chemistry, Wiley, Chichester, UK, 2014. [Google Scholar]
- 2.Selected references:
- 2a. Izquierdo S., Essafi S., del Rosal I., Vidossich P., Pleixats R., Vallribera A., Ujaque G., Lledós A., Shafir A., J. Am. Chem. Soc. 2016, 138, 12747–12750; [DOI] [PubMed] [Google Scholar]
- 2b. Sala O., Santschi N., Jungen S., Lüthi H. P., Iannuzzi M., Hauser N., Togni A., Chem. Eur. J. 2016, 22, 1704–1713; [DOI] [PubMed] [Google Scholar]
- 2c. Frei R., Wodrich M. D., Hari D. P., Borin P.-A., Chauvier C., Waser J., J. Am. Chem. Soc. 2014, 136, 16563–16573; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Arava S., Kumar J. N., Maksymenko S., Iron M. A., Parida K. N., Fristrup P., Szpilman A. M., Angew. Chem. Int. Ed. 2017, 56, 2599–2603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2643–2647; [Google Scholar]
- 2e. Zhang J., Szabó K. J., Himo F., ACS Catal. 2017, 7, 1093–1100; [Google Scholar]
- 2f. Colomer I., Batchelor-McAuley C., Odell B., Donohoe T. J., Compton R. G., J. Am. Chem. Soc. 2016, 138, 8855–8861; [DOI] [PubMed] [Google Scholar]
- 2g. Wu Y., Arenas I., Broomfield L. M., Martin E., Shafir A., Chem. Eur. J. 2015, 21, 18779–18784; [DOI] [PubMed] [Google Scholar]
- 2h. Beaulieu S., Legault C. Y., Chem. Eur. J. 2015, 21, 11206–11211; A recent review: [DOI] [PubMed] [Google Scholar]
- 2i.A. Sreenithya, K. Surya, R. B. Sunoj, WIREs Comput. Mol. Sci 2017, ##e1299, DOI: 1210.1002/wcms.1299.
- 3.
- 3a. Olofsson B., Top. Curr. Chem. 2015, 373, 135–166; [DOI] [PubMed] [Google Scholar]
- 3b. Aradi K., Tóth B. L., Tolnai G. L., Novák Z., Synlett 2016, 27, 1456–1485; [Google Scholar]
- 3c. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052–9070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214–9234. [Google Scholar]
- 4.
- 4a. Ochiai M., Top. Curr. Chem. 2003, 224, 5–68; [Google Scholar]
- 4b. Ochiai M., Kitagawa Y., Toyonari M., ARKIVOC (Gainesville, FL, U.S.) 2003, 4, 43–48; [Google Scholar]
- 4c. Pinto de Magalhães H., Lüthi H. P., Togni A., J. Org. Chem. 2014, 79, 8374–8382; [DOI] [PubMed] [Google Scholar]
- 4d. Pinto de Magalhães H., Lüthi H. P., Togni A., Org. Lett. 2012, 14, 3830–3833; [DOI] [PubMed] [Google Scholar]
- 4e. Norrby P.-O., Petersen T. B., Bielawski M., Olofsson B., Chem. Eur. J. 2010, 16, 8251–8254. [DOI] [PubMed] [Google Scholar]
- 5. Kita Y., Dohi T., Chem. Rec. 2015, 15, 886–906. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lubinkowski J. J., Knapczyk J. W., Calderon J. L., Petit L. R., McEwen W. E., J. Org. Chem. 1975, 40, 3010–3015; [Google Scholar]
- 6b. Beringer F. M., Huang S. J., J. Org. Chem. 1964, 29, 445–448; [Google Scholar]
- 6c. Akiyama T., Imasaki Y., Kawanisi M., Chem. Lett. 1974, 3, 229–230; [Google Scholar]
- 6d. Cadogan J. I. G., Rowley A. G., Sharp J. T., Sledzinski B., Wilson N. H., J. Chem. Soc. Perkin Trans. 1 1975, 1072–1074; [Google Scholar]
- 6e. Graskemper J. W., Wang B., Qin L., Neumann K. D., DiMagno S. G., Org. Lett. 2011, 13, 3158–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Kitamura T., Yamane M., J. Chem. Soc. Chem. Commun. 1995, 983–984; [Google Scholar]
- 7b. Kitamura T., Yamane M., Inoue K., Todaka M., Fukatsu N., Meng Z., Fujiwara Y., J. Am. Chem. Soc. 1999, 121, 11674–11679; [Google Scholar]
- 7c. Sundalam S. K., Nilova A., Seidl T. L., Stuart D. R., Angew. Chem. Int. Ed. 2016, 55, 8431–8434; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8571–8574; For N-arylation via aryne intermediates, see: [Google Scholar]
- 7d. Wang M., Huang Z., Org. Biomol. Chem. 2016, 14, 10185–10188. [DOI] [PubMed] [Google Scholar]
- 8.Selected references:
- 8a. Tolnai G. L., Nilsson U. J., Olofsson B., Angew. Chem. Int. Ed. 2016, 55, 11226–11230; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11392–11396; [Google Scholar]
