

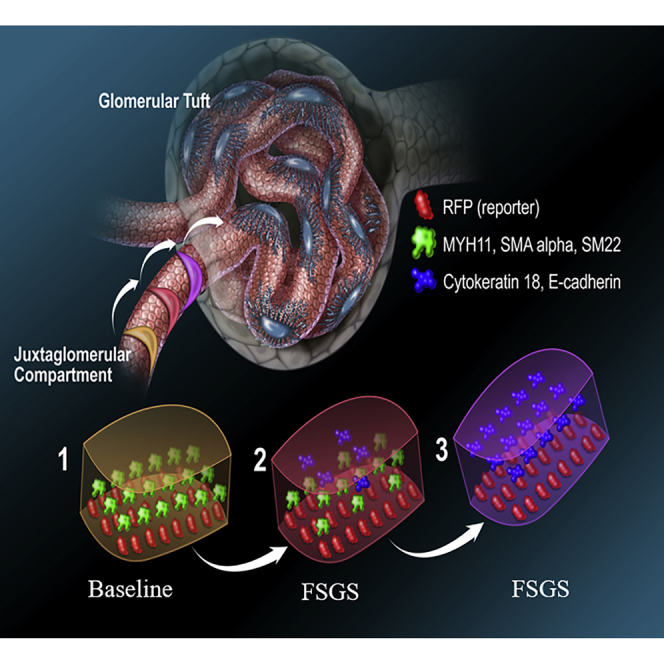
Summary
Wilms' tumor suppressor 1 (WT1) plays an important role in cell proliferation and mesenchymal-epithelial balance in normal development and disease. Here, we show that following podocyte depletion in three experimental models, and in patients with focal segmental glomerulosclerosis (FSGS) and membranous nephropathy, WT1 increased significantly in cells of renin lineage (CoRL). In an animal model of FSGS in RenWt1fl/fl reporter mice with inducible deletion of WT1 in CoRL, CoRL proliferation and migration to the glomerulus was reduced, and glomerular disease was worse compared with wild-type mice. To become podocytes, CoRL undergo mesenchymal-to-epithelial transformation (MET), typified by reduced staining for mesenchymal markers (MYH11, SM22, αSMA) and de novo expression of epithelial markers (E-cadherin and cytokeratin18). Evidence for changes in MET markers was barely detected in RenWt1fl/fl mice. Our results show that following podocyte depletion, WT1 plays essential roles in CoRL proliferation and migration toward an adult podocyte fate.
Keywords: progenitor, regeneration, glomerulus, synaptopodin, p57, enalapril, FSGS, MET, mesenchymal-to-epithelial transformation, membranous nephropathy
Graphical Abstract
Highlights
-
•
WT1 increased in cells of renin lineage in three models of podocyte ablation
-
•
WT1 expression increased in CoRL in human FSGS and membranous nephropathy
-
•
In experimental FSGS, WT1 deletion in CoRL reduced their proliferation and migration
-
•
CoRL undergo mesenchymal-to-epithelial transformation in Wt1 wild-type mice with FSGS
In this article, Shankland and colleagues show that in human and experimental models characterized by reduction in terminally differentiated podocytes, the expression of Wilms' tumor suppressor 1 (WT1) in neighboring cells of renin lineage (CoRL) is increased. CoRL serve as local progenitors for podocytes, and reduction in WT1 in these cells results in decreased proliferation, migration, and mesenchymal-to-epithelial transformation.
Introduction
Kidney podocytes are terminally differentiated glomerular epithelial cells unable to self-renew due to their inability to proliferate (Lasagni et al., 2013, Wanner et al., 2014), and replacement following loss is dependent on local kidney progenitors (Ronconi et al., 2009, Lasagni et al., 2015, Starke et al., 2015). Glomerular parietal epithelial cells (PECs) (Romagnani, 2011, Zhang et al., 2013, Eng et al., 2015, Kuppe et al., 2015, Lazzeri and Romagnani, 2015) and cells of renin lineage (CoRL) (Pippin et al., 2013, Pippin et al., 2014, Lichtnekert et al., 2016) are considered likely adult podocyte progenitors. CoRL serve as progenitors for several kidney cell types (Sequeira Lopez et al., 2004), including podocytes (Grahammer et al., 2013, Pippin et al., 2013, Pippin et al., 2014, Pippin et al., 2015, Shankland et al., 2014, Lichtnekert et al., 2016), PECs (Pippin et al., 2013, Eng et al., 2015), mesangial cells (Thoma, 2014, Starke et al., 2015), and pericytes (Stefanska et al., 2013, Pippin et al., 2015). RAAS inhibition augments the CoRL reservoir in the juxta-glomerular compartment (JGC) through proliferation, enhances migration to the glomerulus, and increases transdifferentiation toward a podocyte fate (Lichtnekert et al., 2016). CoRL and podocytes derive from mesenchymal origins (Costantini and Kopan, 2010, Matsushita et al., 2010, Little and McMahon, 2012, Wang et al., 2013). CoRL maintain mesenchymal characteristics, whereas adult podocytes exhibit epithelial characteristics (Filipovic et al., 2017). Therefore, for CoRL to serve as podocyte progenitors, they must undergo changes resembling a mesenchymal-to-epithelial transition (MET).
The transcription factor Wilms' tumor suppressor protein 1 (WT1) is required for podocyte development and homeostasis (Kann et al., 2015). Missense mutations and conditionally deleting WT1 from podocytes leads to glomerular scarring in humans and mice (Pelletier et al., 1991, Gao et al., 2004, Chau et al., 2011). WT1 is closely linked to the mesenchymal-epithelial balance in development and disease (Miller-Hodges and Hohenstein, 2012), and the homeostasis of different cell types (Ozdemir and Hohenstein, 2014), including the plasticity and cell fate of certain cells (Karki et al., 2014, Wen et al., 2016). WT1 drives MET at the start of nephron development (Essafi et al., 2011, Berry et al., 2015), although in the developing epicardium, WT1 controls the reverse, i.e., epithelial-to-mesenchymal transition (Martinez-Estrada et al., 2010). WT1 enhances proliferation and migration in certain cell types in a context-dependent manner (Wagner et al., 2008, Brett et al., 2013, Graziano et al., 2017). Finally, WT1 and renin co-localize in a fraction of cells in the afferent arterioles. Moreover, WT1 (−KTS) can negatively modulate renin gene transcription through interaction at a binding site within the renin enhancer (Steege et al., 2008).
These reports provided the rationale to test the hypothesis that WT1 is required for CoRL proliferation, migration, and transdifferentiation toward a podocyte fate in the context of podocyte depletion.
Results
WT1 Expression Increases in Renin-Stained Cells Following Podocyte Depletion in Mice and Humans
We began by measuring the temporal expression of WT1 in renin-stained cells in the JGC in RenWt1+/+ mice following podocyte depletion induced by the administration of a cytopathic anti-podocyte antibody. Following podocyte depletion, the percentage of renin cells co-staining for WT1 increased as follows (Figures 1A–1F): from a baseline to D3 of FSGS (6.16% ± 0.3% versus 14.5% ± 3.35%; p = 0.133; Figures 1A and 1B), from baseline to D5 of FSGS (6.16% ± 0.3% versus 20.99% ± 2.20%; p = 0.001; Figures 1A and 1C), from D5 to D14 of FSGS (20.99% ± 2.20% versus 33.63% ± 7.3%; p = 0.001; Figures 1C and 1D), and from baseline to D28 of FSGS (6.16% ± 0.3% versus 26.5% ± 5.24%; p < 0.0001; Figures 1A and 1E).
Figure 1.

WT1 Increases in Renin-Stained Cells in the Juxta-Glomerular Compartment Following Podocyte Depletion in the Cytotoxic Anti-podocyte Antibody Model of FSGS
Confocal microscopy images marked (A–E) show the merge for WT1 (green), renin (red), collagen IV (blue), and DAPI (gray). Collagen IV demarcates glomeruli. Insets show the JGC for each channel, labeled as superscripts. Scale bars, 20 μm.
(A) Baseline (n = 5), WT1 (A1) is limited to podocytes and not detected in renin-stained cells (A2).
(B) D3 (n = 5), WT1 (B1) is detected in the cytoplasm of renin-stained cells (B2).
(C) D5 (n = 5), WT1 (C1) increases in JGC, overlapping with renin (C2), creating a yellow color.
(D) D14 (n = 5), renin-stained cells (D2) co-staining with WT1 (D1) increase in the JGC.
(E) D28 (n = 8), WT1 (E1) and renin (E2) co-staining persists.
(F) Quantitation of the percentage of renin-stained cells in the JGC co-staining for WT1.
Podocyte loss in podocyte TGFβ-Receptor1 transgenic (PodTgfbr1) mice (Figures S1A–S1C) and membranous nephropathy rats (passive Heymann nephritis [PHN] model) (Figures S1D and S1E) was also accompanied by increased WT1 in renin-stained JGC cells. In contrast, WT1 was not detected in renin cells post uninephrectomy in mice, where absolute podocyte number remained normal (Figure S1F).
We confirmed the experimental results in human diseases. In normal human kidney, WT1 was mostly limited to podocytes (Figure 2A). However, de novo WT1 was detected in renin-stained JGC cells in human FSGS (Figure 2B) and membranous nephropathy (Figure 2C). Similar results were obtained with different WT1 antibodies, and no staining was detected when the primary antibody was omitted (not shown).
Figure 2.

WT1 Increases in Renin-Stained Cells in the JGC in Human FSGS and Membranous Nephropathy
Confocal microscopy for WT1, renin, and DAPI in human kidneys. Insets show the JGC for each channel: WT1 (1, green), renin (2, red), DAPI (3, blue), and merged (4, yellow). Scale bars, 20 μm.
(A) Normal human kidney (n = 5), WT1 is limited to podocytes (A1), and not detected in renin-stained cells (A2), hence no merge (A4).
(B) Human FSGS (n = 5), WT1 in the JGC (B1) overlaps with renin-stained cells (B2) creating a yellow color (B4).
(C) Human membranous nephropathy (n = 5), WT1 (C1) and renin in the JGC (C2) produce a yellow color (C4).
These results show that WT1 protein increases in renin-stained cells in the JGC in both experimental and human FSGS and membranous nephropathy in the context of podocyte loss.
Selective Deletion of WT1 in Cells of Renin Lineage Has No Impact on Podocytes or Kidney Function under Non-diseased Conditions
To test if de novo expression of WT1 in CoRL has relevance for their progenitor role to replace lost podocytes, RenCreER tdTomato mice (Pippin et al., 2013, Pippin et al., 2015) were crossed with Wt1fl/fl mice (Martinez-Estrada et al., 2010, DeFilippis and Wagner, 2014) to generate RenCreER tdTomato Wt1fl/fl mice (abbreviated RenWt1fl/fl) so that WT1 could be conditionally and selectively deleted in tdTomato-labeled CoRL (Figure S2). CoRL and their descendants permanently express tdTomato dependent on Cre-mediated recombination, which allows lineage tracing of CoRL. Glomerular cells do not ectopically express renin (and hence Cre) under injury conditions. RenCreER tdTomato Wt1+/+ (abbreviated RenWt1+/+) served as controls, where tdTomato was expressed in CoRL but WT1 remained (Figures S2A, S2F, and S2H). Genotyping discerned the lox and wild-type Wt1 alleles (Figure S2A1), and a flox deletion primer distinguished RenWt1fl/fl mice given tamoxifen versus corn oil (Figure S2A2). Following tamoxifen, Wt1 mRNA from RFP+CoRL isolated using laser capture microscopy was significantly lower in RenWt1fl/fl compared with RenWt+/+ mice (Figure S2B), while renin mRNA levels were similar (Figure S2C). Cre was restricted to the JGC in mice containing the CreER transgene but not in Cre-negative mice (Figures S2D and S2E). TdTomato, detected without the use of an antibody, confirmed similar CoRL labeling efficiency in RenWt1+/+ and RenWt1fl/fl mice given tamoxifen (Figures S2F and S2G). tdTomato was absent in RenCre-negative mice (not shown). Ninety-five percent of renin+ CoRL stained with red fluorescent protein (RFP) antibody, which detected tdTomato in RenWt1+/+ and RenWt1fl/fl mice (not shown). Mice that did not report were excluded, accounting for <1% of all mice given tamoxifen. At baseline, WT1 was in a typical podocyte distribution in RenWt1fl/fl mice, indistinguishable from RenWt1+/+ mice, proving deletion in CoRL had no impact on podocytes (Figures S2H and S2I). WT1 was detected in <5% of CoRL in non-diseased RenWt1+/+ mice but was not detected in CoRL in non-diseased RenWt1fl/fl mice (Figures S2H and S2I). These results show that Wt1 mRNA and WT1 protein were selectively reduced in tdTomato+ CoRL of RenWt1fl/fl but not in RenWT+/+ mice, with no consequences on podocyte health under normal conditions.
Increased WT1 Was Not Seen in JGC of Wt1 Conditional Knockout Mice Following Podocyte Depletion
WT1 overlap with RFP+ was not present in the vast majority of CoRL in the JGC in normal RenWt1+/+and RenWt1fl/fl mice (7 ± 2.5 versus 5.6 ± 2.0, respectively; p = 0.99; Figures 3A, 3D, and 3G). In RenWt1+/+ mice at D28 following podocyte loss, RFP+CoRL in the JGC co-stained with WT1 increased compared with baseline (103 ± 12.36 versus 7 ± 2.5; p < 0.001; Figures 3B and 3G). By contrast, in RenWt1fl/fl mice at D28, only 5.8 ± 1.8 of RFP+ CoRL in the JGC co-stained with WT1 (Figures 3E and 3G). Because enalapril increases RFP+CoRL in the JGC (Lichtnekert et al., 2016), RenWt1+/+ mice were given enalapril for 25 days following podocyte loss. RFP+CoRL in the JGC co-expressing WT1 further increased at D28 following enalapril compared with baseline (419.5 ± 117.79 versus 7 ± 2.5; p < 0.0001; Figures 3A, 3C, and 3G). An increase in WT1 expression was not observed in JGC of RenWt1fl/fl mice after enalapril (Figures 3F and 3G). Similarly, the number of RFP+WT1−CoRL increased in RenWt1+/+ mice following podocyte depletion (Figures S3B, S3C), while the number of RFP+WT1−CoRL in RenWt1fl/fl mice did not increase (Figures S3E, S3F).
Figure 3.

WT1 Does Not Increase in CoRL in RenWt1fl/fl Mice Following Podocyte Depletion in the Cytotoxic Anti-podocyte Antibody Model of FSGS
Confocal microscopy for WT1 (green), RFP (red), collagen IV (blue), and DAPI (gray) in FSGS in RenWt1+/+ (A–C) and RenWt1fl/fl (D–F) mice. Insets show the JGC for each channel, labeled as superscripts. Scale bars, 20 μm.
(A–C) RenWt1+/+ mice. (A) At baseline WT1 is limited to podocytes (A1); RFP is limited to the JGC (A2). (B) At D28, WT1 (B1) merges with RFP (B2) in the JGC, creating a yellow color. WT1 is reduced in glomeruli. (C) At D28, after 25 days of enalapril, WT1 (C1) and RFP (C2) are increased in the JGC.
(D–F) RenWt1fl/fl mice. (D) At baseline, WT1 (D1) is limited to podocytes; RFP (D2) is limited to JGC. (E) At D28, WT1 is reduced in podocytes, and not detected (E1) in RFP+ cells (E2) in the JGC. (F) At D28, after 25 days of enalapril, there is no WT1(F1) in RFP+ cells (F2).
(G) Stacked bar graph shows the number of RFP+CoRL not expressing WT− (red) and co-expressing WT1+ (green) in the JGC. RFP+WT1+ cells increase progressively in diseased RenWt1+/+ but not in RenWt1fl/fl mice (n = 13/group).
These results show the following: (1) although WT1 was not detected in the majority of cells in the JGC under normal conditions, the number of RFP+CoRL co-expressing WT1 increases progressively following podocyte loss in RenWt1+/+ mice; (2) WT1 did not increase in RFP+CoRL in diseased RenWt1fl/fl mice, consistent with successful targeted ablation of WT1 in CoRL; (3) enalapril increased the number of CoRL co-expressing WT1 in RenWt1+/+ mice following podocyte loss but not in RenWt1fl/fl mice.
CoRL Migration to the Glomerulus Is Reduced in RenWt1fl/fl Mice Following Podocyte Depletion
Triple staining was performed for p57 (podocytes), RFP (tdTomato+, CoRL), and collagen IV (demarcates glomeruli) to determine if deleting WT1 reduced CoRL migration from the JGC to the glomerulus following abrupt podocyte loss. RFP+CoRL were rarely detected in glomeruli at baseline of either RenWt1+/+ or RenWt1fl/fl mice (Figures 4A, 4D and 4G). At D28 FSGS in RenWt1+/+ mice, RFP+CoRL were detected in 3.5% ± 2.4% of glomeruli compared with baseline (0.43% ± 0.2%; p = 0.013; Figures 4B and 4G). At D28 FSGS in RenWt1fl/fl mice, RFP+CoRL were barely detected in glomeruli compared with RenWt1+/+ mice (p = 0.001; Figures 4E and 4G). Consistent with our recent report (Lichtnekert et al., 2016), administering enalapril to RenWt1+/+ mice following podocyte depletion increased the migration of RFP+CoRL to 12.95% ± 3.07% of glomeruli compared with untreated (p < 0.001; Figures 4C and 4G). However, enalapril had little impact on RFP+CoRL migration to glomeruli in RenWt1fl/fl mice (Figures 4F and 4G). Staining for renin and Cre was not detected in glomerular cells in disease, consistent with CoRL migration to the glomerulus rather than ectopic renin expression in disease. These results were similar to our previous reports (Pippin et al., 2013).
Figure 4.

RFP+CoRL Migration to the Glomerulus Is Significantly Reduced in Diseased RenWt1fl/fl Mice Compared with Diseased RenWt1+/+ Mice
Confocal microscopy for p57 (green, podocytes), RFP (red, CoRL), collagen IV (blue, demarcates glomerulus), and DAPI (gray, nucleus). Insets show the glomerulus marked with a white box, labeled as superscripts. Scale bars, 20 μm.
(A–C) RenWt1+/+ mice. (A, A1) In baseline RenWt1+/+ mice, RFP+CoRL (solid arrows) are restricted to the JGC and p57 to podocytes. (B, B1) A serial biopsy at D28 in the same RenWt1+/+ mouse from (A). RFP+CoRL are in glomeruli (dashed arrows). (C, C1) At FSGS D28 after enalapril, RFP+CoRL in both the JGC and glomeruli are further increased and a subset in the tuft de novo express the podocyte marker p57 (arrowheads).
(D–F) RenWt1fl/fl mice. (D, D1) In baseline RenWt1fl/fl mice, RFP+CoRL (solid arrows) are restricted to the JGC and p57 to podocytes. (E, E1) At D28, despite the decrease in p57 (podocytes), RFP+CoRL remain restricted to the JGC (arrow). (F, F1) At D28 after enalapril, RFP+CoRL were mostly limited to the JGC.
(G) Quantitation of the percentage of glomeruli with RFP+CoRL increased progressively in diseased RenWt1+/+ mice but not in diseased RenWt1fl/fl mice (n = 13/group).
Figures S4 and 4C show that once RFP+CoRL migrated from the JGC to the glomerulus, a subset de novo co-express podocyte markers P57 (Figure 4C) podocin (Figure S4A–S4A4) and synaptopodin (Figure S4B–S4B4), consistent with CoRL transdifferentiating toward a podocyte fate.
These results show that WT1 is critical for CoRL migration to the glomerulus following podocyte loss and their transdifferentiation. In the absence of WT1, enalapril has reduced efficacy on CoRL migration and transdifferentiation.
WT1 Is Important for CoRL Proliferation Following Podocyte Depletion
To test the role of WT1 in CoRL proliferation, bromodeoxyuridine (BrdU) was administered via intraperitoneal injections every 48 hr, starting at D2. Triple staining was performed for BrdU, WT1, and RFP to determine if proliferation was restricted to RFP+CoRL that co-expressed WT1, and if WT1 is necessary for CoRL proliferation? BrdU was absent in RFP+CoRL at baseline in RenWt1+/+ and RenWt1fl/fl mice (Figures 5A, 5D, and 5G). This was not a false negative, as BrdU was detected in neighboring tubules. Following podocyte depletion in RenWt1+/+ mice, BrdU increased in RFP+WT1+ cells compared with baseline (25.8 ± 9.2 versus 0.8 ± 0.5; p < 0.01; Figures 5B and 5G). In contrast, BrdU was barely detected in RFP+CoRLs in RenWt1fl/fl mice (Figures 5E and 5G). In enalapril-treated RenWt1+/+ FSGS mice, RFP+WT1+BrdU+ cells in the JGC increased compared with untreated mice (131.3 ± 51.7 versus 25.8 ± 9.2; p = 0.0001; Figures 5C and 5G). In enalapril-treated RenWt1fl/fl FSGS mice, RFP+WT1+BrdU+ cells did not increase (Figures 5F and 5G). To test WT1 independent proliferation, we counted RFP+BrdU+WT1− cells. BrdU was only observed in a small portion of RFP+WT1− cells in RenWt1+/+ FSGS mice compared with baseline (5 ± 0.8 versus 0.3 ± 0.5; p < 0.05; Figures 5B, 5C, and 5G).
Figure 5.

RFP+CoRL Proliferation Is Significantly Reduced in Diseased RenWt1fl/fl Mice Compared with Diseased RenWt1+/+ Mice
Confocal microscopy for BrdU (blue), WT1 (green), RFP (red), and DAPI (gray) in experimental FSGS in RenWt1+/+ and RenWt1fl/fl mice. Insets show the JGC for each channel, labeled as superscripts. Scale bars, 20 μm.
(A–C) RenWt1+/+mice. (A) At baseline, a BrdU+ cell (A1) is detected in the adjacent tubule. BrdU is not detected in the JGC. WT1 (A2) is not detected in RFP+ cells (A3). (B) At FSGS D28, BrdU (B1) co-localizes with WT1 (B2) and RFP (B3), creating a white/purple color (B5, solid arrows). A few BrdU+ cells do not express WT1 (B1-B5, dashed arrow) (C) At D28 after enalapril, BrdU+ (C1), WT1+ (C2), RFP+ (C3) cells increase (C5, solid arrows). A few BrdU+ cells do not express WT1 (C1–C5, dashed arrow).
(D–F) RenWt1fl/fl mice. (D) At baseline, neither BrdU (D1) nor WT1 (D2) is detected in RFP+ (D3) cells in the JGC. (E) At D28, a BrdU+ cell (E1, solid arrow) is detected in the adjacent tubule and does not overlap with WT1 (E2) or RFP (E3), hence no merge (E5). (F) At D28 after enalapril, a BrdU+ cell (F1, solid arrow) does not overlap with WT1 (F2) or RFP (F3), hence no merge (F5).
(G) Stacked bar graph shows the number of RFP+BrdU+WT1− (blue) and RFP+BrdU+WT1+ (green) cells in the JGC. RFP+BrdU+WT1+ cells increase progressively in diseased RenWt1+/+ mice but not in RenWt1fl/fl mice (n = 13/group).
Triple staining was also performed for the proliferation marker Ki67, WT1, and RFP (Figure S7). Ki67 was not detected in RFP+CoRL at baseline in RenWt1+/+ and RenWt1fl/fl mice (Figures S7A and S7D) but was detected in neighboring tubules. Following podocyte depletion, Ki67 increased in RFP+ WT1+CoRL (Figure S7B), which was augmented by enalapril (Figure S7C). In RenWt1fl/fl mice, Ki67 was barely detected in RFP+CoRL (Figures S7E and S7F).
These results show that after podocyte depletion, the following occurs: (1) proliferation (BrdU + or Ki67+) of CoRL (RFP+) occurs in cells co-expressing WT1; (2) CoRL proliferation was reduced in RenWt1fl/fl mice; (3) enalapril increased CoRL proliferation in diseased RenWt1+/+ but not in RenWt1fl/fl mice.
Following Podocyte Loss, CoRL that Migrate to the Glomerulus Lose Mesenchymal Markers and De Novo Acquire Epithelial Markers
CoRL and podocytes derive from mesenchymal lineages (Boyle et al., 2008, Kobayashi et al., 2008, Costantini and Kopan, 2010, Little and McMahon, 2012). To determine if WT1 in CoRL is necessary for changes in mesenchymal and epithelial cell markers, and if these changes were augmented by enalapril, additional studies were performed (Figures 6 and S6). Staining for the mesenchymal marker myosin heavy chain 11 (MYH11) was detected in 90% ± 7.5% and 84.1% ± 8.2% of RFP+CoRL in the JGC in baseline RenWt1+/+ and RenWt1fl/fl mice, respectively (Figures 6A, 6D, and 6G). Following podocyte depletion in RenWt1+/+ mice, the percentage of MYH11+RFP+ cells decreased to 67.5% ± 5.3% compared with baseline (p = 0.0059; Figures 6B and 6G). The percentage of MYH11+RFP+ cells did not decrease in RenWt1fl/fl mice with FSGS compared with baseline (82.6% ± 10.6% versus 84.1% ± 8.2%; p = 0.99; Figures 6E and 6G). In enalapril-treated FSGS RenWt1+/+ mice, MYH11+ RFP+CoRL decreased (44.3% versus 67.5%; p = 0.008; Figures 6C and 6G). Giving enalapril to RenWt1fl/fl mice did not reduce MYH11+RFP+CoRL (78.7% versus 82.6%; p > 0.99; Figures 6F and 6G). CoRL that migrated to glomeruli did not stain for MYH11 with either strain (data not shown).
Figure 6.

RFP+CoRL MET Is Significantly Reduced in Diseased RenWt1fl/fl Mice Compared with Diseased RenWt1+/+ Mice
Confocal microscopy for myosin11 (MYH11, green, mesenchymal marker), RFP (red, CoRL), and DAPI (blue, nuclei) in the JGC (A–F). Insets show each channel, labeled as superscripts. Scale bars, 20 μm.
(A–C) RenWt1+/+ mice. (A) At baseline, MYH11 (A1) merges with RFP (A2) in the JGC, creating a yellow color. (B) At FSGS D28, MYH11 (B1) decreases in RFP+ (B2) cells. (C) At D28 after enalapril, MYH11 (C1) decreases, despite an increase in RFP+ cells (C2).
(D–F) RenWt1fl/fl mice. (D) At baseline, MYH11 (D1) merges with RFP (D2). (E) At FSGS D28, MYH11 (E1) in RFP+ cells (E2, solid arrows) persists. (F) At D28 after enalapril, MYH11 (F1) in RFP+ cells (F2, solid arrows) also persists.
(G) The percentage of RFP+CoRL expressing MYH11 decreases in RenWt1+/+ mice with FSGS and is augmented by enalapril. RenWt1fl/fl mice show no significant decrease in MYH11 (n = 13/group).
(H–M) Confocal microscopy for cytokeratin 18 (CK18, green, epithelial marker), RFP (red, CoRL), and DAPI (blue, nuclei) in the JGC (H–M). Insets show each channel, labeled as superscripts. Scale bars, 20 μm.
(H–J) RenWt1+/+ mice. (H) At baseline, CK18 (H1) is barely detected in RFP+ cells (H2). (I) At FSGS D28, CK18 (I1) increases in RFP+ cells (I2), creating a yellow color (solid arrows). (J) At D28 after enalapril, CK18 (J1) further increases in RFP+ cells (J2), creating a yellow color (solid arrows).
(K–M) RenWt1fl/fl mice. (K) At baseline, CK18 (K1) is not detected in RFP+ cells (K2). At FSGS D28 (L) and after enalapril (M), CK18 (L1, M1) does not increase in RFP+ cells (L2, M2).
(N) The percentage of RFP+CK18+cells increases in diseased RenWt1+/+ mice, which is augmented by enalapril. CK18 does not increase significantly in diseased RenWt1fl/fl mice (n = 13/group).
In RFP+CoRL in the JGC, the epithelial cell marker cytokeratin 18 (CK18) was barely detected at baseline in either RenWt1+/+ or RenWt1fl/fl mice (Figures 6H, 6K, and 6N). At D28 FSGS in RenWt1+/+ mice, the percentage of RFP+CK18+CoRL in the JGC increased compared with baseline (16.8% ± 2.4% versus 1.5% ± 0.9%; p < 0.0001; Figures 6I and 6N). The percentage of RFP+CK18+CoRL was further increased by enalapril compared with baseline (27.25% ± 6.08% versus 16.75% ± 2.36%; p < 0.0001; Figures 6J and 6N). In contrast, in RenWt1fl/fl mice, the percentage of RFP+CK18+CoRL did not increase (2.8% ± 3.5% versus 1.5% ± 0.8%; p = 0.606; Figures 6K, 6L, and 6N), and enalapril had no impact (3.7% ± 3.1% versus 2.8% ± 3.5% versus FSGS without enalapril; p > 0.99) (Figures 6K, 6M, and 6N).
Figure S6 confirms these results using additional mesenchymal and epithelial cell markers. The mesenchymal markers αSMA (Figures S6A–S6F) and smooth muscle protein 22 (SM22) (Figures S6G–S6L) were abundant in RFP+CoRL in the JGC at baseline in both strains (Figures S6A, S6D, S6G, and S6J). Co-staining for both mesenchymal markers decreased in RenWt1+/+ mice with FSGS (Figures S6B and S6H), and was further decreased by enalapril (Figures S6C and S6I). No RFP+CoRL in glomeruli co-expressed αSMA or SM22 (Figures 6A and 6C). In contrast, αSMA+/RFP+CoRL and SM22+/RFP+CoRL did not decrease in RenWt1fl/fl mice with FSGS, or with enalapril (Figures S6E, S6F, S6K, and S6L).
The epithelial cell marker E-cadherin was not detected in RFP+ cells in the JGC at baseline in either RenWt1+/+ or RenWt1fl/fl mice (Figures S6M and S6P). Following podocyte depletion in RenWt1+/+ mice, RFP+ cells in the JGC de novo expressed E-cadherin, which was augmented by enalapril (Figures S6N and S6O). In contrast, E-cadherin staining was not detected in the JGC in RenWt1fl/fl mice following podocyte depletion, and enalapril had no effect (Figures S6Q and S6R).
Following podocyte depletion in RenWt1+/+ mice, a subset of CoRL that migrated from the JGC (Figures S5A1–S5A4, S5B1–S5B4, and S5C1–S5C4) to the glomerular tuft no longer expressed αSMA (Figure S5A5–S5A8). There was de novo expression in CoRL in glomeruli of the epithelial markers E-cadherin (Figure S5B5–S5B8) and cytokeratin 18 (Figure S5C5–S5C8), coincident with loss of mesenchymal markers.
These results show that following abrupt podocyte depletion, mesenchymal markers (MYH11, αSMA, SM22) decrease in CoRL, coincident with de novo staining for the epithelial cell markers cytokeratin 18 and E-cadherin (Figure S6S). These events were augmented by enalapril in RenWt1+/+ mice, supporting the notion that CoRL undergo MET in RenWt1+/+ mice with FSGS, but these events are significantly reduced when WT1 is deleted.
Podocyte Replacement Is Lower in Diseased RenWt1fl/fl Mice
The binding of the podocyte-depleting antibody was similar in RenWt1+/+ and RenWt1fl/fl mice (Figures S2J and S2K). Podocyte density, measured by quantitating p57, was similar at baseline in RenWt1+/+ and RenWt1fl/fl mice (Figures 7A, 7B, and 7E). Podocyte density decreased in RenWt1+/+ mice with FSGS compared with baseline (185.2 ± 17.1 × 106 versus 250.7 ± 10.0 × 106 podocytes/μm3 glomerular tuft volume; p < 0.0001; Figures 7C and 7E). Moreover, podocyte density was significantly lower in diseased RenWt1fl/fl mice compared with diseased RenWt1+/+ mice (109.3 ± 10.09 × 106 versus 185.2 ± 17.1 × 106 podocytes/μm3; p < 0.001; Figures 7D and 7E). Similar findings were shown for podocin (Figures 7G–7J). Glomerulosclerosis increased in diseased RenWt1+/+ mice but was significantly higher in diseased RenWt1fl/fl mice (Figure 7F). The urinary albumin to creatinine ratio was also significantly higher in diseased RenWt1fl/fl mice (Figure 7K). In summary, podocyte density was lower in RenWt1fl/fl mice compared with RenWt1+/+ mice, accompanied by higher glomerulosclerosis and albuminuria.
Figure 7.

Podocyte Depletion in the Cytotoxic Anti-podocyte Antibody Model of FSGS Is Higher in Diseased RenWt1fl/fl Mice than in Diseased RenWt1+/+ Mice
(A–F) Podocytes are identified by p57 (brown, nuclear, arrows) and matrix (pink) in a baseline biopsy from RenWt1+/+ (A) and RenWt1fl/fl (B) mice, and a serial biopsy in the same mice at FSGS D28 (C and D). (E) Podocyte density was similar at baseline but lower in diseased RenWt1fl/fl compared with diseased RenWt1+l+mice. (F) Glomerulosclerosis is higher in diseased RenWt1fl/fl compared with diseased RenWt1+/+ mice.
(G–J) Podocin staining (green) is similar in healthy podocytes in baseline RenWt1+/+ (G) and RenWt1fl/fl (H) mice. Podocin decreases at FSGS D28 in RenWt1+/+ mice (I) and is more pronounced in diseased RenWt1fl/fl mice (J). Scale bars, 20 μm.
(K) Urinary albumin to creatinine ratio in RenWt1fl/fl mice (dark bars) is higher than RenWt1+/+ mice (light bars) (n = 13/group).
Discussion
Adult podocytes cannot proliferate and are unable to self-renew following depletion (Barisoni et al., 2000, Pavenstadt et al., 2003). Their replacement in glomerular diseases is reliant on progenitors (Romagnani et al., 2013, Shankland et al., 2014). CoRL plasticity has been reported in podocytes, PECs, kidney pericytes, and mesangial cells (Gomez et al., 2014, Pippin et al., 2015, Starke et al., 2015, Kaverina et al., 2016, Stefanska et al., 2016). To serve as progenitors for podocytes, CoRL must increase in number, migrate from the JGC to the glomerulus, and reprogram from a mesenchymal to an epithelial fate. To better understand the mechanisms underlying these events, we selectively genetically deleted WT1 in CoRL in transgenic mice. Reducing podocytes with a cytopathic anti-podocyte antibody resulted in a significant reduction in CoRL proliferation, migration, and MET, suggesting a critical role for WT1.
We show that WT1 staining is detected in <5%–7% of renin-expressing cells in the JGC in normal mice and rats, and only occasionally in the JGC in normal human kidneys, similar to previous reports (Puelles et al., 2015). The first major result of the studies was that following podocyte depletion in three experimental models, WT1 increased up to 5.5-fold from baseline in renin-expressing cells in the JGC. WT1 also increased in renin-expressing cells in human FSGS and membranous nephropathy. WT1 did not increase following uninephrectomy, serving as a negative control because podocyte number was unchanged.
The role for WT1 is well described in podocyte health (Ozdemir and Hohenstein, 2014, Mazzei and Manucha, 2016), in non-kidney cell transdifferentiation (Ijpenberg et al., 2007), and progenitor function (Martinez-Estrada et al., 2010, Bandiera et al., 2013). To test the hypothesis that WT1 is necessary for CoRL to function as podocyte progenitors, we generated a transgenic mouse where WT1 was inducibly deleted specifically in CoRL. Under non-stressed conditions, deleting WT1 in CoRL had no impact on kidney or podocyte function and morphology.
CoRL migrate from the JGC to the glomerulus or tubulo-interstitium during kidney development and in disease (Sequeira Lopez et al., 2004, Pippin et al., 2013, Pippin et al., 2015, Starke et al., 2015, Zhang et al., 2015, Lichtnekert et al., 2016). Here, RFP+CoRL were rarely detected in glomeruli of non-diseased RenWt1+/+ and RenWt1fl/fl mice. However, RFP+CoRL were readily detected in glomeruli in RenWt1+/+ mice following podocyte depletion, similar to previous reports (Pippin et al., 2013, Lichtnekert et al., 2016). The second major finding was that following podocyte depletion, RFP+CoRL were barely detected in glomeruli of RenWt1fl/fl mice, and migration to the glomerulus was not augmented by enalapril in RenWt1fl/fl mice. These results are consistent with WT1 being necessary for CoRL migration and the enhanced migratory effects of enalapril. Similar results in the cancer field suggest that WT1 enhances cell invasion and migration (Jomgeow et al., 2006, Brett et al., 2013).
CoRL increase in the JGC following podocyte depletion (Pippin et al., 2013, Pippin et al., 2014), increased glomerular volume (Hodgin et al., 2015), hypotension (Castellanos Rivera et al., 2011), and following angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (Lichtnekert et al., 2016). BrdU and Ki67 staining showed that following podocyte depletion in RenWt1+/+ mice, RFP+CoRL proliferated, which was augmented by enalapril (Duim et al., 2015, Lichtnekert et al., 2016). A third major finding was no increase in CoRL proliferation in RenWt1fl/fl mice, and enalapril had no effect. This was of interest, as several groups have shown that WT1 enhances proliferation of tumor cells and cardiac endothelial cells (Li et al., 2015, Wu et al., 2015).
Like podocytes (Boyle et al., 2008, Kobayashi et al., 2008), CoRL derive from mesenchymal origins (Sequeira Lopez et al., 2001, Matsushita et al., 2010, Wang et al., 2013, Ciampi et al., 2016). To become podocytes, CoRL need to decrease mesenchymal proteins and acquire epithelial and podocyte proteins. At baseline, RFP+CoRL in the JGC in both RenWt1+/+ and RenWt1fl/fl mice express mesenchymal markers (MYH11, αSMA, SM22) but not epithelial markers (cytokeratin 18, E-cadherin). Following podocyte depletion in RenWt1+/+ mice, RFP+CoRL in the JGC expressing these mesenchymal markers decreased significantly. This coincided with the de novo expression of cytokeratin 18 and E-cadherin. The MET-like changes in diseased RenWt1+/+ mice were augmented by enalapril. When in the glomerulus, a subset of CoRL co-expressed the podocyte markers p57, podocin, and synaptopodin. The fourth major finding was that following podocyte depletion in RenWt1fl/fl mice, mesenchymal markers in RFP+ CoRL did not decrease, nor did they begin to express epithelial markers, and enalapril had no impact.
These findings are consistent with our previous reports on the importance of disruption of MET underlying the origin of Wilms' tumors (Berry et al., 2015, Hohenstein et al., 2015), the role of Wt1 in this MET programming (Davies et al., 2004, Essafi et al., 2011), and that loss of WT1 blocks MET in the cap mesenchyme (Essafi et al., 2011).
The fifth major finding was that following abrupt podocyte loss, podocyte density remained lower in diseased RenWt1fl/fl compared with diseased RenWt1+/+mice, accompanied by more glomerular scarring and albuminuria. We recognize that CoRL transdifferentiation alone is not sufficient to explain the differences, and other factors including different kidney progenitor populations need to be considered.
Finally, our observation of intra-glomerular CoRL with coincident downregulation of renin expression would be consistent with the negative regulation previously described (Steege et al., 2008). It will be of interest to assess the specific isoform(s) of WT1 expressed and retained in CoRL and the kinetics of renin downregulation and other cell differentiation features altered as a function of this interaction.
In summary, WT1 expression in CoRL is important for the glomerular response to damage. When WT1 is selectively deleted in CoRL in the setting of podocyte loss, their proliferation and migration to the glomerulus, and to some extent their transdifferentiation toward a podocyte fate through MET changes, are markedly reduced.
Experimental Procedures
RenCreER tdTomato Wt1+/+and Wt1fl/fl Reporter Mice
Ren1cCreERxRs-tdTomato-R mice (Pippin et al., 2013) were crossed with a Wt1 conditional knockout mouse (Wt1fl/fl) (Martinez-Estrada et al., 2010). Wt1 was inactivated in RenCreER tdTomato Wt1fl/fl mice (abbreviated RenWt1 fl/fl) by administration of 100 mg/kg tamoxifen on four occasions. Mouse genotype was identified by PCR (Martinez-Estrada et al., 2010). Kidney biopsies were performed to assess CoRL labeling as described in Supplemental Experimental Procedures. RenWt1fl/fl and RenWt1+/+ were housed in the animal care facility of the University of Washington (UW) under specific pathogen-free conditions with food and water available ad libitum. These studies were reviewed and approved by the UW Institutional Animal Care and Use Committee (2968-04).
Experimental Models of Glomerular Disease Accompanied by Podocyte Depletion
The following experimental models of podocyte depletion were used:
-
(1)
A cytotoxic sheep anti-glomerular antibody model of experimental FSGS was induced in RenWt1fl/f and RenWt1+/+mice as previously described (Kaverina et al., 2016, Lichtnekert et al., 2016). On D3, when podocyte number was decreased by 30%–40%, mice were randomized into two groups: group 1 received drinking water; group 2 received the ACE inhibitor enalapril (75 mg/mL). Mice were killed on D28. Urine was collected at baseline, and on days 7, 14, and 28. Amersham Cell Proliferation Labeling Reagent (GE Healthcare Life Sciences, Little Chalfont, UK) was administered to quantitate cell proliferation (Kaverina et al., 2016). Urines were collected for albumin measurements by radial immunodiffusion assay (RID) (Marshall et al., 2011); urine creatinine was measured by colorimetric micro-plate assay (Cayman Chemical Company, Ann Arbor, MI).
-
(2)
Podocyte TGFβ-Receptor1 transgenic (PodTgfbr1) mice given doxycycline underwent TGFβR1-induced podocyte apoptosis and loss of 25% (D7) and 40% (D14), accompanied by glomerulosclerosis (Daehn et al., 2014).
-
(3)
A PHN model of membranous nephropathy induced as previously described (Ohse et al., 2010) underwent progressive podocyte depletion (Petermann et al., 2003).
-
(4)
A uninephrectomy model served as a negative control for podocyte depletion (Pippin et al., 2015).
Human Glomerular Disease
Kidney biopsies from patients with FSGS and membranous nephropathy were obtained from the University of Chicago (UC). The study protocol was approved by the UC Institutional Review Board. Further details are described in Supplemental Experimental Procedures.
Laser Capture Microscopy and qRT-PCR
RFP+CoRL were isolated by LCM, as described (Tretiakova and Hart, 2011), with the Leica Laser Microdissection Systems LMD6500 and LMD7000 (Leica Microsystems Inc., Buffalo Grove, IL).
Immunoperoxidase and Immunofluorescent Staining
Formalin-fixed paraffin-embedded mouse, rat, and human kidney sections or frozen tissue sections (4–20 μm thick) were used. Following standard antigen retrieval steps, Avidin-biotin based, polymer-based, and fluorochrome-based staining was performed as previously described (Kimura et al., 1995, Wagner et al., 2014). Further details are described in Supplemental Experimental Procedures.
Quantitative Analysis
Absolute numbers of renin+, RFP+, WT1+, and RFP+/WT1+ expressing cells were counted in the JGC of fluorescence-stained sections. Results were expressed as a percentage of co-localized RFP+/WT1+ cells per number of renin+ cells. The percentage of proliferating (BrdU+) CoRL co-expressing WT1 in the JGC was measured by dividing the number of RFP+/WT1+/BrdU+-stained cells by the total number of RFP+ cells in the JGC. The total number of glomeruli and glomeruli with RFP+ cells were counted to determine the percentage of glomeruli with RFP+ cells. Absolute numbers of RFP+-, MYH11+, CK18+, MYH11+/RFP+, and CK18+/RFP+-expressing cells were counted in the JGC. Results were expressed as a percentage of MYH11+/RFP+ and CK18+/RFP+ cells per number of RFP+ cells in the JGC. Podocyte number was measured on 150 ± 20 glomeruli per animal on p57/periodic acid-Schiff-stained sections (Zhang et al., 2015, Lichtnekert et al., 2016).
Statistical Analysis
Groups were compared using a one-way or two-way ANOVA for multiple comparisons with Bonferroni post hoc analysis with significant set at p < 0.05. Data are presented as means ± SEM. All data were analyzed in GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA).
Published: September 28, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.08.020.
Supplemental Information
References
- Bandiera R., Vidal V.P., Motamedi F.J., Clarkson M., Sahut-Barnola I., von Gise A., Pu W.T., Hohenstein P., Martinez A., Schedl A. WT1 maintains adrenal-gonadal primordium identity and marks a population of AGP-like progenitors within the adrenal gland. Dev. Cell. 2013;27:5–18. doi: 10.1016/j.devcel.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisoni L., Mokrzycki M., Sablay L., Nagata M., Yamase H., Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. 2000;58:137–143. doi: 10.1046/j.1523-1755.2000.00149.x. [DOI] [PubMed] [Google Scholar]
- Berry R.L., Ozdemir D.D., Aronow B., Lindstrom N.O., Dudnakova T., Thornburn A., Perry P., Baldock R., Armit C., Joshi A. Deducing the stage of origin of Wilms’ tumours from a developmental series of Wt1-mutant mice. Dis. Model. Mech. 2015;8:903–917. doi: 10.1242/dmm.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S., Misfeldt A., Chandler K.J., Deal K.K., Southard-Smith E.M., Mortlock D.P., Baldwin H.S., de Caestecker M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 2008;313:234–245. doi: 10.1016/j.ydbio.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett A., Pandey S., Fraizer G. The Wilms’ tumor gene (WT1) regulates E-cadherin expression and migration of prostate cancer cells. Mol. Cancer. 2013;12:1–28. doi: 10.1186/1476-4598-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos Rivera R.M., Monteagudo M.C., Pentz E.S., Glenn S.T., Gross K.W., Carretero O., Sequeira-Lopez M.L., Gomez R.A. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol. Genomics. 2011;43:1021–1028. doi: 10.1152/physiolgenomics.00061.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau Y.Y., Brownstein D., Mjoseng H., Lee W.C., Buza-Vidas N., Nerlov C., Jacobsen S.E., Perry P., Berry R., Thornburn A. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet. 2011;7:e1002404. doi: 10.1371/journal.pgen.1002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi O., Iacone R., Longaretti L., Benedetti V., Graf M., Magnone M.C., Patsch C., Xinaris C., Remuzzi G., Benigni A., Tomasoni S. Generation of functional podocytes from human induced pluripotent stem cells. Stem Cell Res. 2016;17:130–139. doi: 10.1016/j.scr.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini F., Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell. 2010;18:698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daehn I., Casalena G., Zhang T., Shi S., Fenninger F., Barasch N., Yu L., D'Agati V., Schlondorff D., Kriz W. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Invest. 2014;124:1608–1621. doi: 10.1172/JCI71195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.A., Ladomery M., Hohenstein P., Michael L., Shafe A., Spraggon L., Hastie N. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum. Mol. Genet. 2004;13:235–246. doi: 10.1093/hmg/ddh015. [DOI] [PubMed] [Google Scholar]
- DeFilippis M., Wagner K.D. Management of treatment-resistant depression in children and adolescents. Paediatr. Drugs. 2014;16:353–361. doi: 10.1007/s40272-014-0088-y. [DOI] [PubMed] [Google Scholar]
- Duim S.N., Kurakula K., Goumans M.J., Kruithof B.P. Cardiac endothelial cells express Wilms’ tumor-1: Wt1 expression in the developing, adult and infarcted heart. J. Mol. Cell. Cardiol. 2015;81:127–135. doi: 10.1016/j.yjmcc.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Eng D.G., Sunseri M.W., Kaverina N.V., Roeder S.S., Pippin J.W., Shankland S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015;88:999–1012. doi: 10.1038/ki.2015.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essafi A., Webb A., Berry R.L., Slight J., Burn S.F., Spraggon L., Velecela V., Martinez-Estrada O.M., Wiltshire J.H., Roberts S.G. A wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell. 2011;21:559–574. doi: 10.1016/j.devcel.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic N., Vukojevic K., Bocina I., Saraga M., Durdov M.G., Kablar B., Saraga-Babic M. Immunohistochemical and electron microscopic features of mesenchymal-to-epithelial transition in human developing, postnatal and nephrotic podocytes. Histochem. Cell Biol. 2017;147:481–495. doi: 10.1007/s00418-016-1507-7. [DOI] [PubMed] [Google Scholar]
- Gao F., Maiti S., Sun G., Ordonez N.G., Udtha M., Deng J.M., Behringer R.R., Huff V. The Wt1+/R394W mouse displays glomerulosclerosis and early-onset renal failure characteristic of human Denys-Drash syndrome. Mol. Cell. Biol. 2004;24:9899–9910. doi: 10.1128/MCB.24.22.9899-9910.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R.A., Belyea B., Medrano S., Pentz E.S., Sequeira-Lopez M.L. Fate and plasticity of renin precursors in development and disease. Pediatr. Nephrol. 2014;29:721–726. doi: 10.1007/s00467-013-2688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahammer F., Wanner N., Huber T.B. Podocyte regeneration: who can become a podocyte? Am. J. Pathol. 2013;183:333–335. doi: 10.1016/j.ajpath.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Graziano A.C., Cardile V., Avola R., Vicario N., Parenti C., Salvatorelli L., Magro G., Parenti R. Wilms’ tumor gene 1 silencing inhibits proliferation of human osteosarcoma MG-63 cell line by cell cycle arrest and apoptosis activation. Oncotarget. 2017;8:13917–13931. doi: 10.18632/oncotarget.14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgin J.B., Bitzer M., Wickman L., Afshinnia F., Wang S.Q., O'Connor C., Yang Y., Meadowbrooke C., Chowdhury M., Kikuchi M. Glomerular aging and focal global glomerulosclerosis: a podometric perspective. J. Am. Soc. Nephrol. 2015;26:3162–3178. doi: 10.1681/ASN.2014080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein P., Pritchard-Jones K., Charlton J. The yin and yang of kidney development and Wilms’ tumors. Genes Dev. 2015;29:467–482. doi: 10.1101/gad.256396.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijpenberg A., Perez-Pomares J.M., Guadix J.A., Carmona R., Portillo-Sanchez V., Macias D., Hohenstein P., Miles C.M., Hastie N.D., Munoz-Chapuli R. Wt1 and retinoic acid signaling are essential for stellate cell development and liver morphogenesis. Dev. Biol. 2007;312:157–170. doi: 10.1016/j.ydbio.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Jomgeow T., Oji Y., Tsuji N., Ikeda Y., Ito K., Tsuda A., Nakazawa T., Tatsumi N., Sakaguchi N., Takashima S. Wilms’ tumor gene WT1 17AA(-)/KTS(-) isoform induces morphological changes and promotes cell migration and invasion in vitro. Cancer Sci. 2006;97:259–270. doi: 10.1111/j.1349-7006.2006.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M., Ettou S., Jung Y.L., Lenz M.O., Taglienti M.E., Park P.J., Schermer B., Benzing T., Kreidberg J.A. Genome-wide analysis of Wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J. Am. Soc. Nephrol. 2015;26:2097–2104. doi: 10.1681/ASN.2014090940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S., Surolia R., Hock T.D., Guroji P., Zolak J.S., Duggal R., Ye T., Thannickal V.J., Antony V.B. Wilms’ tumor 1 (Wt1) regulates pleural mesothelial cell plasticity and transition into myofibroblasts in idiopathic pulmonary fibrosis. FASEB J. 2014;28:1122–1131. doi: 10.1096/fj.13-236828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina N.V., Eng D.G., Schneider R.R., Pippin J.W., Shankland S.J. Partial podocyte replenishment in experimental FSGS derives from nonpodocyte sources. Am. J. Physiol. Renal Physiol. 2016;310:F1397–F1413. doi: 10.1152/ajprenal.00369.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K., Nagai R., Sakai T., Aikawa M., Kuro-o M., Kobayashi N., Shirato I., Inagami T., Oshi M., Suzuki N. Diversity and variability of smooth muscle phenotypes of renal arterioles as revealed by myosin isoform expression. Kidney Int. 1995;48:372–382. doi: 10.1038/ki.1995.305. [DOI] [PubMed] [Google Scholar]
- Kobayashi A., Valerius M.T., Mugford J.W., Carroll T.J., Self M., Oliver G., McMahon A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppe C., Grone H.J., Ostendorf T., van Kuppevelt T.H., Boor P., Floege J., Smeets B., Moeller M.J. Common histological patterns in glomerular epithelial cells in secondary focal segmental glomerulosclerosis. Kidney Int. 2015;88:990–998. doi: 10.1038/ki.2015.116. [DOI] [PubMed] [Google Scholar]
- Lasagni L., Lazzeri E., Shankland S.J., Anders H.J., Romagnani P. Podocyte mitosis - a catastrophe. Curr. Mol. Med. 2013;13:13–23. doi: 10.2174/1566524011307010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagni L., Angelotti M.L., Ronconi E., Lombardi D., Nardi S., Peired A., Becherucci F., Mazzinghi B., Sisti A., Romoli S. Podocyte regeneration driven by renal progenitors determines glomerular disease remission and can be pharmacologically enhanced. Stem Cell Reports. 2015;5:248–263. doi: 10.1016/j.stemcr.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzeri E., Romagnani P. Podocyte biology: differentiation of parietal epithelial cells into podocytes. Nat. Rev. Nephrol. 2015;11:7–8. doi: 10.1038/nrneph.2014.218. [DOI] [PubMed] [Google Scholar]
- Li X., Ottosson S., Wang S., Jernberg E., Boldrup L., Gu X., Nylander K., Li A. Wilms’ tumor gene 1 regulates p63 and promotes cell proliferation in squamous cell carcinoma of the head and neck. BMC Cancer. 2015;15:342. doi: 10.1186/s12885-015-1356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtnekert J., Kaverina N.V., Eng D.G., Gross K.W., Kutz J.N., Pippin J.W., Shankland S.J. Renin-angiotensin-aldosterone system inhibition increases podocyte derivation from cells of renin lineage. J. Am. Soc. Nephrol. 2016;27:3611–3627. doi: 10.1681/ASN.2015080877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little M.H., McMahon A.P. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb. Perspect. Biol. 2012;4:1–46. doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.B., Krofft R.D., Blonski M.J., Kowalewska J., Logar C.M., Pippin J.W., Kim F., Feil R., Alpers C.E., Shankland S.J. Role of smooth muscle protein SM22alpha in glomerular epithelial cell injury. Am. J. Physiol. Renal Physiol. 2011;300:F1026–F1042. doi: 10.1152/ajprenal.00187.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Estrada O.M., Lettice L.A., Essafi A., Guadix J.A., Slight J., Velecela V., Hall E., Reichmann J., Devenney P.S., Hohenstein P. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010;42:89–93. doi: 10.1038/ng.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K., Morello F., Wu Y., Zhang L., Iwanaga S., Pratt R.E., Dzau V.J. Mesenchymal stem cells differentiate into renin-producing juxtaglomerular (JG)-like cells under the control of liver X receptor-alpha. J. Biol. Chem. 2010;285:11974–11982. doi: 10.1074/jbc.M109.099671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzei L., Manucha W. Growing evidence suggests WT1 effects in the kidney development are modulated by Hsp70/NO interaction. J. Nephrol. 2016;1:1–18. doi: 10.1007/s40620-016-0302-9. [DOI] [PubMed] [Google Scholar]
- Miller-Hodges E., Hohenstein P. WT1 in disease: shifting the epithelial-mesenchymal balance. J. Pathol. 2012;226:229–240. doi: 10.1002/path.2977. [DOI] [PubMed] [Google Scholar]
- Ohse T., Vaughan M.R., Kopp J.B., Krofft R.D., Marshall C.B., Chang A.M., Hudkins K.L., Alpers C.E., Pippin J.W., Shankland S.J. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am. J. Physiol. Renal Physiol. 2010;298:F702–F711. doi: 10.1152/ajprenal.00428.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir D.D., Hohenstein P. Wt1 in the kidney–a tale in mouse models. Pediatr. Nephrol. 2014;29:687–693. doi: 10.1007/s00467-013-2673-7. [DOI] [PubMed] [Google Scholar]
- Pavenstadt H., Kriz W., Kretzler M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Pelletier J., Bruening W., Kashtan C.E., Mauer S.M., Manivel J.C., Striegel J.E., Houghton D.C., Junien C., Habib R., Fouser L. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- Petermann A.T., Krofft R., Blonski M., Hiromura K., Vaughn M., Pichler R., Griffin S., Wada T., Pippin J., Durvasula R., Shankland S.J. Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 2003;64:1222–1231. doi: 10.1046/j.1523-1755.2003.00217.x. [DOI] [PubMed] [Google Scholar]
- Pippin J.W., Sparks M.A., Glenn S.T., Buitrago S., Coffman T.M., Duffield J.S., Gross K.W., Shankland S.J. Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am. J. Pathol. 2013;183:542–557. doi: 10.1016/j.ajpath.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippin J.W., Glenn S.T., Krofft R.D., Rusiniak M.E., Alpers C.E., Hudkins K., Duffield J.S., Gross K.W., Shankland S.J. Cells of renin lineage take on a podocyte phenotype in aging nephropathy. Am. J. Physiol. Renal Physiol. 2014;306:F1198–F1209. doi: 10.1152/ajprenal.00699.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippin J.W., Kaverina N.V., Eng D.G., Krofft R.D., Glenn S.T., Duffield J.S., Gross K.W., Shankland S.J. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am. J. Physiol. Renal Physiol. 2015;309:F341–F358. doi: 10.1152/ajprenal.00438.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles V.G., Douglas-Denton R.N., Cullen-McEwen L.A., Li J., Hughson M.D., Hoy W.E., Kerr P.G., Bertram J.F. Podocyte number in children and adults: associations with glomerular size and numbers of other glomerular resident cells. J. Am. Soc. Nephrol. 2015;26:2277–2288. doi: 10.1681/ASN.2014070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnani P. Parietal epithelial cells: their role in health and disease. Contrib. Nephrol. 2011;169:23–36. doi: 10.1159/000313943. [DOI] [PubMed] [Google Scholar]
- Romagnani P., Lasagni L., Remuzzi G. Renal progenitors: an evolutionary conserved strategy for kidney regeneration. Nat. Rev. Nephrol. 2013;9:137–146. doi: 10.1038/nrneph.2012.290. [DOI] [PubMed] [Google Scholar]
- Ronconi E., Sagrinati C., Angelotti M.L., Lazzeri E., Mazzinghi B., Ballerini L., Parente E., Becherucci F., Gacci M., Carini M. Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol. 2009;20:322–332. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira Lopez M.L., Pentz E.S., Robert B., Abrahamson D.R., Gomez R.A. Embryonic origin and lineage of juxtaglomerular cells. Am. J. Physiol. Renal Physiol. 2001;281:F345–F356. doi: 10.1152/ajprenal.2001.281.2.F345. [DOI] [PubMed] [Google Scholar]
- Sequeira Lopez M.L., Pentz E.S., Nomasa T., Smithies O., Gomez R.A. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev. Cell. 2004;6:719–728. doi: 10.1016/s1534-5807(04)00134-0. [DOI] [PubMed] [Google Scholar]
- Shankland S.J., Pippin J.W., Duffield J.S. Progenitor cells and podocyte regeneration. Semin. Nephrol. 2014;34:418–428. doi: 10.1016/j.semnephrol.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke C., Betz H., Hickmann L., Lachmann P., Neubauer B., Kopp J.B., Sequeira-Lopez M.L., Gomez R.A., Hohenstein B., Todorov V.T., Hugo C.P. Renin lineage cells repopulate the glomerular mesangium after injury. J. Am. Soc. Nephrol. 2015;26:48–54. doi: 10.1681/ASN.2014030265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steege A., Fahling M., Paliege A., Bondke A., Kirschner K.M., Martinka P., Kaps C., Patzak A., Persson P.B., Thiele B.J. Wilms’ tumor protein (-KTS) modulates renin gene transcription. Kidney Int. 2008;74:458–466. doi: 10.1038/ki.2008.194. [DOI] [PubMed] [Google Scholar]
- Stefanska A., Peault B., Mullins J.J. Renal pericytes: multifunctional cells of the kidneys. Pflugers Arch. 2013;465:767–773. doi: 10.1007/s00424-013-1263-7. [DOI] [PubMed] [Google Scholar]
- Stefanska A., Eng D., Kaverina N., Pippin J.W., Gross K.W., Duffield J.S., Shankland S.J. Cells of renin lineage express hypoxia inducible factor 2alpha following experimental ureteral obstruction. BMC Nephrol. 2016;17:1–29. doi: 10.1186/s12882-015-0216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma C. Glomerular disease: to the rescue–migrating renin lineage cells heal the injured glomerular mesangium. Nat. Rev. Nephrol. 2014;10:424. doi: 10.1038/nrneph.2014.115. [DOI] [PubMed] [Google Scholar]
- Tretiakova M., Hart J. Laser microdissection for gene expression study of hepatocellular carcinomas arising in cirrhotic and non-cirrhotic livers. Methods Mol. Biol. 2011;755:233–244. doi: 10.1007/978-1-61779-163-5_19. [DOI] [PubMed] [Google Scholar]
- Wagner N., Michiels J.F., Schedl A., Wagner K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: expression in tumour vessels in vivo. Oncogene. 2008;27:3662–3672. doi: 10.1038/sj.onc.1211044. [DOI] [PubMed] [Google Scholar]
- Wagner K.D., Cherfils-Vicini J., Hosen N., Hohenstein P., Gilson E., Hastie N.D., Michiels J.F., Wagner N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014;5:5852. doi: 10.1038/ncomms6852. [DOI] [PubMed] [Google Scholar]
- Wang H., Gomez J.A., Klein S., Zhang Z., Seidler B., Yang Y., Schmeckpeper J., Zhang L., Muramoto G.G., Chute J. Adult renal mesenchymal stem cell-like cells contribute to juxtaglomerular cell recruitment. J. Am. Soc. Nephrol. 2013;24:1263–1273. doi: 10.1681/ASN.2012060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner N., Hartleben B., Herbach N., Goedel M., Stickel N., Zeiser R., Walz G., Moeller M.J., Grahammer F., Huber T.B. Unraveling the role of podocyte turnover in glomerular aging and injury. J. Am. Soc. Nephrol. 2014;25:707–716. doi: 10.1681/ASN.2013050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Q., Wang Y., Tang J., Cheng C.Y., Liu Y.X. Sertoli cell Wt1 regulates peritubular myoid cell and fetal Leydig cell differentiation during fetal testis development. PLoS One. 2016;11:e0167920. doi: 10.1371/journal.pone.0167920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Wang S., Xu C., Tyler A., Li X., Andersson C., Oji Y., Sugiyama H., Chen Y., Li A. WT1 enhances proliferation and impedes apoptosis in KRAS mutant NSCLC via targeting cMyc. Cell Physiol. Biochem. 2015;35:647–662. doi: 10.1159/000369726. [DOI] [PubMed] [Google Scholar]
- Zhang J., Pippin J.W., Krofft R.D., Naito S., Liu Z.H., Shankland S.J. Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am. J. Physiol. Renal Physiol. 2013;304:F1375–F1389. doi: 10.1152/ajprenal.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Yanez D., Floege A., Lichtnekert J., Krofft R.D., Liu Z.H., Pippin J.W., Shankland S.J. ACE-inhibition increases podocyte number in experimental glomerular disease independent of proliferation. J. Renin Angiotensin Aldosterone Syst. 2015;16:234–248. doi: 10.1177/1470320314543910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.