

Abstract
As a key molecule involved in cell recognition, calreticulin (CRT) may be expressed on the surface of (pre-) apoptotic cells and provide the signal that is recognized by dendritic cells (DCs) or other antigen presenting cells (APCs), which results in phagocytosis. Within the APCs, tumor-associated antigens (TAAs) may be subsequently presented to T lymphocytes, which triggers a specific antitumor immune response. It has been hypothesized that CRT is able to act as the immunologic adjuvant and translocate itself and TAAs to the cell surface and induce a specific antitumor immune response. In the present study, CRT was demonstrated to translocate itself and mucin 1 (MUC1), a breast cancer antigen, to the surface of 4T1 cells and the MUC1-CRT-coated cells were able to induce apoptosis in a time-dependent manner. When DCs were infected with adenovirus containing MUC1-CRT, an increase in T cell proliferation and cytokine production was exhibited. These results suggest that CRT may act as an immunologic adjuvant with MUC1 and induce a strong immune response.
Keywords: calreticulin, mucin 1, dendritic cells, breast cancer, tumor-immunity
Introduction
Surgical resection, combination chemotherapy and radiotherapy have been acknowledged to improve patient prognosis as the multimodal treatment for advanced cancer (1,2). However, overall survival has remained low in advanced cancer and is a challenging obstruction to overcome (3). Globally, various clinical trials have investigated the associations between tumor and host immune responses induced by immunotherapy (4,5). Previous results have suggested that immunotherapy has indicated potent antitumor activity in melanoma, non-small cell lung cancer and other tumors (6). Given its superior efficacy and innovation, tumor immunotherapy was named as the most important scientific breakthrough by Science magazine in 2013 and is expected to become the ‘big bang’ in cancer treatment, in comparison with surgery, chemotherapy, radiation therapy and targeted therapy (7).
One important factor of tumor generation is that mutated tumor cells are able to escape from immunological surveillance by lowering the expression of membrane marker molecules, which have an essential role in the process of cell recognition and phagocytosis (8,9). Restoring or stimulating the immune response of the body against cancer has been expected to result in effective tumor prevention and treatment. Numerous studies have demonstrated that, with the exception of causing apoptosis, when tumor cells were treated with chemotherapeutic agents (including anthracyclines and platinum compounds), cancer cells are able to release ATP and high mobility group box 1 protein, causing an antitumor immune response via the process known as tumor immunogenic apoptosis (10–12). Previous results have indicated that calreticulin (CRT), an antigen-presenting component, is a key molecule involved in antigen recognition during cancer chemotherapy (13). In 2007, Obeid et al (14) discovered that when murine colon carcinoma CT-26 cells were treated with the antitumor agent, anthracycline, this caused translocation of CRT from the endoplasmic reticulum (ER) to the cell surface, thereby acting as a phagocytic signal for dendritic cells (DCs).
CRT is a highly conserved 60-kDa Ca2+ binding protein, which is ubiquitous in mammalian cells and is predominantly located in the ER lumen (15). CRT has various biological functions that are relevant to its subcellular localization, such as chaperone activity, lectin binding, Ca2+ homeostasis regulation, cell adhesion signaling and removal of apoptotic cells (16). Furthermore, a previous study has revealed that CRT translocation from the ER to the cell surface was the key step involved in the recognition and clearance of apoptotic cells by phagocytosis (17). Additionally, Zeng et al (18) indicated that as a specific marker on the surface of (pre-)apoptotic cells, CRT may be recognized by DCs or other antigen presenting cells (APCs), which may lead to the collective phagocytosis of apoptotic cells. Subsequently, within the APCs, tumor-associated antigens (TAAs) or tumor-specific antigens (TSA) may be processed, presented to cluster of differentiation (CD)4+ and CD8+ T lymphocytes and trigger a specific antitumor immune response (19,20). It has been hypothesized that CRT may be used as an immunologic adjuvant to translocate itself and TAA to the cell surface and induce a potent antitumor immune response.
Breast cancer is the most common cancer that causes severe cancer-related fatality in women across Europe and the USA (21–23). In the present study, mucin 1 (MUC1), a type I transmembrane glycoprotein that is overexpressed in breast cancer cells, was used as a TAA (24,25). In vitro studies have demonstrated that the expression of MUC1 is involved in the invasion and resistance to genotoxic anticancer reagents, suggesting its close association with the poor prognosis of patients with breast cancer (26,27). Furthermore, previous results have revealed that MUC1 is a diagnostic or prognostic marker and may be a therapeutic target in breast cancer (28).
The present study focused on the ability of CRT to promote MUC1 localization on the cell surface and the ability of MUC1-CRT-infected DCs to induce a potent specific immunological effect. The present findings may lead to an improved antitumor immunotherapy modality against breast cancer.
Materials and methods
Experimental animals
Ethical approval from the Medical Animal Care and Welfare Committee of China Three Gorges University (Yichang, China) was obtained prior to animal use in the present study. A total of 17 male BALB/c mice (18±2 g, 4–6 weeks old) were purchased from the Laboratory Animal Center of China Three Gorges University. All mice were housed in specific pathogen-free conditions, with free access to food and water. The ambient temperature was maintained at 22±2°C with a humidity of 50–60% and a 12 h light/dark cycle.
Pharmacological agents and chemicals
Scientific TurboFect transfection reagents were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA; cat. no. R0532). Mitoxantrone (MIT) was purchased from Jiangsu Aosaikang Pharmaceutical Co., Ltd. (Jiangsu, China). Mouse granulocyte-macrophage colony-stimulating factor (mGM-CSF) and mouse interleukin-4 (mIL-4) were purchased from PeproTech, Inc. (Rocky Hill, NJ, USA; cat. no. 315-03 and 500-p45, respectively). Anti-mouse CD80-fluorescein isothiocyanate (FITC), anti-mouse CD86-FITC, anti-mouse CD11c-FITC, anti-mouse CD8-Alexa Fluor 700 and anti-mouse CD4-Pacific Blue antibodies were purchased from eBioscience, Inc. (San Diego, CA, USA; cat. no. 11-0801, 11-0862, 11-0114, 56-0081-80, and 48-0041, respectively). Anti-mouse CD3-peridinin chlorophyll protein complex (PerCP), anti-mouse CD4-Pacific Blue and anti-mouse CD8-Alexa Flour 700 antibody were purchased from Abcam (Cambridge, UK; cat. no. ab106215). Streptomycin and penicillin were purchased from Sigma-Aldrich (Merck KGaA Darmstadt, Germany). Adenoviruses containing MUC1 and CRT-MUC1 were constructed by Hanheng Biotechnology Co., Ltd. (Shanghai, China). Interferon (IFN)-γ and tumor necrosis factor (TNF)-α ELISA kits were purchased from Wuhan Boshide Biological Technology Company (Wuhan, China; cat. no. BMS233, and 740001, respectively). RPMI-1640 medium, calf serum and fetal bovine serum (FBS) were purchased from Gibco (Thermo Fisher Scientific, Inc.). All primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
Cell culture
BALB/c mice were sacrificed with carbon dioxide (CO2) and death was confirmed by cervical dislocation. Bone marrow dendritic cells (BMDCs) were flushed from the femurs and tibiae of BALB/c mice and cultured in RPMI-1640 medium with 10 ng/ml mGM-CSF and 10 ng/ml mIL-4. Following 48 h of culture in an atmosphere containing 5% CO2 at a temperature of 37°C, adherent cells were collected and fresh medium containing mGM-CSF (10 ng/ml) and mIL-4 (10 ng/ml) was added. Once cultured for 120 h, cells were seeded at 1×106 cells/ml and flow cytometry (EPICS XL-4) was performed to identify BMDCs using anti-mouse CD80-FITC, anti-mouse CD86-FITC and anti-mouse CD11c-FITC antibodies. The murine breast cancer cell line 4T1 (ATCC, Manassas, VA, USA) was maintained in our laboratory and cultured in complete media (RPMI-1640 medium supplemented with 10 mmol/l L-glutamine, 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin) in a humidified incubator containing 5% CO2 at 37°C.
Detection of MUC1-CRT subcellular localization in pre-apoptotic 4T1 cells by fluorescence microscopy
Overlap polymerase chain reaction (PCR) was used to amplify CRT (GeneBank no. NM_007591.3), MUC1 (GeneBank no. NM_013605.2) and MUC1-CRT using primers (Table I). The reaction mixture (25 µl) consisted of 2.5 µl of each primer, 4 µl reaction buffer, 500 µM dNTPs and 3% DMSO. Cycling conditions were as follows: 98°C for 3 min, 37–44°C for 3 min 20 sec, 72°C for 3 min for two cycles, then added 2 µl of MUC1-CRT_F/R and cycled at 98°C for 30 sec, 65°C for 30 sec, 72°C for 3 min, 24 times. Subsequently, CRT and MUC1 were linked to pEGFP-c1 vector to construct pEGFP-CRT, pEGFP-MUC1 and pEGFP-MUC1-CRT plasmids, respectively. Fluorescence microscopy was used to observe the subcellular localization of MUC1-CRT. 4T1 cells were seeded into 12-well culture plates (2×104 cells/well in 1 ml RPMI-1640 medium). pEGFP-CRT, pEGFP-MUC1 and pEGFP-MUC1-CRT were transiently transfected to 4T1 cells according to the manufacturer's protocol and treated with MIT (8 µg/ml) for 12 h. Finally, the cells were washed with PBS, enclosed in 50% glycerol (diluted in 0.01 mol/l PBS, pH 8.0) and observed by fluorescence and phase-contrast microscopy (magnification, ×400; TE2000S; Nikon Corporation, Tokyo, Japan).
Table I.
Primer pairs.
| Primer name | Sequences (5′-3′) | Restriction enzyme |
|---|---|---|
| CRT_F | GGTTCTGTCGACGACCCTGCCATCTATTTC | SalI |
| CRT_R | TACGGATCCCTACAGCTCATCCTTGGC | BamHI |
| MUC1_F | AATAGTCGACCCGGACACCAGGCCGGCCCC | SalI |
| MUC1_R | ATATGGATCCGGCCGAGGTGACACCATGGG | BamHI |
| MUC1-CRT_F | GGTCACGCGTAGAACCGCCGGCCGAGG | MluI |
| MUC1-CRT_R | GGTTCTACGCGTGACCCTGCCATCTATTTC | MluI |
| MUC1-q_F | TCTTTCCAACCCAGGACACC | – |
| MUC1-q_R | TCCTCATAGGGGCTACGCTT | – |
| GAPDH-q_F | CGCTCTCTGCTCCTCCTGTT | – |
| GAPDH-q_R | CCATGGTGTCTGAGCGATGT | – |
The restriction size is underlined. F, forward; R, reverse; q, quantitative polymerase chain reaction; MUC1, mucin; CRT, calreticulin.
Detection of MUC1 and CRT in apoptotic 4T1 cells
DNA fragmentation and fluorescence microscopy were used to detect whether CRT may promote MUC1 translocation to the cell surface of apoptotic 4T1 cells and whether CRT and MUC1 jointly promote a phagocytic signal in apoptotic 4T1 cells, which were induced by MIT. DNA fragmentation assay was performed as described by Cao et al (29), and non-treated cells were used as negative control. pEGFP-CRT, pEGFP-MUC1 and pEGFP-MUC1-CRT were transiently transfected into 4T1 cells and treated with MIT (10 µg/ml) for 24 h. The supernatant of lysed cells was extracted to precipitate DNA fragments and loaded onto a 2% (w/v) agarose gel for electrophoresis, and the results were visualized using a GEL-doc™ XR+System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). O' Gene Ruler 1 kb plus DNA ladder and Gel red (Thermo Fisher Scientific, Inc.) were used. Fluorescence microscopy was conducted to detect whether the apoptotic behavior observed was dose and/or time-dependent. pEGFP-MUC1-CRT was transiently transfected into 4T1 cells and treated with MIT (0, 2, 4 or 8 µg/ml) for 12 or 24 h, respectively, cells were washed with PBS, enclosed in 50% glycerol (diluted in 0.01 mol/l PBS, pH 8.0) and were observed by fluorescence and phase-contrast microscopy (magnification, ×400; TE2000S; Nikon Corporation).
T cell proliferation in MUC1-CRT-infected BMDCs
BMDCs were seeded onto 12-well culture plates (1×104 cells/well in 1 ml RPMI-1640 medium) in triplicate and infected with adenoviruses (multiplicity of infection, 100) containing MUC1 and MUC1-CRT, respectively. Successful infection was verified by reverse transcription-quantitative PCR (RT-qPCR) using the primers listed in Table I. In brief, RNA were extracted from BMDCs with TRLzol according to the manufacturer's protocol (Thermo Fisher Scientific, Inc.) and used as a template. A total of 1 ng/µl RNA and 1 µl oligodT was used for RT, and incubated in an Opticn2 Thermal Cycler (MJ Research Inc., St. Bruno, Canada) at 65°C for 5 min. The final reaction volume (20 µl) comprised 8 µl 5X reaction buffer, 10 mM dNTP Mix, RevertAid and RiboLock RNAase inhibitor mixture (Thermo Fisher Scientific, Inc.; cat. no. K1691). The cycling conditions were as follows: 1 h at 42°C, 5 min at 70°C. qPCR was performed with SYBR PrimeScript kit (RR086A; Takara Bio, Inc., Otsu, Japan) using the Opticn2 Thermal Cycler. Primers used for amplifying MUC1 were MUC1-q_F/R and primers for GAPDH were GAPDH-q_F/R (Table I). The PCR cycling conditions were as follows: 95°C for 5 min, 94°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec, 78°C for 1 sec for 30 cycles and a final extension at 72°C for 10 min. Expressions were quantified using the 2−ΔΔCq method (30). Splenocytes were harvested and single cell suspensions were prepared and co-incubated with lentivirus-infected BMDCs at 37°C in an atmosphere containing 5% CO2 for 72 h. The cell suspension was subsequently centrifuged at 400 × g for 5 min at 4°C. The supernatant was discarded and the pellet washed 3 times with PBS. The pellet was resuspended in flow cytometry staining buffer (eBioscience, Inc.; cat. no. 00-4222-26) so that the final cell concentration was 1×107 cells/ml. CD3+/CD4+/CD8+ T cells were detected by flow cytometry using anti-CD3-PerCP, anti-CD4-Pacific Blue and anti-CD8-Alexa Fluor 700 antibodies (all 1:1,000), and non-treated cells were used as negative control. Briefly, cells were resuspended in 1 ml PBS with formaldehyde in a final concentration of 4%, fixed for 10 min at 37°C, and washed twice with PBS. Cells were blocked with blocking IgG (1:100; eBioscience, Inc.; cat. no. 14-9161-73) at 4°C for 15 min, and antibodies and flow cytometry staining buffer were added to cells, mixed gently and incubated for 30 min on ice in the dark. The cells were washed twice in flow cytometry staining buffer and centrifuged at 400 × g for 5 min at room temperature. Cells were subsequently re-suspended in 300 µl flow cytometry staining buffer and detected by BD LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and results were analyzed by FlowJo 10.7 (FlowJo, LLC, Ashland, OR, USA).
Cytokine production induced by MUC1-CRT-infected BMDCs
For the measurement of cytokine production, splenic lymphocytes were separated and collected by lymphocyte separation medium (eBiscience, Inc.) according to the manufacturer's protocol. Cells were washed with PBS, and enclosed in 50% glycerol (diluted in 0.01 mol/l PBS, pH 8.0). Purification was verified by light field microscopy at ×400 magnification. BMDCs were infected with adenoviruses as outlined previously and co-incubated with lymphocytes at 37°C in 5% CO2 for 72 h. Culture supernatants (100 µl/well) were used to analyze IFN-γ and TNF-α levels using the respective ELISA kits (Wuhan Boshide Biological Engineering Company, Ltd., Wuhan, China; cat. no. BMS233 and 740001) in accordance with the manufacturer's protocol. Non-treated cells were used as control, and cells treated with mock vehicle were used as null control (Nco).
