

Abstract
The present study aimed to elucidate the role of Notch signaling in the development of myocardial infarction (MI) concomitant with diabetes in vivo and in vitro and evaluated the therapeutic effect of the Notch signaling in vitro. Streptozotocin-induced diabetic rats were subjected to 25 min of ischemia and 2 h of reperfusion. Cardiac troponin T (cTnT) and creatine kinase-MB (CK-MB) isoenzyme levels were detected. Infarct size was measured by 2,3,5-triphenyltetrazolium chloride staining. Myocardial apoptosis and fibrosis were examined by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling and Masson Trichrome staining, respectively. The mRNA and protein levels of Notch signaling components, including Notch1, Notch4, Delta-like 1, Jagged1, Mastermind-like protein 1 and p300, were quantified by reverse transcription-quantitative polymerase chain reaction and western blotting analyses, respectively. H9c2 cells were treated with/without 33 mM high glucose (HG) and/or subjected to hypoxia in the presence/absence of Jagged1. Cell viability and apoptosis were determined by MTT assay and Annexin V-fluorescein isothiocyanate/propidium iodide assay. Levels of the Notch signaling pathway members were examined. The present findings revealed that diabetes elevated CK-MB and cTnT, increased infarct size, induced myocardial apoptosis and inhibited the Notch signaling pathway in vivo after ischemia/reperfusion. Ischemia/reperfusion augmented the severity of MI in diabetic rats. Furthermore, HG reduced cell viability and induced cell apoptosis in H9c2 cells after hypoxia exposure, which was inhibited by Jagged1. We also found that HG inhibited Notch signaling in H9c2 cells after hypoxia, whereas Jagged1 exerted its cardioprotective effect on hypoxic injury (in HG environments or not) by activating the Notch signaling pathway. In conclusion, these findings suggest that diabetes promoted the progression of MI in vivo and in vitro via the inhibition of the Notch signaling pathway. Jagged1 may protect against MI in in vitro models by activating Notch signaling.
Keywords: cardioprotection, diabetes mellitus, Jagged1, myocardial infarction, Notch signaling
Introduction
Cardiovascular diseases (CVDs), such as myocardial infarction (MI), atherosclerosis, vascular dysfunction, arrhythmias, hypertension, stroke and heart failure, lead to increased mortality in humans and the mortality rates are expected to increase to >23.6 million by 2030 (1,2). Interruption of the blood supply to the heart causes injury to cardiomyocytes and leads to MI (3). Diabetes mellitus (DM), or simply diabetes, is a metabolic disorder in which diabetic patients cannot appropriately regulate their blood glucose levels (4). Type 1 DM (T1DM) is characterized by the destruction of β cells due to an autoimmune response; whereas T2DM is characterized by obesity, insulin resistance and relative insulin deficiency (5,6). Greater than 90% of diabetic patients are diagnosed with T2DM (5). DM is known to increase the morbidity and mortality of patients with CVDs; the CVD-associated mortality rates are 2–8 times higher in diabetic patients than in non-diabetic patients (7). Pathogenic factors associated with CVD in T2DM include smoking, obesity, hypertension, stress, insufficient sleep, hyperglycemia, dyslipidemia, physical inactivity, clotting factors and pro-inflammatory cytokines (8). Therefore, it is of great importance that the mechanisms involved in DM-related CVDs are explored and clarified.
Notch signaling pathway is an evolutionarily conserved pathway that regulates cell proliferation, death, differentiation, cell-to-cell communication and angiogenesis (9,10). There are five Notch ligands (Jagged1, Jagged2, DLL1, DLL3 and DLL4) and four Notch receptors (Notch1, Notch2, Notch3 and Notch4) in mammals (11,12). Previous evidence has shown that Notch activation contributes to β-cell proliferation and dedifferentiation (13). Notch has a central role in diabetes (14) and the Notch signaling pathway is associated with cardioprotection in cardiac injury, as Notch signaling activation alleviates ischemic injury and modulates cardiac repair after MI (15). However, few studies have focused on the role of the Notch signaling pathway in the development of MI in diabetes.
The present study investigated whether diabetes influenced the development of MI using an in vivo model and an in vitro model. Following this, we assessed the activation of Notch signaling in DM-MI and evaluated the therapeutic efficiency of Notch signaling in the treatment of DM-related MI. The findings of our study may increase understanding pertaining to the prevention and control of MI in diabetic patients.
Materials and methods
Animals
A total of 90 male Sprague-Dawley (SD) rats (aged 8 weeks, 220–250 g) were purchased from Beijing Vital River Laboratories (Beijing, China). Our study was approved by the Ethics Committee of China Medical University (Shenyang, China) and the procedures were performed according to National Institutes of Health Guidelines for Use and Care of Animals. Rats were fed with water and standard chow ad libitum with a 12-h light/dark cycle; however, 12 h prior to the experiments, the rats were only permitted free access to water.
Experimental groups
SD rats were randomly assigned to five equal groups (n=18), as follows: i) Vehicle group: Rats were treated with sodium citrate buffer (pH 4.4) by intraperitoneal (i.p.) injection; ii) streptozotocin (STZ) group: Diabetes was induced in rats by i.p. injection of STZ (65 mg/kg bodyweight; Solarbio Science & Technology Co., Ltd., Beijing, China) dissolved in sodium citrate buffer. Blood glucose was measured 8 days after STZ injection. Diabetic rats (blood glucose, ≥396 mg/dl) were subsequently used; iii) STZ+Sham group: Diabetic rats underwent a sham operation; iv) Vehicle+ischemia/reperfusion (I/R) group: 8 days after the injection of sodium citrate buffer, rats were anaesthetized with 10% chloral hydrate (3.5 ml/kg bodyweight) and MI was induced (25 min of ischemia followed by 2 h of reperfusion) by left anterior descending coronary artery (LAD) ligation, as previously described (16); and v) STZ+I/R group: Diabetic rats were subjected to ischemia and reperfusion. Following reperfusion, the rats were sacrificed. Blood and heart samples were obtained.
Measurement of cardiac troponin T (cTnT) and creatine kinase-MB (CK-MB) isoenzyme
Serum samples were separated by centrifugation at 1,111 × g at 4°C for 10 min and used for ELISA assay measurement. cTnT and CK-MB were measured using respective cTnT (SED232Ra; USCN, Wuhan, China) and CK-MB (H197; Nanjing Jiancheng, Nanjing, China) ELISA kits, according to the manufacturer's instructions. Absorbance was determined at 450 nm using an ELx800 absorbance microplate reader (BioTek Instruments, Inc., Winooski, VA, USA). cTnT and CK-MB levels were calculated against standard curves.
Infarct size
Infarct size was evaluated using 2,3,5-triphenyltetrazolium chloride (TTC) staining. At the end of the reperfusion, the hearts were excised and immersed in 10% KCl solution. The hearts were frozen at −20°C and sectioned into five slices (2 mm) that were perpendicular to the long axis of the heart. Subsequently, the slices were stained with 1% TTC (Solarbio Science and Technology Co., Ltd.) at 37°C for 10–15 min in the dark and images were captured using a D3000 digital camera (Nikon Corporation, Tokyo, Japan). Infarct area and non-infarct area were analyzed using Image-Pro Plus version 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)
Myocardial apoptosis was determined by TUNEL (Roche Diagnostics GmbH, Basel, Switzerland). Heart tissues were embedded in paraffin and cut into 5-µm-thick sections. Then, the sections were deparaffinized in xylene twice and rehydrated in ethanol. Sections were washed with PBS and permeabilized with 0.1% Triton X-100. Endogenous peroxidase activity was blocked by H2O2. Sections were incubated with 50 µl TUNEL reaction mixture for 1 h at 37°C and then with 50 µl Converter-POD for 30 min. Following this, the sections were treated with DAB substrate, counterstained with hematoxylin and dehydrated. Images were captured under a DP73 microscope (Olympus Corp., Tokyo, Japan) at a magnification of ×400.
Masson Trichrome staining
Paraffin-embedded heart tissues were sectioned into 5-µm-thick sections and subjected to Masson Trichrome staining to evaluate the degree of myocardial fibrosis. Images of the stained sections were captured using a DP73 microscope at a magnification of ×200.
Cell culture and treatment groups
H9c2 cardiomyocytes were obtained from the cell bank at the Chinese Academy of Sciences (Shanghai, China). Cells were cultured at 37°C in Dulbecco's modified Eagle medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% FBS (HyClone, Logan, UT, USA) and placed in a 5% CO2 incubator. Cells were divided into six groups: i) Control group, in which un-treated H9c2 cells were cultured for 72 h under normoxic conditions and served as the controls; ii) hypoxia group, in which cells were cultured under normoxic conditions and then exposed to hypoxia (95% N2 and 5% CO2) 3 h prior to the end of cell culture; iii) high glucose (HG) group, in which cells were incubated with 33 mM glucose for 72 h; iv) hypoxia+HG group, in which cells were cultured with HG for a total of 72 h in a normoxic and then a hypoxic environment; v) hypoxia+Jagged1 group, in which, after Jagged1 (5 µg/ml) (GL Biochem, Shanghai, China) pretreatment, cells were cultured under normoxia and then subjected to hypoxic conditions; and vi) hypoxia+HG+Jagged1 group, in which, after Jagged1 pretreatment, cells were cultured in a normoxic and then a hypoxic environment in the presence of HG.
MTT assay
In order to determine cell viability, cells (3×103 cells/well) were cultured in 96-well plates at 37°C followed by the aforementioned treatment. The cells were incubated with MTT (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) (5 mg/ml) for 4 h at 37°C and then the medium was replaced with DMSO (Sigma-Aldrich; Merck KGaA). Absorbance was measured at 490 nm (BioTek Instruments, Inc.). Untreated cells cultured under normoxic conditions served as the controls.
Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI)
In order to determine the rate of apoptosis, following the indicated treatment, H9c2 cells were collected by centrifugation (309 × g for 5 min at room temperature) and washed with PBS twice. Cells were gently re-suspended in binding buffer containing Annexin V-FITC and PI (KeyGEN Biotech Co., Ltd., Nanjing, China) and incubated at room temperature for 15 min in the dark. Stained cells were analyzed using an Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues and cells using the RNA pure Extraction Kit (BioTeke Corp., Beijing, China), according to the manufacturer's instructions. RNA sample concentration was measured by UV spectrophotometry and RNAs were reverse-transcribed using M-MLV Reverse Transcriptase (BioTeke Corp.). Primers were synthesized by Sangon Biotech Co., Ltd., (Shanghai, China) and the primer sequences were as follows: Notch1, forward 5′-TGGCTCCATCGTCTACCTG-3′ and reverse 5′-GGCTCCACCGTCTCACTCT-3′; Notch4, forward 5′-GGACTAGGAAATCCCGAACC-3′ and reverse 5′-AACCTCCCGAGCATCAGC-3′; Jagged1, forward 5′-GTGCCGCCATAGGTAGAGT-3′ and reverse 5′-CCAGCCAACCACAGAAAC-3′; Delta-like 1 (DLL1), forward 5′-GGGACGATGTTCGGATAA-3′ and reverse 5′-TCGGCACAGGTAGGAGTT-3′; Mastermind-like protein 1 (MAML1), forward 5′-AGCAACAGTTTCAGCGTCAT-3′ and reverse 5′-GCACAGCAGCAGAAGGTC-3′; p300, forward 5′-ATGATGCCTCGGATGACA-3′ and reverse 5′-GACACTGGTGCTTGACTGC-3′; and β-actin, forward 5′-GGAGATTACTGCCCTGGCTCCTAGC-3′ and reverse 5′-GGCCGGACTCATCGTACTCCTGCTT-3′. Amplification was carried out on an Exicycler™ 96 Real-Time Quantitative Thermal Block (Bioneer Corp., Daejeon, Korea) and the amplification conditions were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec, 60°C for 20 sec and 72°C for 30 sec. Results were normalized to β-actin and analyzed using the 2−ΔΔCq formula (17).
Western blotting
Heart tissues and H9c2 cells were lysed using RIPA lysis buffer containing phenylmethylsulfonyl fluoride (both Beyotime Institute of Biotechnology, Haimen, China). Lysate supernatant was collected after centrifugation (10,005 × g, 10 min at 4°C). The protein concentration in the supernatant was determined via a bicinchoninic acid kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. Protein samples (40 µg per lane) were separated by 6/8/10% SDS-PAGE (6% SDS-PAGE, Notch4 and p300; 8% SDS-PAGE, Notch1, MAML1 and Jagged1; and 10% SDS-PAGE, DLL1) according to the molecular weight of proteins and transferred to polyvinylidene difluoride membranes (EMD Millipore, Bedford, MA, USA). Membranes were incubated with non-fat milk to eliminate non-specific binding and subsequently incubated with antibodies against Notch1 (cat. no. sc-373891), Notch4 (cat. no. sc-393893), Jagged1 (cat. no. sc-6011), p300 (cat. no. sc-585; all 1:200; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA), DLL1 (bs-7345R) and MAML1 (bs-12396R) (both 1:500; both BIOSS, Beijing, China) at 4°C overnight. Following washing 4 times (5 min each time) with Tris buffered saline with Tween-20, the membranes were incubated with horseradish peroxidase-conjugated donkey anti-goat, goat anti-mouse and goat anti-rabbit secondary antibodies (A0181/A0216/A0208; 1:5,000; Beyotime Institute of Biotechnology) for 45 min at 37°C. Protein bands were visualized using enhanced chemiluminescence regent (Beyotime Institute of Biotechnology). Results were analyzed using Gel-Pro analyzer version 4.0 software (Media Cybernetics) and expression levels were normalized to β-actin.
Statistical analysis
Data were expressed as the mean ± standard deviation and analyzed using one-way analysis of variance, followed by Bonferroni's multiple comparison test. P<0.05 was considered to indicate a statistically significant difference.
Results
Diabetes increases the levels of cardiac markers in diabetic rats after MI
To establish a rat model of DM and MI, STZ-induced diabetic rats were subjected to LAD ligation to induce MI 8 days after STZ injection. We measured serum levels of cardiac markers, cTnT (Fig. 1A) and CK-MB (Fig. 1B), using respective ELISA kits. Results showed that diabetes did not influence serum levels of cTnT and CK-MB in comparison with the control rats. cTnT and CK-MB levels in the STZ+I/R group were markedly higher than those in the STZ+Sham group. Compared with the Vehicle+I/R group, STZ injection increased the levels of cTnT and CK-MB, although the difference was not statistically significant.
Figure 1.
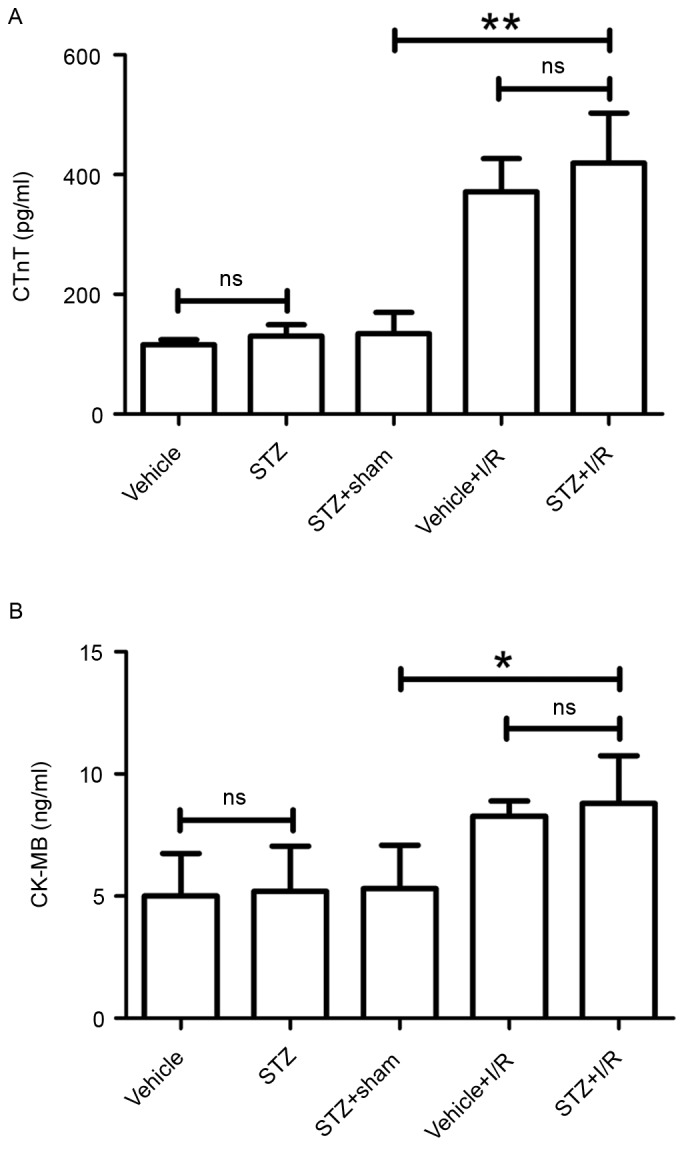
Determination of myocardial infarction by the levels of cTnT and CK-MB isoenzyme. (A) cTnT levels were measured using a commercial ELISA kit. Results are presented as pg/ml. (B) CK-MB levels were detected using a commercial ELISA kit. Results are presented as ng/ml. Data are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01. cTnT, cardiac troponin T; CK-MB, creatine kinase-MB; STZ, streptozotocin; I/R, ischemia/reperfusion; ns, not significant.
Diabetes increases infarct size in diabetic rats after MI
At the end of reperfusion, hearts were excised from the rats in each group and subjected to TTC staining (Fig. 2). We found that no infarct was observed in the rats of the Vehicle, STZ and STZ+Sham groups, respectively. Compared with the STZ+Sham group, I/R induced a larger infarct size in diabetic rats from nondetectable to 30.13±6.90%. Compared with the rats in the Vehicle+I/R group, diabetes led to a significant increase in infarct size from 22.46±4.67 to 30.13±6.90%.
Figure 2.
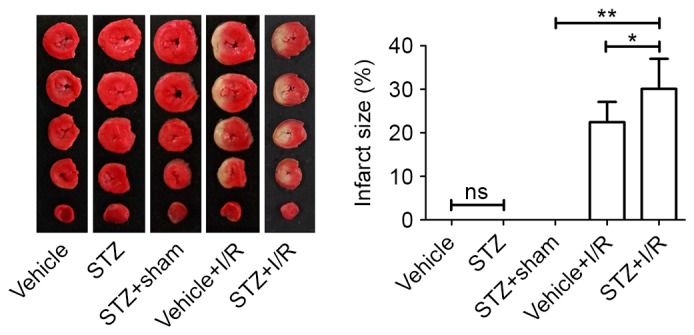
Measurement of infarct size. Following reperfusion, we excised the hearts from the rats and 2,3,5-triphenyltetrazolium chloride staining was performed to assess infarct size. Data are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01. STZ, streptozotocin; I/R, ischemia/reperfusion; ns, not significant.
Effect of diabetes on myocardial apoptosis and fibrosis
Diabetic rats showed an increased myocardial apoptotic rate (TUNEL-positive) (7.89±2.38%) compared with the control rats (4.44±1.86%); however, the difference was not statistically significant (Fig. 3). Ischemia followed by reperfusion promoted myocardial apoptosis in the myocardium of diabetic rats (STZ+I/R group, 19.87±3.07% vs. STZ+Sham group, 8.73±2.09%). In comparison with that of the Vehicle+I/R group (14.86±2.71%), the apoptotic rate in the STZ+I/R group (19.87±3.07%) was significantly increased. Myocardial fibrosis was subsequently examined by Masson Trichrome staining (Fig. 4), which demonstrated that diabetes slightly induced myocardial fibrosis and I/R significantly increased the degree of myocardial fibrosis in diabetic rats.
Figure 3.

Myocardial apoptosis in the myocardium. Paraffin-embedded rat heart tissues were sectioned and myocardial apoptosis was measured by Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling. Apoptotic cell rate (%) was calculated. Scale bar, 50 µm. Magnification, ×400. Data are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01. STZ, streptozotocin; I/R, ischemia/reperfusion; ns, not significant.
Figure 4.

Myocardial fibrosis in the myocardium. Heart tissues were embedded and cut into sections (5-µm thick). Myocardial fibrosis was examined by Masson Trichrome staining. Scale bar, 100 µm. Magnification, ×200. STZ, streptozotocin; I/R, ischemia/reperfusion.
Inactivation of Notch signaling in diabetic rats after I/R
We measured the activation of Notch signaling. The results showed that diabetes increased the mRNA expression levels of all Notch signaling members assessed, including Notch1, Notch4, DLL1, Jagged1, MAML1 and p300 (Fig. 5). We further confirmed, via western blotting analysis, that the protein levels were also upregulated (Fig. 6). Compared with the STZ+Sham group, the mRNA and protein expression levels of all Notch signaling members were markedly decreased in rats from the STZ+I/R group. STZ+I/R group rats exhibited lower mRNA and protein expression levels of Notch signaling members compared with the Vehicle+I/R rats. In particular, Notch1 and p300 mRNA and protein expression levels were significantly downregulated in the STZ+I/R group.
Figure 5.

Role of the Notch signaling pathway in DM-MI in vivo. Total RNA was isolated from myocardial tissues and mRNA levels of Notch1, Notch4, DLL1, Jagged1, MAML1 and p300 were determined by reverse transcription-quantitative polymerase chain reaction. β-actin served as an internal control. Data are expressed as the mean ± standard deviation. **P<0.01. STZ, streptozotocin; I/R, ischemia/reperfusion; DM, diabetes mellitus; MI, myocardial infarction; DLL1, Delta-like 1; MAML1, Mastermind-like protein 1; ns, not significant.
Figure 6.

Role of the Notch signaling pathway in DM-MI in vivo. Total protein was extracted from myocardial tissues and Notch1, Notch4, DLL1, Jagged1, MAML1 and p300 protein expression levels were determined by western blotting. β-actin served as a loading control. Data are expressed as the mean ± standard deviation. **P<0.01. STZ, streptozotocin; I/R, ischemia/reperfusion; DM, diabetes mellitus; MI, myocardial infarction; DLL1, Delta-like 1; MAML1, Mastermind-like protein 1; ns, not significant.
Jagged1 reverses the low viability and high apoptosis rate of H9c2 cells induced by HG and hypoxia exposure
Hypoxia exposure or HG treatment alone resulted in significantly reduced H9c2 cell viability (Fig. 7A) and significantly increased apoptosis rate (Fig. 7B), when compared with the respective controls. Following exposure to both hypoxia and HG, cell viability was significantly reduced and cell apoptosis rate was significantly increased compared with the Hypoxia group or HG group. In the Hypoxia+Jagged1 group, Jagged1 promoted cell survival and inhibited hypoxia-induced cell apoptosis. Jagged1 notably improved the loss in cell viability and myocardial cell apoptosis induced by hypoxia plus HG.
Figure 7.

Effect of Jagged1 on H9c2 cell viability and apoptosis. (A) Following the indicated treatments, cell viability was measured by MTT assay. (B) Harvested cells were subjected to Annexin V-FITC/PI assay. Apoptotic cell rate was analyzed using a flow cytometer. Data are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01. FITC, fluorescein isothiocyanate; PI, propidium iodide; HG, high glucose.
Jagged1 activates the Notch signaling pathway in H9c2 cells exposed to hypoxia and HG
As shown in Fig. 8, hypoxia exposure or HG treatment induced the activation of Notch signaling pathway compared with the control group. However, hypoxia plus HG decreased the upregulated levels of Notch1, Notch4, DLL1 and Jagged1 in H9c2 cells induced by hypoxia or HG. Compared with the Hypoxia group, Jagged1 further elevated Notch1, Notch4, DLL1 and Jagged1 levels in H9c2 cells. In the Hypoxia+HG+Jagged1 group, Jagged1 reversed inactivation of the Notch signaling pathway induced by hypoxia plus HG.
Figure 8.

Effect of Jagged1 on the Notch signaling pathway in vitro. (A) Gene expression levels of Notch1, Notch4, DLL1 and Jagged1 were measured by reverse transcription-quantitative polymerase chain reaction. Results were normalized to β-actin expression. (B) Protein expression levels of Notch1, Notch4, DLL1 and Jagged1 were measured by western blotting and normalized to β-actin. Data are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01. HG, high glucose; DLL1, Delta-like 1; ns, not significant.
Discussion
Previous studies have shown that hearts from diabetic animals are more sensitive to ischemic injury than non-diabetic animals (18,19). In the present study, we initially investigated whether diabetes influences myocardial injury via the Notch signaling in diabetic rats and in vitro. We subsequently studied whether regulation of Notch signaling improves DM-related MI using an in vitro model.
CK-MB is an isoenzyme of CK and a serum marker of myocardial injury. Troponins are the components of contractile apparatus in cardiac and skeletal muscle (20). cTnT is a cardiac-specific biomarker of MI and cTnT is released from damaged myocardial tissues during MI (21,22). In the present study, we found that I/R surgery induced increases in serum levels of CK-MB and cTnT in diabetic rats. STZ injection slightly elevated serum CK-MB and cTnT in I/R rats, indicating the aggravation of MI, which is consistent with previous evidence (23). We subsequently measured infarct size in each group using TTC staining. The results showed that diabetes did not influence infarct size. I/R significantly elevated infarct size in diabetic rats and diabetes led to a larger infarct size in rats after I/R, which is in accordance with previous research (24). The results suggest that diabetes may aggravate myocardial I/R injury by influencing myocardial injury markers and infarct size.
Previous studies have revealed that myocardial apoptosis has a critical role in the pathogenesis of heart disease, including myocardial infarction and heart failure (25,26). In our study, we demonstrated that I/R significantly promoted myocardial apoptosis in diabetic rats and diabetes induced myocardial apoptosis in I/R rats. This suggests that diabetes may aggravate myocardial I/R injury via its pro-apoptotic effect in myocardium. Excessive fibrosis is known to decrease cardiac contractility and disrupt cardiac function after MI (27). We found that I/R significantly induced myocardial fibrosis in diabetic rats.
Ligand-mediated Notch activation results in the release of Notch IC, which is an intracellular receptor domain of Notch, into the nucleus to regulate the expression of target genes (28). MAML1 is a co-activator of Notch signaling. MAML1 directly interacts with Notch, p300 (a co-activator with histone acetyltransferase activity) and histones to regulate Notch IC-mediated gene transcription (28,29). Recent evidence has shown that Notch signaling activation has a beneficial effect on myocardial injury and protects the myocardium against myocardial injury (15,30). However, whether Notch signaling is involved in the progression of DM-related MI remains unknown. We found that Notch signaling was activated in diabetic rats, which is consistent with the findings of previous studies (31,32). Furthermore, diabetes inhibited Notch signaling in rats after the I/R procedure, suggesting that diabetes may aggravate myocardial I/R injury by inhibiting the Notch signaling pathway.
Yang et al (33) revealed that Jagged1 attenuates ischemia-induced cardiac injury in isolated hearts during I/R. Additionally, Boopathy et al (34) found that Jagged1-containing hydrogels contribute to cardiac repair and improve cardiac function when compared with scrambled peptide hydrogels. Pei et al (35) have reported that Jagged1 alleviates myocardial I/R injury in mice. However, no reports have clarified the effect of Jagged1 on myocardial I/R injury in vivo and in vitro. In the present study, H9c2 cells subjected to hypoxia were used as an in vitro I/R model. Cell viability was examined by MTT assay and cell apoptosis was assessed by Annexin V-FITC/PI assay. We found that hypoxia plus HG significantly reduced H9c2 cell viability and increased the cell apoptosis rate compared with the HG group or Hypoxia group. Jagged1 treatment notably promoted cell survival and inhibited cell apoptosis in H9c2 cardiomyocytes subjected to hypoxia or hypoxia+HG. Jagged1 markedly reversed the pro-apoptotic effect and inhibitory effect on cell survival. The results suggest that Jagged1 may alleviate hypoxic injury by promoting cardiomyocyte survival and inhibiting cardiomyocyte apoptosis.
Following these investigations, we measured the expression of Notch signaling components (Notch1, Notch4, DLL1 and Jagged1) using RT-qPCR and western blotting analyses. Our data showed that hypoxia plus HG further inhibited the activation of Notch pathway in comparison with the Hypoxia group or HG group, which was consistent with our in vivo studies. However, Jagged1 significantly activated the Notch signaling pathway in H9c2 cells subjected to hypoxia or hypoxia+HG, which suggests that Jagged1 may alleviate hypoxic injury by activating the Notch signaling pathway.
In conclusion, diabetes promoted the progression of MI by inhibiting the Notch signaling pathway in the present in vivo and in vitro models. Furthermore, it was demonstrated that Jagged1 may alleviate I/R injury and hypoxic injury by activating Notch signaling. Our study provides novel data on potential treatment strategies for MI in patients with diabetes.
Acknowledgements
This study was supported by grants from the Science and Technology Project of Department of Education, Liaoning Province (grant no. L2014321) and the Open Foundation of Key Laboratory for Myocardial Ischemia, Ministry of Education (grant no. KF201515).
References
- 1.Bonnefont-Rousselot D. Resveratrol and cardiovascular diseases. Nutrients. 2016;8 doi: 10.3390/nu8050250. pii: E250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Du Y, Xu X, Chu M, Guo Y, Wang J. Air particulate matter and cardiovascular disease: The epidemiological, biomedical and clinical evidence. J Thorac Dis. 2016;8:E8–E19. doi: 10.3978/j.issn.2072-1439.2015.11.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elkhader BA, Abdulla AA, Omer Ali MA. Correlation of smoking and myocardial infarction among sudanese male patients above 40 years of age. Pol J Radiol. 2016;81:138–140. doi: 10.12659/PJR.894068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenny GP, Sigal RJ, McGinn R. Body temperature regulation in diabetes. Temperature (Austin) 2016;3:119–145. doi: 10.1080/23328940.2015.1131506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saisho Y. Postprandial C-peptide to glucose ratio as a marker of β cell function: Implication for the management of type 2 diabetes. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17050744. pii: E744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi B, Huang G, Zhou Z. Different role of zinc transporter 8 between type 1 diabetes mellitus and type 2 diabetes mellitus. J Diabetes Investig. 2016;7:459–465. doi: 10.1111/jdi.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundy SM, Howard B, Smith S, Jr, Eckel R, Redberg R, Bonow RO. Prevention conference VI: Diabetes and cardiovascular disease: Executive summary: Conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation. 2002;105:2231–2239. doi: 10.1161/01.CIR.0000013952.86046.DD. [DOI] [PubMed] [Google Scholar]
- 8.Joseph JJ, Donner TW. Long-term insulin glargine therapy in type 2 diabetes mellitus: A focus on cardiovascular outcomes. Vasc Health Risk Manag. 2015;11:107–116. doi: 10.2147/VHRM.S50286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toonen JA, Ronchetti A, Sidjanin DJ. A Disintegrin and metalloproteinase10 (ADAM10) regulates NOTCH signaling during early retinal development. PLoS One. 2016;11:e0156184. doi: 10.1371/journal.pone.0156184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lubecka K, Kurzava L, Flower K, Buvala H, Zhang H, Teegarden D, Camarillo I, Suderman M, Kuang S, Andrisani O, et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis. 2016;37:656–668. doi: 10.1093/carcin/bgw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JJ, Gao XT, Yang L, Fu W, Liang L, Li JC, Hu B, Sun ZJ, Huang SY, Zhang YZ, et al. Disruption of Notch signaling aggravates irradiation-induced bone marrow injury, which is ameliorated by a soluble Dll1 ligand through Csf2rb2 upregulation. Sci Rep. 2016;6:26003. doi: 10.1038/srep26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crabtree JS, Singleton CS, Miele L. Notch signaling in neuroendocrine tumors. Front Oncol. 2016;6:94. doi: 10.3389/fonc.2016.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bar Y, Efrat S. The NOTCH pathway in β-cell growth and differentiation. Vitam Horm. 2014;95:391–405. doi: 10.1016/B978-0-12-800174-5.00015-6. [DOI] [PubMed] [Google Scholar]
- 14.Kim W, Shin YK, Kim BJ, Egan JM. Notch signaling in pancreatic endocrine cell and diabetes. Biochem Biophys Res Commun. 2010;392:247–251. doi: 10.1016/j.bbrc.2009.12.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Hiroi Y, Liao JK. Notch signaling as an important mediator of cardiac repair and regeneration after myocardial infarction. Trends Cardiovasc Med. 2010;20:228–231. doi: 10.1016/j.tcm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckle T, Grenz A, Köhler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F, Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–H2540. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, Lei S, Liu Z, Ng KF, Wong GT, et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med. 2013;63:291–303. doi: 10.1016/j.freeradbiomed.2013.05.043. [DOI] [PubMed] [Google Scholar]
- 19.Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: Phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology. 2011;114:1364–1372. doi: 10.1097/ALN.0b013e31820efafd. [DOI] [PubMed] [Google Scholar]
- 20.Carretón E, Morchón R, González-Miguel J, Juste MC, Simón F, Montoya-Alonso JA. Utility of cardiac biomarkers during adulticide treatment of heartworm disease (Dirofilaria immitis) in dogs. Vet Parasitol. 2013;197:244–250. doi: 10.1016/j.vetpar.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Starnberg K, Jeppsson A, Lindahl B, Hammarsten O. Revision of the troponin T release mechanism from damaged human myocardium. Clin Chem. 2014;60:1098–1104. doi: 10.1373/clinchem.2013.217943. [DOI] [PubMed] [Google Scholar]
- 22.Pawula M, Altintas Z, Tothill IE. SPR detection of cardiac troponin T for acute myocardial infarction. Talanta. 2016;146:823–830. doi: 10.1016/j.talanta.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Xia ZY, Dou J, Zhang L, Xu JJ, Zhao B, Lei S, Liu HM. Protective effect of ginsenoside Rb1 against myocardial ischemia/reperfusion injury in streptozotocin-induced diabetic rats. Mol Biol Rep. 2011;38:4327–4335. doi: 10.1007/s11033-010-0558-4. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Liu Z, Wang J, Wong GT, Cheung CW, Zhang L, Chen C, Xia Z, Irwin MG. Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc Diabetol. 2013;12:133. doi: 10.1186/1475-2840-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo KQ, Long HB, Xu BC. Reduced apoptosis after acute myocardial infarction by simvastatin. Cell Biochem Biophys. 2015;71:735–740. doi: 10.1007/s12013-014-0257-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Wang Z, Feng SJ, Xu L, Shi HX, Chen LL, Yuan GD, Yan W, Zhuang W, Zhang YQ, et al. PEDF improves cardiac function in rats with acute myocardial infarction via inhibiting vascular permeability and cardiomyocyte apoptosis. Int J Mol Sci. 2015;16:5618–5634. doi: 10.3390/ijms16035618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tony H, Meng K, Wu B, Yu A, Zeng Q, Yu K, Zhong Y. MicroRNA-208a dysregulates apoptosis genes expression and promotes cardiomyocyte apoptosis during ischemia and its silencing improves cardiac function after myocardial infarction. Mediators Inflamm. 2015;2015:479123. doi: 10.1155/2015/479123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallberg AE, Pedersen K, Lendahl U, Roeder RG. P300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol Cell Biol. 2002;22:7812–7819. doi: 10.1128/MCB.22.22.7812-7819.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saint Just, Ribeiro M, Hansson ML, Wallberg AE. A proline repeat domain in the Notch co-activator MAML1 is important for the p300-mediated acetylation of MAML1. Biochem J. 2007;404:289–298. doi: 10.1042/BJ20061900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felician G, Collesi C, Lusic M, Martinelli V, Ferro MD, Zentilin L, Zacchigna S, Giacca M. Epigenetic modification at Notch responsive promoters blunts efficacy of inducing notch pathway reactivation after myocardial infarction. Circ Res. 2014;115:636–649. doi: 10.1161/CIRCRESAHA.115.304517. [DOI] [PubMed] [Google Scholar]
- 31.Min XH, Yu T, Qing Q, Yuan YH, Zhong W, Chen GC, Zhao LN, Deng N, Zhang LF, Chen QK. Abnormal differentiation of intestinal epithelium and intestinal barrier dysfunction in diabetic mice associated with depressed Notch/NICD transduction in Notch/Hes1 signal pathway. Cell Biol Int. 2014;38:1194–1204. doi: 10.1002/cbin.10323. [DOI] [PubMed] [Google Scholar]
- 32.Gao F, Yao M, Cao Y, Liu S, Liu Q, Duan H. Valsartan ameliorates podocyte loss in diabetic mice through the Notch pathway. Int J Mol Med. 2016;37:1328–1336. doi: 10.3892/ijmm.2016.2525. [DOI] [PubMed] [Google Scholar]
- 33.Yang H, Sun W, Quan N, Wang L, Chu D, Cates C, Liu Q, Zheng Y, Li J. Cardioprotective actions of Notch1 against myocardial infarction via LKB1-dependent AMPK signaling pathway. Biochem Pharmacol. 2016;108:47–57. doi: 10.1016/j.bcp.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boopathy AV, Martinez MD, Smith AW, Brown ME, García AJ, Davis ME. Intramyocardial delivery of notch ligand-containing hydrogels improves cardiac function and angiogenesis following infarction. Tissue Eng Part A. 2015;21:2315–2322. doi: 10.1089/ten.tea.2014.0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pei H, Song X, Peng C, Tan Y, Li Y, Li X, Ma S, Wang Q, Huang R, Yang D, et al. TNF-α inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic Biol Med. 2015;82:114–121. doi: 10.1016/j.freeradbiomed.2015.02.002. [DOI] [PubMed] [Google Scholar]