- 8b. Reitti M., Villo P., Olofsson B., Angew. Chem. Int. Ed. 2016, 55, 8928–8932; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9074–9078; [Google Scholar]
- 8c. Dey C., Lindstedt E., Olofsson B., Org. Lett. 2015, 17, 4554–4557; [DOI] [PubMed] [Google Scholar]
- 8d. Ghosh R., Stridfeldt E., Olofsson B., Chem. Eur. J. 2014, 20, 4772. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Lindstedt E., Stridfeldt E., Olofsson B., Org. Lett. 2016, 18, 4234–4237; [DOI] [PubMed] [Google Scholar]
- 9b. Sundalam S. K., Stuart D. R., J. Org. Chem. 2015, 80, 6456–6466; [DOI] [PubMed] [Google Scholar]
- 9c. Ghosh R., Lindstedt E., Jalalian N., Olofsson B., ChemistryOpen 2014, 3, 54–57; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Lindstedt E., Ghosh R., Olofsson B., Org. Lett. 2013, 15, 6070–6073; [DOI] [PubMed] [Google Scholar]
- 9e. Jalalian N., Ishikawa E. E., L. F. Silva Jr. , Olofsson B., Org. Lett. 2011, 13, 1552–1555; Metal-catalyzed reactions: [DOI] [PubMed] [Google Scholar]
- 9f. Alvarado J., Fournier J., Zakarian A., Angew. Chem. Int. Ed. 2016, 55, 11625–11628; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11797–11800; [Google Scholar]
- 9g. Kuriyama M., Hamaguchi N., Onomura O., Chem. Eur. J. 2012, 18, 1591–1594; [DOI] [PubMed] [Google Scholar]
- 9h. Salvador T. K., Arnett C. H., Kundu S., Sapiezynski N. G., Bertke J. A., Raghibi Boroujeni M., Warren T. H., J. Am. Chem. Soc. 2016, 138, 16580–16583. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Roughley S. D., Jordan A. M., J. Med. Chem. 2011, 54, 3451–3479; [DOI] [PubMed] [Google Scholar]
- 10b. Satam V. S., Pedada S. R., Kamaraj P., Antao N., Singh A., Hindupur R. M., Pati H. N., Thompson A. M., Launay D., Martin D., Org. Process Res. Dev. 2017, 21, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beringer F. M., Brierley A., Drexler M., Gindler E. M., Lumpkin C. C., J. Am. Chem. Soc. 1953, 75, 2708–2712. [Google Scholar]
- 12.See supporting information for details.
- 13.
- 13a. Dong Y., Lipschutz M. I., Tilley T. D., Org. Lett. 2016, 18, 1530–1533; [DOI] [PubMed] [Google Scholar]
- 13b. Medina J. M., Mackey J. L., Garg N. K., Houk K. N., J. Am. Chem. Soc. 2014, 136, 15798–15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Jalalian N., Petersen T. B., Olofsson B., Chem. Eur. J. 2012, 18, 14140–14149; [DOI] [PubMed] [Google Scholar]
- 14b. Chan L., McNally A., Toh Q. Y., Mendoza A., Gaunt M. J., Chem. Sci. 2015, 6, 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Panda N., Mattan I., Nayak D. K., J. Org. Chem. 2015, 80, 6590–6597; [DOI] [PubMed] [Google Scholar]
- 15b. Hong F., Chen Y., Lu B., Cheng J., Adv. Synth. Catal. 2016, 358, 353–357; For similar iodine(III) reactions yielding ortho-iodinated products, see: [Google Scholar]
- 15c. Wu Y., Izquierdo S., Vidossich P., Lledós A., Shafir A., Angew. Chem. Int. Ed. 2016, 55, 7152–7156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7268–7272; [Google Scholar]
- 15d. Jia Z., Gálvez E., Sebastián R. M., Pleixats R., Álvarez-Larena Á., Martin E., Vallribera A., Shafir A., Angew. Chem. Int. Ed. 2014, 53, 11298–11301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11480–11483; [Google Scholar]
- 15e. Ochiai M., Ito T., Takaoka Y., Masaki Y., J. Am. Chem. Soc. 1991, 113, 1319–1323. [Google Scholar]
- 16. Bhunia A., Yetra S. R., Biju A. T., Chem. Soc. Rev. 2012, 41, 3140–3152. [DOI] [PubMed] [Google Scholar]
- 17. Sandtorv A. H., Stuart D. R., Angew. Chem. Int. Ed. 2016, 55, 15812–15815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16044–16047. [Google Scholar]
- 18. Ling L., Liu K., Li X., Li Y., ACS Catal. 2015, 5, 2458–2468. [Google Scholar]
- 19.All results with M06-2X are given in the ESI, as well as B3LYP-D3 results in H2O.