Statistical analysis
All data were presented as mean ± standard deviation. Statistical analysis was assessed between groups using the Student's two-tailed t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Plasmid construction
Overlap PCR was used to amplify CRT, MUC1 and MUC1-CRT using the primers listed in Table I, which yielded 1,200, 60 and 1,275 bp fragments, respectively. Each fragment was linked to the pEGFP-c1 vector to construct pEGFP-CRT, pEGFP-MUC1 and pEGFP-MUC1-CRT plasmids, which produced fragments 5,916, 4,776 and 5,991 bp, respectively (data not shown).
CRT promotes MUC1 to translocate to the cell surface of pre-apoptotic 4T1 cells
In order to determine whether CRT was able to promote MUC1 localization on the cell surface of 4T1 cells, fluorescence and phase-contrast microscopy was performed. pEGFP-CRT, pEGFP-MUC1 and pEGFP-MUC1-CRT were constructed, transiently transfected into 4T1 cells, treated with MIT (8 µg/ml) for 12 h and analyzed using fluorescence and phase-contrast microscopy. As indicated by the green fluorescence distribution in Fig. 1, the subcellular localization of MUC1-CRT and CRT was similar and they were observed to be primarily localized in the cell cytoplasm of pre-apoptotic 4T1 cells. Furthermore, no obvious morphological changes were exhibited. Additionally, MUC1 appeared to be localized in cytoplasm and nuclei. Previous studies have indicated that the antitumor agent, MIT, may induce apoptosis in murine CT-26 and B16-F1 cells and translocation of CRT from the ER to the cell surface (11,29). The present results suggested that CRT has the ability to translocate itself and MUC1 to the surface of 4T1 cells.
Figure 1.

Detection of the subcellular localization of MUC1-CRT in 4T1 cells. Fluorescence microscopy was performed to elucidate the subcellular localization of MUCI-CRT in 4T1 cells. (A) pEGFP-CRT, (B) pEGFP-MUC1 and (C) pEGFP-MUC1-CRT were transiently transfected into 4T1 cells, treated with mitoxantrone (8 µg/ml) and cultured for 12 h prior to fluorescence microscopy analysis. Magnification, ×400. MUC1, mucin 1; CRT, calreticulin.
MUC1-CRT-coated 4T1 cells induce a potent apoptosis signal
In order to detect whether CRT is able to associate with TAA to induce a potent apoptotic signal, fluorescence microscopy analysis and DNA fragmentation assay were performed. DNA fragmentation assay demonstrated that, following treatment with MIT (10 µg/ml) for 24 h, typical DNA ladders for apoptosis were observed in the 4T1 control cells and the pEGFP-MUC1 cells. Conversely, the intensity of the DNA ladder was increased in MUC1-CRT-transfected 4T1 cells and pEGFP-MUC1-CRT cells (Fig. 2). Furthermore, pEGFP-MUC1-CRT was transiently transfected into 4T1 cells and treated with MIT (0, 2, 4 or 8 µg/ml) for 12 and 24 h, respectively. The results indicated that MUC1-CRT transfected 4T1 cells exhibited increased apoptotic bodies in a MIT dose- and time-dependent manner (Fig. 3). Both of these results suggest that CRT was able to work with MUC1 as a phagocytic signal on the surface of apoptotic 4T1 cells to induce apoptosis.
Figure 2.
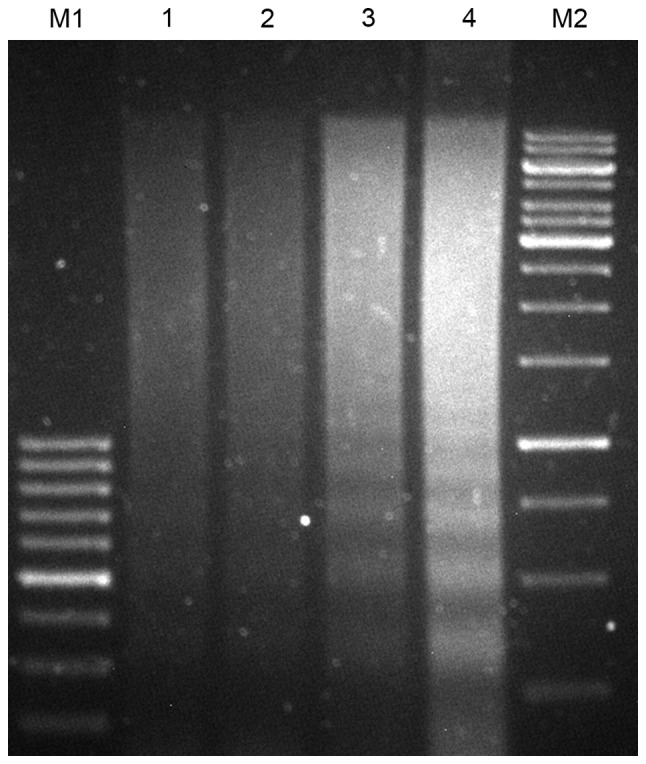
Detection of CRT and MUC1 promoting a phagocytic signal on the cell surface of apoptotic 4T1 cells by DNA fragmentation analysis. DNA fragmentation was induced by mitoxantrone (10 µg/ml) for 24 h. Lane 1, 4T1 control cells; lane 2, 4T1 cells transiently transfected with pEGFP-MUC1; lane 3, 4T1 cells transiently transfected with pEGFP-CRT; lane 4, 4T1 cells transiently transfected with pEGFP-MUC1-CRT; M1 and M2, DNA markers. MUC1, mucin 1; CRT, calreticulin.
Figure 3.

Fluorescence microscopy analysis of the apoptotic behavior of CRT-MUC1-coated 4T1 cells. pEGFP-MUC1-CRT was transiently transfected into 4T1 cells and treated with (A) 0 µg/ml, (B) 2 µg/ml, (C) 4 µg/ml and (D) 8 µg/ml mitoxantrone for 12 h, or with (E) 0 µg/ml, (F) 2 µg/ml, (G) 4 µg/ml and (H) 8 µg/ml mitoxantrone for 24 h and the results were observed by fluorescence microscopy. Fluorescence microscopy was used to detect that the apoptotic behavior of CRT-MUC1-coated 4T1 cells treated with mitoxantrone was dose- and time-dependent. Magnification, ×400. MUC1, mucin 1; CRT, calreticulin.
MUC1-CRT-infected BMDCs are able to induce an immunological effect
BMDCs caused marked T cell proliferation as a function of the ratio. In the present study, BMDCs were flushed and subsequently cultured. Purification was detected by flow cytometry using anti-mouse CD80-FITC, anti-mouse CD86-FITC and anti-mouse CD11c-FITC antibodies (data not shown). Adenoviruses containing MUC1-CRT and MUC1 were used to infect splenocytes and infection was verified by qPCR (data not shown) with the primers listed in Table I. As indicated in Fig. 4, MUC1-CRT-infected BMDCs exhibited 37.1% CD3+ T cells and 41.1% CD8+ T cells, whereas MUC1-infected BMDCs exhibited a significantly decreased number of viable CD3+ T cells (17.5%; P<0.001) and CD8+ T cells (31.0%; P<0.05), which suggested that MUC1-CRT-infected BMDCs induced increased T cell proliferation and antitumor immunity.
Figure 4.

T cell proliferation induced by MUC1-CRT-infected dendritic cells. Splenocytes were incubated with adenovirus-infected dendritic cells and CD3+/CD4+/CD8+ T cells were detected by flow cytometry using anti-CD3-PerCP, anti-CD4-Pacific Blue and anti-CD8-Alexa Flour 700 antibodies. (A and B) Control, (C and D) pEGFP-MUC1 and (E and F) pEGFP-MUC1-CRT. A, C and E, CD3 T cells; B, D and F, CD4 and CD8 T cells. CD, cluster of differentiation; MUC1, mucin; PerCP, peridinin chlorophyll protein complex; CRT, calreticulin.
IFN-γ and TNF-α are critical cytokines for tumor control (31). IFN-γ is produced predominantly by natural killer (NK) cells, whereas TNF-α is produced primarily by macrophages (32). As part of the innate immune response, IFN-γ and TNF-α may be produced by CD4+ T cells once antigen-specific immunity has developed (33). To validate whether MUC1-CRT-infected BMDCs increased the production of IFN-γ and TNF-α by immune cells, the levels of IFN-γ and TNF-α in culture supernatant were determined by ELISA. MUC1-CRT-infected BMDCs exhibited significantly increased levels of IFN-γ (951 pg/ml) when compared with MUC1-infected BMDCs (732 pg/ml; P<0.05; Fig. 5A). Furthermore, MUC1-CRT-infected BMDCs exhibited significantly increased levels of TNF-α (1,388 pg/ml) when compared with MUC1-infected BMDCs (1,169 pg/ml; P<0.05; Fig. 5B). The results suggest that MUC1-CRT-infected BMDCs induced the production of cytokines. Furthermore, these findings indicated that MUC1-CRT-infected BMDCs induced a potent immunological effect.
Figure 5.
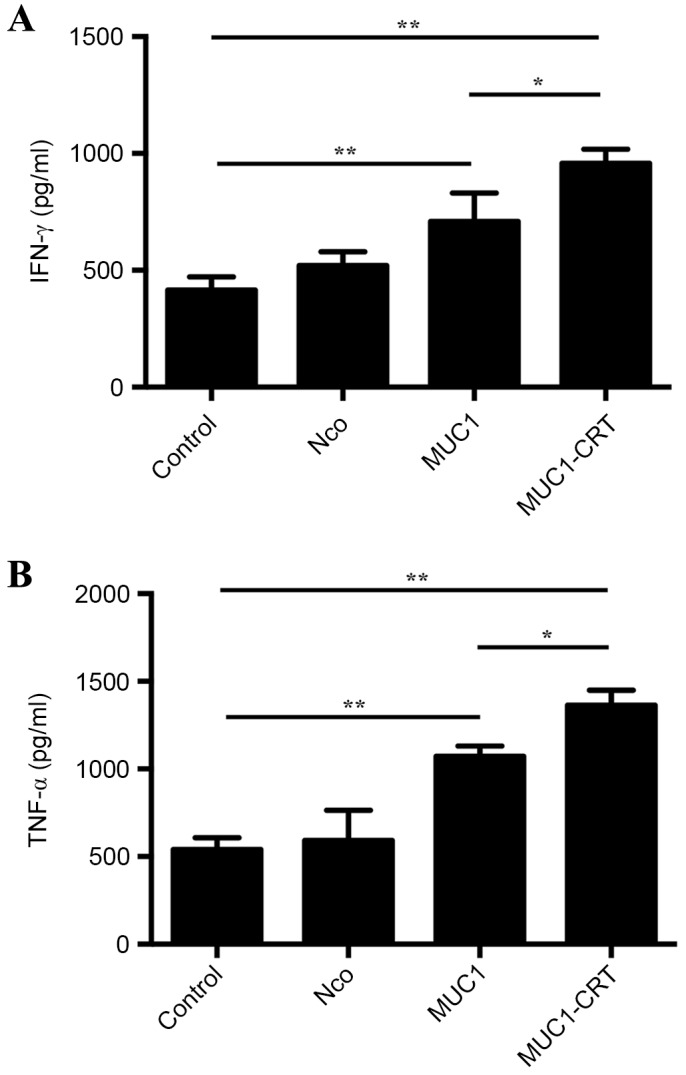
IFN-γ and TNF-α production by MUC1-CRT-infected dendritic cells. Dendritic cells were infected with adenoviruses containing MUC1 and MUC1-CRT, respectively, and further co-incubated with lymphocytes. Non-treated cells were used as control, and cells treated with mock vehicle were used as Nco. Culture supernatants were used to analyze (A) IFN-γ and (B) TNF-α levels by ELISA assay. Data are presented as the mean ± standard deviation (n=3). *P<0.05 and **P<0.005. INF-γ, interferon-γ; TNF-α, tumor necrosis factor-α; MUC1, mucin 1; CRT, calreticulin; Nco, null control.
Discussion
The majority of recent cancer treatment modalities aim to destroy tumor cells directly or indirectly. Since failure of the cancer immune-surveillance function in the host immune system is thought to give rise to multiple, if not all cancer types (34–36), partial or complete restoration of the immune-surveillance function may prove to be one of the most promising approaches for cancer immunotherapy.
DCs are important immune cells for uptaking, processing and presenting tumor antigens and DC-based vaccines are an attractive approach to treat cancer (37–39). Clinical trials have provided evidence that DC vaccines are able to elicit immunological responses; however, few complete tumor remissions have been reported (40). In order to improve the efficacy of DC vaccines, restoring or stimulating the immune response of the body against cancer may be an effective approach.
Previous studies have demonstrated that MIT is able to induce apoptosis in CT-26 and B16-F1 cells and promote CRT membrane translocation to the cell surface and aggregation (11,29). A recent report indicated the same phenomenon in fungi cells. Moreover, a previous study indicated that MIT stimulated the relocation of CRT in human and yeast cells, suggesting that the CRT pathway is phylogenetically conserved (41). MIT is a derivative of anthracyclines, which were believed to be the only agents that cause CRT cell surface localization (42). In 2014, Sukkurwala et al (41) demonstrated that tumor cells treated with platinum compounds combined with C-X-C motif chemokine ligand 8 were able to stimulate relocation of CRT. Similarly, in the present study, treatment with MIT (8 µg/ml) induced CRT localization to the cell surface. Notably, CRT was also able to promote MUC1 translocation to the cell surface and jointly act as an apoptotic signal to induce a potent apoptosis reaction. The exposure of MUC1-CRT on the surface of cancer cells facilitates their uptake by DCs and the subsequent presentation of TAAs to T cells.
Previous findings have identified that CRT-coated apoptotic cells may be used as an antigen to inoculate mice and elicit a specific antitumor effect against homogeneous tumor cells (43–45), suggesting the potential importance of CRT cell surface localization in mediating an antitumor immune response. T cell immunity is initiated by the interaction of naive T cells with DCs and mature DCs are potent T cell stimulators (46). Furthermore, T cell proliferation is a critical step in the induction of the immune response. In the present study, the ability of BMDCs to induce T cell proliferation was investigated and the results indicated that MUC1-CRT-infected BMDCs exhibited a significantly increased number of CD3+ and CD8+ T cells compared with MUC1-infected BMDCs, suggesting that MUC1-CRT-infected BMDCs were therefore able to potentiate functional proliferation of T cells efficiently. ELISA assay results indicated that the levels of IFN-γ and TNF-α were significantly increased in MUC1-CRT-infected BMDCs when compared with MUC1-infected BMDCs. Previous results have indicated that IFN-γ and TNF-α are essential immune effectors released by activated immunocytes (32,33,47). Therefore, the supernatant concentration of IFN-γ and TNF-α may indirectly represent the active extent of the immune system in vivo.
To conclude, the findings of the present study indicated that CRT may promote CRT-mediated antitumor immunity by acting as an immunological adjuvant and associating with MUC1 to induce potent immunogenicity. Overall, the present results indicated a novel role for CRT in mediating antitumor immunity and provide a novel concept and approach for tumor immune prevention and treatment.
Acknowledgements
The present study was supported by the Nature Science Foundation of China (grant nos. 81201766 and 81550028) and the Hubei Office of Education Foundation (grant no. Q20151204).
References
- 1.Marsh Rde W, Alonzo M, Bajaj S, Baker M, Elton E, Farrell TA, Gore RM, Hall C, Nowak J, Roy H, et al. Comprehensive review of the diagnosis and treatment of biliary tract cancer 2012. Part II: Multidisciplinary management. J Surg Oncol. 2012;106:339–345. doi: 10.1002/jso.23027. [DOI] [PubMed] [Google Scholar]
- 2.Weber GF, Rosenberg R, Murphy JE, zum Büschenfelde Meyer C, Friess H. Multimodal treatment strategies for locally advanced rectal cancer. Expert Rev Anticancer Ther. 2012;12:481–494. doi: 10.1586/era.12.3. [DOI] [PubMed] [Google Scholar]
- 3.Ahn YH, Hong SO, Kim JH, Noh KH, Song KH, Lee YH, Jeon JH, Kim DW, Seo JH, Kim TW. The siRNA cocktail targeting interleukin 10 receptor and transforming growth factor-β-receptor on dendritic cells potentiates tumor antigen-specific CD8(+) T cell immunity. Clin Exp Immunol. 2015;181:164–178. doi: 10.1111/cei.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karbach J, Neumann A, Brand K, Wahle C, Siegel E, Maeurer M, Ritter E, Tsuji T, Gnjatic S, Old LJ, et al. Phase I clinical trial of mixed bacterial vaccine (Coley's toxins) in patients with NY-ESO-1 expressing cancers: Immunological effects and clinical activity. Clin Cancer Res. 2012;18:5449–5459. doi: 10.1158/1078-0432.CCR-12-1116. [DOI] [PubMed] [Google Scholar]
- 5.Tsuboi A, Oka Y, Kyo T, Katayama Y, Elisseeva OA, Kawakami M, Nishida S, Morimoto S, Murao A, Nakajima H, et al. Long-term WT1 peptide vaccination for patients with acute myeloid leukemia with minimal residual disease. Leukemia. 2012;26:1410–1413. doi: 10.1038/leu.2011.343. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. In tratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 7.Breakthrough of the year 2013, corp-author. Notable developments. Science. 2013;342:1435–1441. doi: 10.1126/science.342.6165.1444. [DOI] [PubMed] [Google Scholar]
- 8.Kepp O, Tesniere A, Schlemmer F, Michaud M, Senovilla L, Zitvogel L, Kroemer G. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis. 2009;14:364–375. doi: 10.1007/s10495-008-0303-9. [DOI] [PubMed] [Google Scholar]
- 9.Rescigno M, Avogadri F, Curigliano G. Challenges and prospects of immunotherapy as cancer treatment. Biochem Biophys Acta. 2007;1776:108–123. doi: 10.1016/j.bbcan.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Tarr JM, Young PJ, Morse R, Shaw DJ, Haigh R, Petrov PG, Johnson SJ, Winyard PG, Eggleton P. A mechanism of release of calreticulin from cells during apoptosis. J Mol Biol. 2010;401:799–812. doi: 10.1016/j.jmb.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 11.Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, Menger L, Michaud M, Zitvogel L, Kroemer G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann N Y Acad Sci. 2010;1209:77–82. doi: 10.1111/j.1749-6632.2010.05740.x. [DOI] [PubMed] [Google Scholar]
- 12.Païdassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, Ling WL, Arlaud GJ, Frachet P. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol. 2011;408:277–290. doi: 10.1016/j.jmb.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 13.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 14.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 15.Verneret M, Tacnet-Delorme P, Osman R, Awad R, Grichine A, Kleman JP, Frachet P. Relative contribution of c1q and apoptotic cell-surface calreticulin to macrophage phagocytosis. J Innate Immun. 2014;6:426–434. doi: 10.1159/000358834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser SA, Karimi R, Michalak M, Hudig D. Perforin lytic activity is controlled by calreticulin. J Immunol. 2000;164:4150–4155. doi: 10.4049/jimmunol.164.8.4150. [DOI] [PubMed] [Google Scholar]
- 17.Kuraishi T, Manaka J, Kono M, Ishii H, Yamamoto N, Koizumi K, Shiratsuchi A, Lee BL, Higashida H, Nakanishi Y. Identification of calreticulin as a marker for phagocytosis of apoptotic cells in Drosophila. Exp Cell Res. 2007;313:500–510. doi: 10.1016/j.yexcr.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 18.Zeng G, Aldridge ME, Tian X, Seiler D, Zhang X, Jin Y, Rao J, Li W, Chen D, Langford MP, et al. Dendritic cell surface calreticulin is a receptor for NY-ESO-1: Direct interactions between tumor-associated antigen and the innate immune system. J Immunol. 2006;177:3582–3589. doi: 10.4049/jimmunol.177.6.3582. [DOI] [PubMed] [Google Scholar]
- 19.Gardai SJ, McPhilips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Obeid M, Tesniere A, Panaretakis T, Tufi R, Joza N, van Endert P, Ghiringhelli F, Apetoh L, Chaput N, Flament C, et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev. 2007;220:22–34. doi: 10.1111/j.1600-065X.2007.00567.x. [DOI] [PubMed] [Google Scholar]
- 21.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 22.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 23.De Angelis R, Sant M, Coleman MP, Francisci S, Baili P, Pierannunzio D, Trama A, Visser O, Brenner H, Ardanaz E, et al. Cancer survival in Europe 1999–2007 by country and age: Results of EUROCARE-5-a population-based study. Lancet Oncol. 2014;15:23–34. doi: 10.1016/S1470-2045(13)70546-1. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Yi H, Yao Y, Liao X, Xie Y, Yang J, Yan Z, Wang L, Lu S, Kuang Y, et al. The cytoplasmic domain of MUC1 induces hyperplasia in the mammary gland and correlates with nuclear accumulation of β-catenin. PLoS One. 2011;6:e19102. doi: 10.1371/journal.pone.0019102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad R, Rajabi H, Kosugi M, Joshi MD, Alam M, Vasir B, Kawano T, Kharbanda S, Kufe D. MUC1-C oncoprotein promotes STAT3 activation in an autoinductive regulatory loop. Sci Signal. 2011;4:ra9. doi: 10.1126/scisignal.2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hisatsune A, Nakayama H, Kawasaki M, Horie I, Miyata T, Isohama Y, Kim KC, Katsuki H. Anti-MUC1 antibody inhibits EGF receptor signaling in cancer cells. Biochem Biophys Res Commun. 2011;405:377–381. doi: 10.1016/j.bbrc.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 27.Gheybi E, Amani J, Salmanian AH, Mashayekhi F, Khodi S. Designing a recombinant chimeric construct contain MUC1 and HER2 extracellular domain for prediagnostic breast cancer. Tumour Biol. 2014;35:11489–11497. doi: 10.1007/s13277-014-2483-y. [DOI] [PubMed] [Google Scholar]
- 28.Zanetti JS, Soave DF, Oliveira-Costa JP, da Silveira GG, Ramalho LN, Garcia SB, Zucoloto S, Ribeiro-Silva A. The role of tumor hypoxia in MUC1-positive breast carcinomas. Virchows Arch. 2011;459:367–375. doi: 10.1007/s00428-011-1142-6. [DOI] [PubMed] [Google Scholar]
- 29.Cao C, Han Y, Ren Y, Wang Y. Mitoxantrone-mediated apoptotic B16-F1 cells induce specific anti-tumor immune response. Cell Mol Immunol. 2009;6:469–475. doi: 10.1038/cmi.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bustin SA, editor. A-Z of Quantitative PCR. La Jolla, CA: International University Line; 2004–2006. [Google Scholar]
- 31.Wang L, Zhao Y, Liu Y, Akiyama K, Chen C, Qu C, Jin Y, Shi S. IFN-γ and TNF-α synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFkB signaling. Stem Cells. 2013;31:1383–1395. doi: 10.1002/stem.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katanov C, Lerrer S, Liubomirski Y, Leider-Trejo L, Meshel T, Bar J, Feniger-Barish R, Kamer I, Soria-Artzi G, Kahani H, et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: Prominent roles for TNF-α and the NF-kB pathway. Stem Cell Res Ther. 2015;6:87. doi: 10.1186/s13287-015-0080-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi IK, Li Y, Oh E, Kim J, Yun CO. Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-γ- and TNF-α-co-producing T cell-mediated antitumor immunity. PLoS One. 2013;8:e67512. doi: 10.1371/journal.pone.0067512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow MT, Möller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22:23–32. doi: 10.1016/j.semcancer.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Bruyns C, Gérard C, Velu T. Cancer escape from immune surveillance: How can it be overcome by gene transfer? Eur J Cancer. 1994;30:1176–1181. doi: 10.1016/0959-8049(94)90479-0. [DOI] [PubMed] [Google Scholar]
- 36.Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H, Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell Mol Life Sci. 2013;70:4431–4448. doi: 10.1007/s00018-013-1339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shixiang Y, Xi S, Junliang L, Shanyi Z, Xingke X, Meiguang Z, Kai W, Fangcheng L. Antitumor efficacy of a photodynamic therapy-generated dendritic cell glioma vaccine. Med Oncol. 2011;28(Suppl 1):453–461. doi: 10.1007/s12032-010-9713-y. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Fan H, Wu Y, Chen B. Potent in vivo anti-tumor activity of isolated CD62L(low) lymph node cells sensitized in vivo with tumor lysate-pulsed DC-based vaccines. Cytotherapy. 2005;7:353–362. doi: 10.1080/14653240500241925. [DOI] [PubMed] [Google Scholar]
- 39.Hirschowitz EA, Foody T, Kryscio R, Dickson L, Sturgill J, Yannelli J. Autologous dendritic cell vaccines for non-small-cell lung cancer. J Clin Oncol. 2004;22:2808–2815. doi: 10.1200/JCO.2004.01.074. [DOI] [PubMed] [Google Scholar]
- 40.Jung NC, Kim HJ, Kang MS, Lee JH, Song JY, Seo HG, Bae YS, Lim DS. Photodynamic therapy-mediated DC immunotherapy is highly effective for the inhibition of established solid tumors. Cancer Lett. 2012;324:58–65. doi: 10.1016/j.canlet.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 41.Sukkurwala AQ, Martins I, Wang Y, Schlemmer F, Ruckenstuhl C, Durchschlag M, Michaud M, Senovilla L, Sistigu A, Ma Y, et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21:59–68. doi: 10.1038/cdd.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wemeau M, Kepp O, Tesnière A, Panaretakis T, Flament C, De Botton S, Zitvogel L, Kroemer G, Chaput N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010;1:e104. doi: 10.1038/cddis.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia GM, Kepp O, Piacentini M, Froehlich KU, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499–1509. doi: 10.1038/cdd.2008.67. [DOI] [PubMed] [Google Scholar]
- 44.Brusa D, Garetto S, Chiorino G, Scatolini M, Migliore E, Camussi G, Matera L. Post-apoptotic tumors are more palatable to dendritic cells and enhance their antigen cross-presentation activity. Vaccine. 2008;26:6422–6432. doi: 10.1016/j.vaccine.2008.08.063. [DOI] [PubMed] [Google Scholar]
- 45.Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3–12. doi: 10.1038/sj.cdd.4402269. [DOI] [PubMed] [Google Scholar]
- 46.Rosenblatt J, Wu Z, Vasir B, Zarwan C, Stone R, Mills H, Friedman T, Konstantinopoulos PA, Spentzos D, Ghebremichael M, et al. Generation fo tumor-specific T lymphocytes using dendritic cell/tumor fusions and CD3/CD28. J Immunother. 2010;33:155–166. doi: 10.1097/CJI.0b013e3181bed253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin P, Zhao Y, Liu H, Chen J, Ren J, Jin J, Bedognetti D, Liu S, Wang E, Marincola F, Stroncek D. Interferon-γ and tumor necrosis factor-α polarize bone marrow stromal cells uniformly to a Th1 phenotype. Sci Rep. 2016;6:26345. doi: 10.1038/srep26345. [DOI] [PMC free article] [PubMed] [Google Scholar]