- 20. Zhao J., Li S., J. Org. Chem. 2017, 82, 2984–2991. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Dixon L. I., Carroll M. A., Gregson T. J., Ellames G. J., Harrington R. W., Clegg W., Org. Biomol. Chem. 2013, 11, 5877–5884; [DOI] [PubMed] [Google Scholar]
- 21b. Okuyama T., Takino T., Sato K., Ochiai M., J. Am. Chem. Soc. 1998, 120, 2275–2282. [Google Scholar]
- 22.All calculations were performed with 2 hydroxides. Free hydroxides are omitted for clarity in Figure 1.
- 23. Clot E., Norrby P.-O., in Innovative Catalysis in Organic Synthesis Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 167–191. [Google Scholar]
- 24. Neumann C. N., Hooker J. M., Ritter T., Nature 2016, 534, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Ochiai M., Yamamoto S., Suefuji T., Chen D.-W., Org. Lett. 2001, 3, 2753–2756; [DOI] [PubMed] [Google Scholar]
- 25b. Ochiai M., Oshima K., Masaki Y., J. Am. Chem. Soc. 1991, 113, 7059–7061. [Google Scholar]
- 26. McMullin C. L., Jover J., Harvey J. N., Fey N., Dalton Trans. 2010, 39, 10833–10836. [DOI] [PubMed] [Google Scholar]
- 27. Nieves-Quinones Y., Singleton D. A., J. Am. Chem. Soc. 2016, 138, 15167–15176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The diphenyl ether obtained in the presence of piperidine in CH2Cl2 is not formed via arynes; see the supporting information.
- 29.
- 29a. Tripathy S., LeBlanc R., Durst T., Org. Lett. 1999, 1, 1973–1975; [Google Scholar]
- 29b. Reich H. J., Green D. P., Phillips N. H., J. Am. Chem. Soc. 1989, 111, 3444–3445. [Google Scholar]
- 30.Triaryliodonium intermediates have been reported previously, see:
- 30a. Reich H. J., Cooperman C. S., J. Am. Chem. Soc. 1973, 95, 5077–5078; [Google Scholar]
- 30b. Wittig G., Rieber M., Justus Liebigs Ann. Chem. 1949, 562, 187–192. [Google Scholar]
- 31. Malamidou-Xenikaki E., Spyroudis S., Synlett 2008, 2725–2740. [Google Scholar]
- 32.As phenolates are regiospecifically arylated without byproduct formation (cf Scheme 2 a and ref. 9e), intermediate D cannot be involved in the byproduct formation.
- 33.We hypothesized that an alcohol coordinated to a diaryliodonium salt would be acidic enough to be deprotonated by piperidine.
- 34.
- 34a. Dess D. B., Martin J. C., J. Am. Chem. Soc. 1991, 113, 7277–7287; [Google Scholar]
- 34b. Su J. T., Goddard W. A., J. Am. Chem. Soc. 2005, 127, 14146–14147; [DOI] [PubMed] [Google Scholar]
- 34c. Satam V., Harad A., Rajule R., Pati H., Tetrahedron 2010, 66, 7659–7706. [Google Scholar]
- 35.Considering the heterogeneous nature of the reaction, the actual concentration might be lower than 0.1 m, in which case the standard state correction should be even larger.
- 36. Plata R. E., Singleton D. A., J. Am. Chem. Soc. 2015, 137, 3811–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sunoj R. B., Anand M., Phys. Chem. Chem. Phys. 2012, 14, 12715–12736. [DOI] [PubMed] [Google Scholar]
- 38. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 39. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 40. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 41. Bergner A., Dolg M., Küchle W., Stoll H., Preuß H., Mol. Phys. 1993, 80, 1431–1441. [Google Scholar]
- 42.
- 42a. Clark T., Chandrasekhar J., Spitznagel G. W., Schleyer P. V. R., J. Comput. Chem. 1983, 4, 294–301; [Google Scholar]
- 42b. Krishnan R., Binkley J. S., Seeger R., Pople J. A., J. Chem. Phys. 1980, 72, 650–654; [Google Scholar]
- 42c. McLean A. D., Chandler G. S., J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- 43.
- 43a. Cossi M., Scalmani G., Rega N., Barone V., J. Chem. Phys. 2002, 117, 43; [Google Scholar]
- 43b. Mennucci B., Tomasi J., J. Chem. Phys. 1997, 106, 5151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary